

Abstract
Alternate DNA structures that deviate from B-form double-stranded DNA such as G-quadruplex (G4) DNA can be formed by sequences that are widely distributed throughout the human genome. G-quadruplex secondary structures, formed by the stacking of planar quartets composed of four guanines that interact by Hoogsteen hydrogen bonding, can affect cellular DNA replication and transcription, and influence genomic stability. The unique metabolism of G-rich chromosomal regions that potentially form quadruplexes may influence a number of biological processes including immunoglobulin gene rearrangements, promoter activation and telomere maintenance. A number of human diseases are characterized by telomere defects, and it is proposed that G-quadruplex structures which form at telomere ends play an important role in telomere stability. Evidence from cellular studies and model organisms suggests that diseases with known defects in G4 DNA helicases are likely to be perturbed in telomere maintenance and cellular DNA replication. In this minireview, we discuss the connections of G-quadruplex nucleic acids to human genetic diseases and cancer based on the recent literature.
Keywords: BLM, Bloom’s syndrome, FANCJ′, Fanconi anemia, G4 DNA, genomic instability, G-quadruplex, helicase, human disease, telomere, Werner syndrome, WRN
G-quadruplex DNA structures commonly referred to as G4 DNA have attracted considerable interest in recent years [1–3]. G4 DNA structures deviate from the canonical B-form Watson–Crick double helix by their Hoogsteen hydrogen bonding between four guanines which form a quartet, which stack to form the quadruplex. G-quadruplexes are stabilized by K+ or Na+ ions at physiological temperature and pH in vitro. The unconventional structure of G4 DNA has led researchers to believe that its unique properties are likely to impact cellular DNA metabolism. Despite evidence that G4 structures form readily in vitro under physiological conditions, the existence of G4 nucleic acid in living cells is still controversial. However, evidence suggesting that G-quadruplexes may form in vivo in G-rich regions of the human genome has cast a new light on the importance of studying G4 DNA metabolic processes. Several excellent reviews have covered various aspects of G4 structure and metabolism (see [4,5] and the accompanying minireviews in this series [6,7]). Aspects of telomere G-quadruplex structure and metabolism have also been discussed ([8] and the references cited therein), as well targeting telomeres and other DNA structures with G-quadruplex ligands [9,10]. The unique geometry of the G4 structure is thought to allow specific recognition by small molecules through various binding modes in a manner analogous to that of double-helical DNA intercalators. Several reviews have summarized the potential use of G4 DNA as a drug target [11–13], and are not discussed further here. We focus the discussion largely on the importance of G-quadruplexes in human disease and summarize evidence from the literature for the relevance of these alternate DNA structures in molecular and cellular pathways of DNA metabolism that influence genomic integrity. A distinguishing feature of this minireview is the strong emphasis on helicases that resolve G-quadruplex nucleic acids.
Existence of sequences predicted to form G4 structures in the human genome
Using algorithms to predict the probability of DNA sequences to form G-quadruplex structures, it has been estimated that there are over 300 000 potential quadruplex sequences (PQS) in the human genome [14,15]. However, PQS are not randomly distributed. G-rich sequences are enriched at the 5′-end of human genes (5′-untranslated region, first exon and first intron), but not the coding regions [16], consistent with the notion that PQS contributes to the regulation of gene expression [17]. The abundance of sequences in oncogenes predicted to form G4 structures may influence expression or contribute to genomic instability [18]. Oncogenes that promote cell proliferation and tumorigenesis contain significantly higher PQS than tumor suppressor genes that maintain chromosomal integrity [16].
The G-rich human immunoglobulin heavy-chain switch regions are targeted for class switch recombination to promote antibody diversity [19,20]. Interestingly, the post-transcriptional structure at the immunoglobulin switch region forms a G-loop, as detected by electron microscopy, suggesting a possible role of the G-rich element in DNA rearrangement in switch regions [21]. However, the belief that G-quadruplexes form at single-stranded G-rich loops at switch regions is controversial [22].
Human repeat elements and G-rich triplet repeat disorders
The human genome contains tandem repeat blocks known as microsatellites (1–9 bp) or minisatellites (10–100 bp) that may be prone to expansion or contraction. Such repeat elements contribute to genomic instability and have been implicated in a number of human diseases (for review, see [23]). Expansion of intragenic triplet repeats is observed in human neurodegenerative diseases including ataxias and Fragile X syndrome (FXS). FXS is one of the most common forms of inherited mental retardation [24], and is the result of expansion of a CGG trinucleotide repeat in the 5′-untranslated region of the FMR1 gene. When the CGG expansion repeats exceed 200 they become hypermethylated, causing transcriptional silencing of the FMR1 gene, resulting in the loss of its encoded protein FMRP, which is a selective RNA-binding protein implicated in regulating dendritic mRNA transport and local protein synthesis at synapses. Because the unstable trinucleotide repeats can form alternate DNA structures, it is proposed that such structures are responsible for the expanded CGG repeats that interfere with cellular nucleic acid processes.
Fry and Loeb first reported that FXS CGG repeats could form stable G4 DNA in monovalent cation solution [25], and this was further supported by NMR evidence [26] and results from DNA polymerase arrest assays [27]. CGG repeat sequences that can readily form G4 DNA structures in vitro can be unwound by specific G4 DNA helicases such as WRN [28] and FANCJ [29] that are mutated in the genetic diseases Werner syndrome (WS) and Fanconi anemia (FA) complementation group J, respectively (see ‘Helicase Disorders’ section). However, CGG repeats can also form hairpin structures [30], which might be responsible for repeat expansion in genomes [31]. The importance of G4 and other alternate DNA structures in the genomic instability of FXS and other triplet repeat disorders is an active area of investigation [23,32,33]. Further development of antibodies that specifically recognize sequence- or structure-specific G4 and other alternate DNA structures will help to advance the field’s understanding of the mechanism of repeat tract instability. Recently, a single-chain antibody selective for G-quadruplex DNA was developed and found to regulate gene expression in human cells in a manner that correlated with the predicted formation of G-quadruplex structures [34]. However, the existence of G4 structures in vivo is still a matter of debate and additional experimental evidence is needed to establish their existence and importance.
Instability of G-rich telomere structures and human disease
Telomeres are specialized nucleoprotein complexes that reside at the physical ends of linear eukaryotic chromosomes and exist in all vertebrates (for review, see [35,36]). In most organisms studied, telomeres contain lengthy stretches of tandem-repeated simple DNA sequences composed of a G-rich strand and a C-rich strand (called ‘terminal repeats’). These terminal repeats are highly conserved because all vertebrates appear to have the same telomeric DNA repeat of (TTAGGG)n for several kilobases.
The single-stranded G-rich 3′ overhang folds back and base pairs with the complementary sequence at the loop junction to form a t-loop structure. The area where the 3′ G-strand extension invades the duplex telomeric repeats is called a ‘D loop’ (displacement loop).
A growing number of proteins have been discovered that bind to the telomeric tandem DNA repeats, including the telomere protein complex known as shelterin and telomeric-associated factors, some of which have already been shown to play important roles in the maintenance of genomic stability and/or DNA repair [37]. Together, shelterin proteins and telomere-associated factors have important functions in the protection, replication and stabilization of the chromosome ends.
Two mechanisms to maintain telomere length have been described in human tumor cells [38,39]. The first requires a specialized enzyme telomerase which is able to copy as a reverse transcriptase the short TTAGGG motif at the 3′-end of telomeres. Telomerase activity in most human tissues is tightly regulated, leading to gradual telomere shortening with cell divisions [35]. Shortening beyond a critical length causes telomere uncapping, manifested by the activation of a DNA damage response and cell-cycle arrest. The second mechanism is observed in tumors (∼ 15%) as well as in immortalized cell lines lacking telomerase activity and involves recombination between telomeres, a mechanism known as alternative lengthening of telomeres (ALT).
Shorter telomere length is a risk factor for the development of cancer [40,41]. Clinical data revealed that telomere length is shorter in subjects with different types of cancer, including cancers of the head, neck, breast, bladder, prostate, lung and kidney [42]. In addition, telomerase is activated in ∼ 90% of cancer cells. However, a sizeable fraction of cancerous cells employ the ALT pathway to transfer tandem repeats between telomeres or with extrachromosomal telomere-repeat-containing sequences, resulting in more heterogeneous telomere lengths [43].
Although telomere structure and/or function appears to be defective in certain human diseases, the precise nature of the molecular defects at chromosome ends remains under active investigation. Different forms of anemia, hypertension, coronary heart diseases, chronic human immunodeficiency virus infection, ulcerative colitis and chronic liver disease are among the diseases with defects in telomerase or telomere stability [40,44]. Bone marrow failure syndromes represent a diverse group of diseases with similar phenotypes, including dyskeratosis congenita, aplastic anemia and myelodysplatic syndromes [45]. The fact that patients with bone marrow failure syndromes have shortened telomeres led researchers to screen these patients for mutations in telomerase and components of the shelterin complex. These efforts resulted in the identification of mutations in hTERT [46], hTERC [47], the telomerase-associated proteins dyskerin [46], NOP10 [48], NHP2 [49] and the shelterin component TINF2 [50,51]. These findings support the hypothesis that dysfunctional telomeres due to mutations in telomere maintenance genes promote exhaustion of the stem cell compartment and defects in cell types that have a high turnover rate such as the hematopoietic system.
Telomeric 3′ G-rich tails play an important role in the protection, replication and stabilization of chromosome ends
Human telomeric DNA can spontaneously assemble into a number of different G-quadruplex conformations, as demonstrated by NMR and X-ray crystallography [52–55] or atomic absorption spectroscopy, confirmed by fluorescence resonance energy transfer and CD spectral analysis [56]. However, the existence of G-quadruplex structures in vivo has been debated. Studies on the telomeres of ciliated protozoa have provided the most direct evidence that G4 DNA forms at telomeres in vivo [57], consistent with their ability to fold spontaneously into G4 DNA in vitro under physiological conditions [58,59]. Owing to the high concentration of telomeric sequences in the transcriptionally active macronucleus, ciliated protozoa were among the first model organisms characterized for telomere composition, structure and behavior. Demonstration that G4 DNA forms in vivo has proven difficult, although several studies do provide strong support for its existence at telomeres. The development of antibodies specific for telomeric G4 DNA structures enabled researchers to visualize antiparallel G4 structures by immunostaining in the model organism Stylonychia [60]. The staining of Stylonichia telomeres with the anti-G4 DNA serum was shown to depend on expression of the natural telomere end-binding proteins (TEBP)-α and TEBP-[61]. TEBP-is required for recruitment of TEBP-β to telomeres, and TEBP-β was observed to catalyze G4 DNA formation in vitro. Importantly, this work showed that the staining with the anti-G4 serum was not simply due to stabilization of the G4 fold by the antibody, but rather depended on the natural (i.e. G4) structure of the telomere in vivo. Another piece of supporting evidence that a G4 DNA structure can form at the telomere end in vivo was provided in a recent article by Zhang et al. which showed that the same amino acids found in a proposed divalent cation-binding motif of Saccharomyces cerevisiae Est1 protein are required for it to promote G4 DNA formation in vitro are required for the protein to stimulate telomere extension by telomerase [62], arguing that G4 DNA also forms at yeast telomeres and can play a positive role in extension of telomeres by telomerase.
If telomeres do indeed form G4 structures in vivo, it will be of interest to determine whether special G-quadruplex configurations at telomeres play a role in their stability in mammalian cells. Biochemical studies suggest that G-quadruplexes preferentially form at the 3′-end of telomeric DNA rather than internal positions [63]. The stable G4 structure may pose a challenge to the replication or DNA repair machinery, or the ability of telomerase to elongate the 3′ single-stranded tail assuming a G-quadruplex topology [64]. However, research from the Bryan laboratory has demonstrated that Tetrahymean thermophila telomerase can readily extend telomere sequences in vitro that are in a G4 configuration [65].
The notion that telomeric overhangs may form G-quadruplexes in cells is supported by indirect experimental strategies which are able to overcome challenges such as reduced telomere concentration and the potentially transient nature of G4 structures. Exposing human cells to G4 DNA-binding compounds such as telomestatin (TMS) induces the dissociation of shelterin proteins (e.g. POT1, TRF2) or telomere-associated proteins (e.g. TOPIII) from their telomeric sites [66–68]. TMS may compete with such proteins for binding to G4 DNA or stabilize a G4 structure that is not favorably bound by the telomere-interacting protein, leading to telomere uncapping in ALT cells [66,69]. TMS can also effectively reduce proliferation of telomerase positive tumor cells by inhibiting telomerase [68,70]. In future, small molecules that specifically bind G4 DNA structures may be useful for the treatment of cancer and other diseases.
Human proteins that bind G-rich DNA
Certain DNA helicases genetically linked to human diseases characterized by chromosomal instability have been shown to catalytically unwind G4 DNA substrates in vitro (Table 1). In addition, a number of human nuclear proteins have been reported to preferentially bind G-rich DNA that can form quadruplexes in vitro (Table 2). For example, the nuclear proteins poly[ADP-ribose]polymerase 1 (PARP-1), Ku70/Ku86 and heterogeneous nuclear ribonucleoprotein A1 all are associated with the G-rich promoter of the KRAS oncogene which is one of the most frequently mutated oncogenes in human cancer [71,72]. It is proposed that KRAS-associated proteins regulate its expression, and may represent targets for anti-cancer strategies to inhibit KRAS gene expression. Human nucleolin can bind G4 DNA with high affinity [73,74] and stabilize G4 DNA in the C-MYC promoter, thereby inhibiting C-MYC promoter-driven transcription as measured by luciferase assays [75].
Table 1.
Biochemical properties of G4 helicases and phenotypes of helicase deficiencies
| Protein | Organism | G4 DNA | G4 RNA | Phenotype | Ref. |
|---|---|---|---|---|---|
| WRN | Human | 3′→5′ a | ND b | Premature aging, genomic instability | [28,107,115] |
| BLM | Human | 3′→5′ | ND | Cancer, elevated sister-chromatid exchanges, genomic instability |
[114,154] |
| FANCJ | Human | 5′→3′ | ND | Fanconi anemia, breast cancer, defective interstrand cross-link repair and slow S phase progression |
[29,98,104] |
| G4R1/RHAU | Human | Yes c | Yes c | Abnormal mRNA deadenylation and decay |
[139–141] |
| Pif1 | Human | ND | ND | Possible telomere defects | [152,155] |
| Yeast | 5′→3′ | ND | Mitochondrial DNA and G-rich DNA instability, longer telomeres, defects in Okazaki fragment metabolism |
[151,153,156,157] | |
| Dna2 | Human | 5′→3′ | ND | Cell-cycle delay and aberrant cell division, genomic and mitochondrial DNA instability |
[158,159] |
| Yeast | 5′→3′ | ND | Defects in Okazaki fragment metabolism | [149,160,161] | |
| Sgs1 | Yeast | 3′→5′ | ND | Defective double-strand break repair, defective restart of stalled replication forks, inappropriate processing of meiotic recombination intermediates, telomere instability |
[162,163] |
Table 2.
Human proteins that interact with G-quadruplex DNA in vitro. hnRNP, heterogeneous nuclear ribonucleoprotein A1
| Protein | Function | Ref. |
|---|---|---|
| Topo I | Promotes G-quadruplex formation and binds G-quadruplex |
[164] |
| GQN1 (Nuclease) | Cleaves G-quadruplex specifically | [165] |
| hnRNP A1/UP1 | Binds and unfolds G-quadruplex (e.g. G-quadruplex in KRAS promoter) |
[166–169] |
| hnRNP D/BD2 | Unfolds human telomeric G-quadruplex | [170] |
| Pot1 | Disrupts telomeric G-quadruplex | [81] |
| Insulin and insulin-like growth factor-2 (IGF-2) |
Binds G-quadruplex formed in human insulin gene promoter |
[171,172] |
| Nucleolin | Binds and stabilizes G-quadruplex formed in c-MYC promoter |
[75] |
| MutSα (MSH2/MSH6) | Binds G-quadruplex | [29,79] |
| RPA | Binds and unfolds human telomeric intramolecular G-quadruplex, but not intermolecular G-quadruplex |
[173,174] |
An example of a human nuclear DNA repair protein that preferentially binds G-rich repetitive DNA is the MutSα heterodimer (MSH2/MSH6) [21]. Association of MutSα with the G-rich repetitive switch regions was demonstrated by chromatin immunoprecipitation assay [21]. Deletion of Msh2 or Msh6 in mice resulted in decreased switch recombination and reduced heterogeneity of switch junctions [76–78]. MutSα was also shown to bind the G-loop of transcribed S regions, as detected by electron microscopy, further suggesting a potential role of the MSH2/MSH6 protein complex in PQS metabolism of G-rich elements [79].
Understanding the biological importance of preferential G-rich DNA binding by the MutSα complex and other protein factors (Table 2) is an active area of investigation. Whereas some G-rich proteins stabilize the quadruplex structure, other proteins can destabilize certain G4 structures in vitro in an ATP-dependent manner (helicases, discussed below) or ATP-independent manner (e.g. replication protein A; RPA) [80]. The human shelterin protein POT1 has been shown to disrupt telomeric G-quadruplexes allowing telomerase extension in vitro [81].
Supporting evidence that certain genetic helicase disorders have defects in G-quadruplex DNA metabolism
In the previous sections, we highlighted some diseases with G-rich repeat tract instability or telomere defects that may arise from the instability or improper metabolism of G-quadruplex structures. It is plausible that G4 structures in certain regions of the genome contribute to poor maintenance of genomic stability characteristic of DNA repair disorders or certain cancers. The chromosomal instability disorders WS, Bloom’s syndrome (BS) and FACGJ are all characterized by autosomal recessive mutations in genes encoding DNA helicases that unwind G-quadruplex DNA in vitro (Table 1). We briefly discuss each of these disorders, highlighting evidence that defects in G4 metabolism may contribute to the cellular phenotypes of individuals with these genetic helicase diseases.
Fanconi anemia complementation group J
FA is a genetic disease characterized by bone marrow failure and a predisposition to cancer [82–84]. Some FA patients also show congenital abnormalities, growth and endocrine abnormalities, infertility and hematologic manifestations [82]. The incidence of FA has been estimated at 1–5 per million, with a carrier frequency of 1 in 300. The cellular characteristics of FA are chromosome instability and hypersensitivity to genotoxic stress by DNA interstrand cross-linking (ICL) agents such as cisplatin and mitomycin C.
FA results from mutations in one or more of 13 complementation genes (FA-A, -B, -C, -D1, -D2, -E, -F, -G, -I, -J, -L, -M and -N). One of the more recently identified FA genes implicated in FA complementation group J is FANCJ [85–87]. FANCJ was originally designated BACH1 (BRCA1 associated C-terminal helicase), a protein that binds to the BRCT repeats of the tumor suppressor BRCA1 [88]. Like other FA mutant cell lines, FANCJ-deficient cells are sensitive to ICL agents as evidenced by their reduced cell viability and accumulation of 4N DNA content, representing cells arrested in either late S or G2/M [85,87]. Following cellular exposure to ICL agents, FA-J mutant cells are characterized by elevated chromosomal aberrations such as chromosome breaks, quadriradials and triradials [87,89].
Although FANCJ is proposed to function downstream of FANCD2 monoubiquitination in the FA pathway, the precise role(s) of FANCJ in cross-link repair or the replicational stress response remains poorly understood. The demonstration that two FANCJ mutations detected in women with early-onset breast cancer result in the synthesis of enzymatically defective proteins suggests that FANCJ is a breast cancer tumor suppressor gene [90]. Notably, the 2533C→T nonsense mutation in exon 17, resulting in a premature stop codon (R798X) has been reported in a high percentage of FA-J patients, as well as in breast cancer patients [91]. Several missense mutations have been reported in FANCJ that may be associated with inherited breast cancer cases [91–97].
Evidence from genetic studies of Caenorhabditis elegans suggests that a FANCJ ortholog prevents genomic instability in G-rich tracts [98]. We summarize some of the key findings from this and related work, which set the stage for recent studies suggesting that human FANCJ resolves PQS to maintain chromosomal stability.
Biology of FANCJ-like proteins suggest a role in the metabolism of G-rich DNA structures
Convincing evidence for a defect in the metabolism of G-rich sequences in vivo was provided by the Lansdorp group who characterized the genomic instability of the C. elegans dog-1 mutant, an ortholog of mammalian FANCJ. Nematodes mutated in dog-1 showed germline as well as somatic deletions in genes containing polyguanine tracts [99]. In 2008, the Boulton laboratory further characterized the genomic instability of dog-1 mutants and reported that mutations in other FA genes of the worm do not interfere with G/C tract maintenance [100]. In the same year, the Tijsterman laboratory also reported massive genome rearrangements at G4 DNA sites in dog-1 animals [101]. Taken together, the results in C. elegans strongly suggest that DOG-1 plays an important role in maintaining genetic integrity by preventing the accumulation of aberrant DNA structures that form in G-rich tracts. However, there is as yet no biochemical evidence that DOG-1 is a helicase or has the ability to resolve G-quadruplex structures.
Evidence that DOG-1 plays a critical role in genome maintenance through its action at G-rich loci led researchers to search for mammalian orthologs of DOG-1. A gene named rtel that encodes a protein sharing sequence homology with DOG-1 was first identified by genomic mapping of loci that control telomere length differences between Mus musculus and Mus spretus [102]. rtel knockout mice were embryonic lethal and cells derived from these mice exhibited a rapid reduction in proliferative capacity upon differentiation, accompanied by an increased incidence of chromosomal abnormalities and telomere loss [102]. Interestingly, rtel mutant worms do not display G-rich sequence deletions, suggesting that RTEL and DOG-1 have distinct functions and operate in different pathways [103]. rtel-1 mutant worms and RTEL1-depleted human cells share phenotypes characteristic of yeast srs2 mutants: lethality upon deletion of the sgs1/BLM homolog, hyperrecombination, and DNA damage sensitivity [103].
In vitro, purified human RTEL1 antagonizes homologous recombination by promoting the disassembly of D loop recombination intermediates in a reaction dependent upon ATP hydrolysis [103]. However, RTEL1 does not dismantle RAD51 protein filaments. Taken together, these results suggest that RTEL not only regulates telomere metabolic events at the chromosome end, but may also act as an anti-recombinase to dismantle recombination intermediates and allow completion of repair by homologous recombination. Although RTEL shares sequence homology with FANCJ, whether mammalian RTEL could resolve G-quadruplex structures is not known. Clearly, biochemical studies of DOG-1, RTEL and other proteins sharing sequence homology with FANCJ [98] should be informative to understand their mechanism(s) of action.
Evidence that human FANCJ unwinds G4 DNA in vitro and has a role in the metabolism of G-rich sequences in cells
Human FANCJ was demonstrated to efficiently unwind G-quadruplex DNA substrates flanked by a 5′ single-stranded (ss)DNA tail in an ATP hydrolysis-dependent manner in vitro [29], and shown to resolve a variety of G4 DNA structures composed of human genomic DNA sequence such as those observed in telomeres and Fragile X triplet repeats [29,104]. Unwinding of G4 DNA by FANCJ in vitro is stimulated by the human single-stranded DNA binding protein replication protein A (RPA), but is inhibited by MutSα, a mismatch repair protein complex that binds G-rich repetitive DNA with high affinity [29]. FANCJ unwinding of G4 DNA structures but not duplex DNA is also inhibited by TMS, a G4-specific DNA binding compound [29]. FANCJ-depleted human cells are sensitive to TMS, and show elevated DNA damage and apoptosis upon exposure to the drug [29]. Double-strand breaks marked by γ-H2AX foci were observed to accumulate in FANCJ-depleted cells exposed to TMS, suggesting that FANCJ prevents replication-associated DNA damage by removing G-quadruplex structures which can no longer be bound with high affinity by TMS. The role of FANCJ in the metabolism of PQS appears to be unique among the FA proteins because neither FA-D2 nor FA-A mutant cells are hypersensitive to TMS [29]. The fact that FA-D2 and FA-A cells are not sensitive to TMS suggests that FANCJ is operative in at least these other FA mutant cell lines, and so G4 DNA would not be predicted to be increased. The strong co-localization of FANCJ with RPA after nucleotide depletion by hydroxyurea exposure [105] suggests that the two proteins collaborate in a situation of replicational stress that might arise when the replication fork is impeded by an obstacle such as a PQS.
It has been reported that FANCJ helicase activity is manifest in the S phase and is required for normal S-phase progression [106], suggesting that FANCJ removes structural barriers to replication. Indeed, cells that are deficient in FANCJ accumulate deletions at genomic sequences with a G4 DNA signature [104]. Comparative genome hybridization was performed on genomic DNA extracted from FA-J patient cell lines encoding a truncated FANCJ protein, and human FA-D2 cell line or normal human fetal fibroblast cells. From the genomic deletions identified as unique to FA-J cells, a bias toward breakpoint regions adjacent to DNA sequences with a G4 DNA signature was identified. This bias was most pronounced among larger deletions (∼ 36 kb or more), where nearly 75% possess sequences with the G4 signature in the vicinity of the breakpoint [104]. The results suggest that FANCJ-deficient cells accumulate genomic deletions in the vicinity of sequences with the G4 signature. A model is suggested that in FANCJ-deficient cells the replication fork stalls at a G-rich region that has a propensity to form G4 DNA. Conceptually, because FANCJ is not present to unwind the G4 DNA, the accumulation of stalled replication forks lead to chromosomal instability.
RecQ helicase disorders Werner syndrome and Bloom’s syndrome
WS and BS are autosomal recessive diseases with mutations in the RecQ helicase genes WRN and BLM, respectively [107,108]. Both diseases are characterized by chromosomal instability. WS is a premature aging disorder that displays many clinical symptoms of age-associated diseases such as cancer, atherosclerotic cardiovascular disease, diabetes mellitus (type II) and osteoporosis [109]. Unlike normal individuals, WS patients are highly susceptible to early onset of sarcomas and mesenchymal tumors. The genomic instability of WS has been described as variegated translocation mosaicism punctuated by chromosomal deletions and rearrangements as well as elevated spontaneous mutations [110,111].
In contrast to WS, the hallmark of BS is elevated sister-chromatid exchange [112]. BS is associated with a very high incidence of different types of cancers, solid tumors and leukemia, as well as sarcoma (e.g. osteosarcomas). BS is characterized by skin disorders, proportional dwarfism, immunodeficiency and male sterility [113]. Both BS and WS cells are hypersensitive to various agents that induce DNA damage or interfere with replication, including topoisomerase I inhibitors or DNA ICL agents. BS and WS cells also progress more slowly through the S phase than wild-type cells, suggesting that cell-cycle or replication checkpoints may be activated in these cells, even in the absence of cellular stress. Although both WRN and BLM encode DNA helicases, the precise roles of these enzymes to confer genomic stability and insure proper cellular DNA replication are still not well understood.
Preferential unwinding of G-quadruplex DNA structures in vitro by the WRN and BLM helicases
Only a limited number of DNA helicases have been shown to unwind G4 structures (Table 1). Among these, WRN and BLM helicases have been found to efficiently unwind a variety of G4 DNA structures in vitro, including those composed of G-rich sequences found in the human genome [114,115]. The ability of BLM to preferentially unwind G4 DNA relative to HJ substrates reflects an increased G4 binding affinity that is mediated by the RecQ C-terminal (RQC) domain located just after the helicase core domain [116]. However, an RQC motif is not universally required for G4 unwinding activity because the Superfamily 2 helicase FANCJ lacks a recognizable RQC domain but efficiently unwinds G-quadruplex structures [29,104]. Moreover, human RECQ1, which contains a sequence resembling the RQC motif in WRN and BLM, fails to unwind G-quadruplexes despite its efficient ability to unwind duplex and three-stranded D-loop structures [29,117]. This suggests that not all RQC domains are the same. Some (like those in Sgs1, WRN and BLM) can bind G-rich DNA that forms G4 DNA structures in vitro, whereas the RQC domain in RECQ1 may not be able to. This view is consistent with the fact that the RQC domain from any given protein generally binds more than one nucleic acid structure [116,118].
Directionality of unwinding distinguishes the action of WRN and BLM helicases from FANCJ on G-quadruplex DNA structures
One distinction between FANCJ and the human RecQ helicases (BLM, WRN) that unwind G4 structures is the requirement by FANCJ for a 5′ ssDNA tail in the G4 substrate [29,104], whereas WRN and BLM require a 3′ ssDNA tail to unwind at least some G4 substrates [114,115] (Fig. 1), with the possible exception of a d(CGG) Fragile X triplet repeat G4 DNA substrate in which a structure with a 5′ tail was unwound by WRN [28]. The tail requirements likely reflect the directionality of DNA unwinding because FANCJ has been shown to unwind duplex DNA substrates 5′ to 3′ with respect to the strand that it is presumed to translocate, whereas WRN and BLM unwind with a 3′ to 5′ directionality.
Fig. 1.
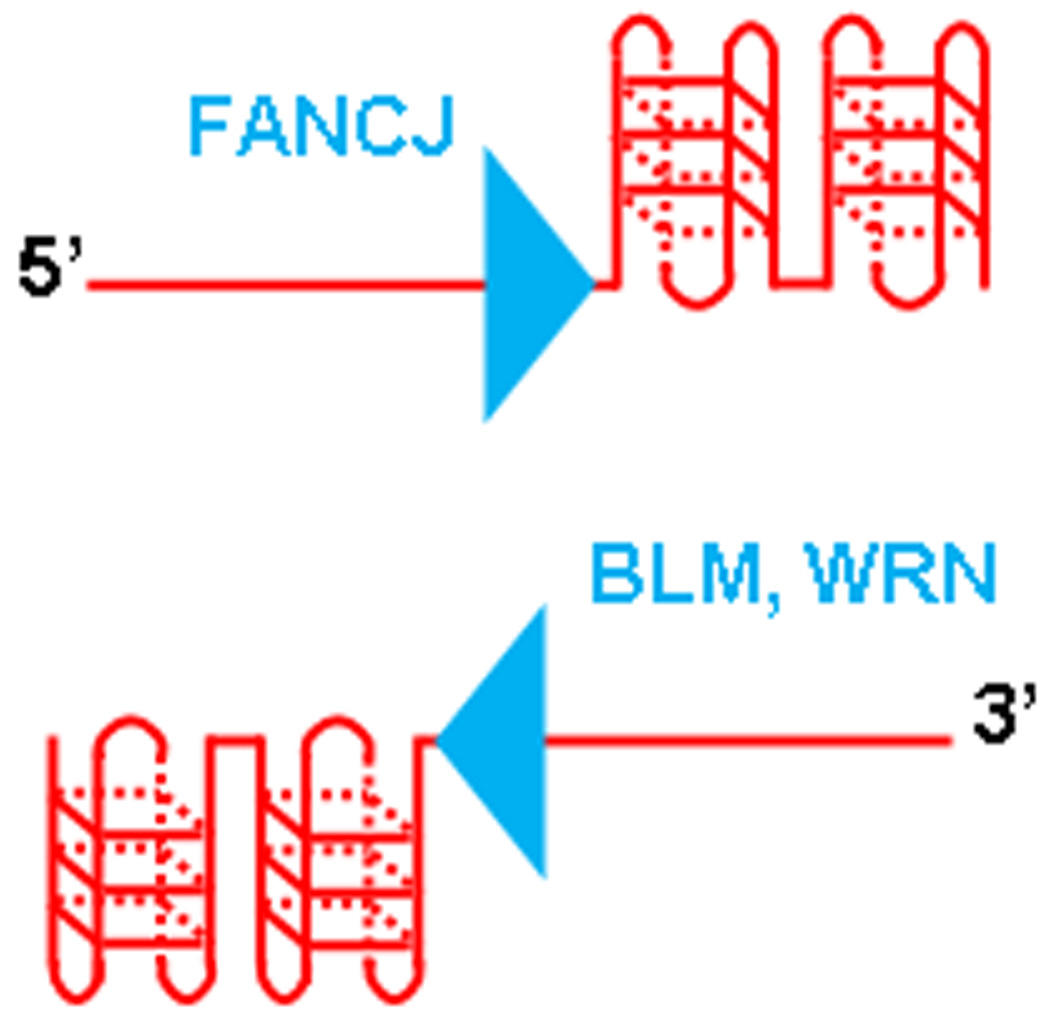
Disease-causing mutations in the FANCJ or WRN and BLM helicases affect G4 DNA unwinding enzymes with opposite polarities of action. The FANCJ helicase mutated in FA complementation group J requires a 5′ ssDNA tail to unwind the adjacent G4 structure. The WRN and BLM helicases mutated in Werner syndrome and Bloom’s syndrome, respectively, require a 3′ ssDNA tail to unwind the adjacent G4 structure.
The interaction between WRN helicase and DNA polymerase δ specifically facilitates DNA synthesis through quadruplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence [119], suggesting a role of WRN helicase for enabling replication progression through genomic DNA roadblocks such as secondary structure. In this regard, WRN (and BLM) seem better suited to operate in concert with a DNA polymerase synthesizing nascent DNA complementary to the template when a single-stranded loading site 3′ to the G-quadruplex is provided. The same directionality of helicase and polymerase translocation may enable DNA synthesis through G-rich tracts prone to form quadruplexes during cellular processes of replication or DNA repair.
As suggested in a review by Maizels [64], leading strand synthesis arrested by the G4 structure may result in a duplex state or stalled DNA polymerase and associated factors immediately adjacent to the G-quadruplex structure that would not permit loading of the WRN or BLM helicases. Single-stranded DNA that accumulates from unwinding at the replication fork catalyzed by the replication fork leading helicase (MCM) would be coated by RPA. The demonstration that RPA stimulates FANCJ unwinding of G4 structures [29] suggests that FANCJ may be able to load onto RPA-coated tails and unwind the adjacent quadruplex structure. Perhaps FANCJ helicase enables smooth replication of G4 structures by ‘turning its shoulder’ as it unwinds the G4 structure to enable the DNA polymerase enzyme advancing from the opposite direction to proceed unimpeded past the FANCJ helicase as it unwinds the G4 obstacle (Fig. 2). Presumably, this would involve a conformational change enabling the polymerase and helicase to pass one another in a coordinate fashion. Shoulder turning by FANCJ and/or the DNA polymerase could be useful in lagging strand synthesis because FANCJ could then move along to the next Okazaki fragment where it might also be needed. For leading strand synthesis, FANCJ might work at gaps on the leading strand template left far behind the advancing fork. Because the unwound G4 structure may form again relatively quickly, the efficiency of polymerase synthesis using the unwound G-rich region as a template might be important. In such a scenario, FANCJ helicase may have to unwind G4, turn its shoulder, and allow polymerase to move past it and catalyze DNA synthesis.
Fig. 2.
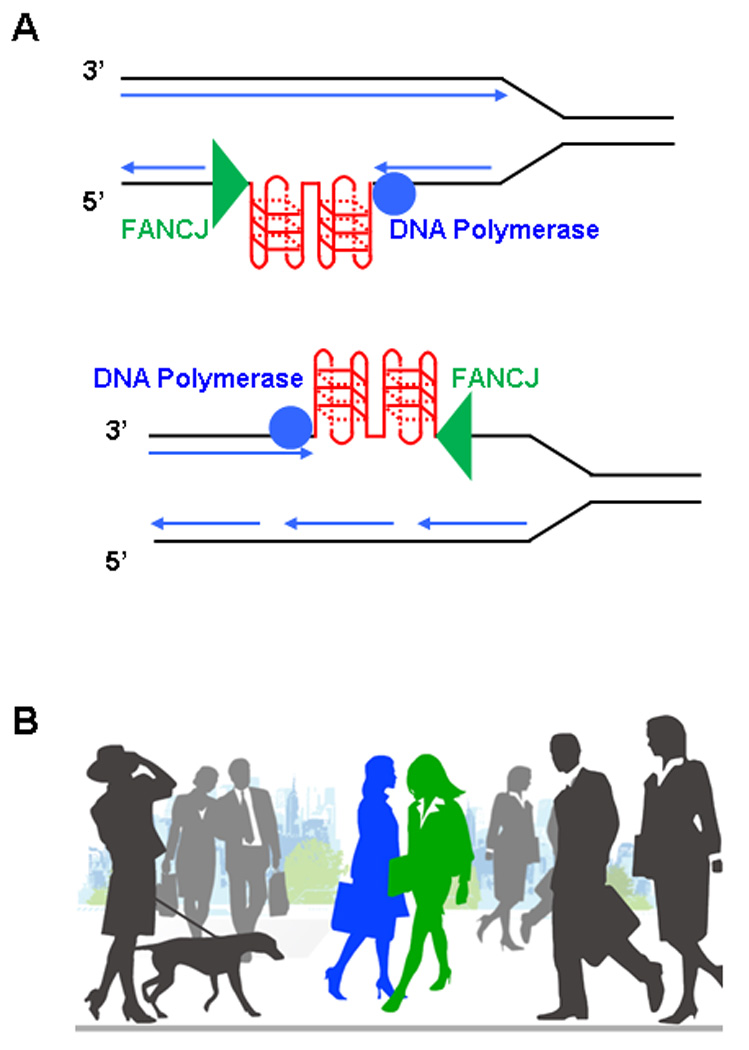
Coordinate action of FANCJ helicase and DNA polymerase during replication. (A) Lagging or leading strand synthesis during replication can be inhibited by G-quadruplex DNA structures. The action of FANCJ helicase is proposed to unwind the G4 block, enabling the DNA polymerase to synthesize the complementary strand. Because FANCJ is a 5′ to 3′ helicase with respect to the strand that it is presumed to translocate, it would have to coordinate its action with the DNA polymerase that is advancing from the opposite direction. (B) Two pedestrians on a busy city sidewalk may proceed to pass each other by turning their shoulders. This analogy may apply to FANCJ and a DNA polymerase that undergo conformational changes to get by one another in an efficient manner.
A reconstituted system that measures DNA synthesis through a G-quadruplex forming sequence with G4 DNA helicases and replication-associated factors would help to address how these replication roadblocks are tolerated. Investigation of protein interactions between helicases that resolve G-quadruplexes and cellular factors such as those that directly bind G-rich sequences (Table 1) may help to better understand the metabolism of G4 structures during cellular DNA replication or transcription, processes that are likely to be affected by G4 DNA.
The yeast RecQ helicase Sgs1 was suggested to have a role in gene expression through its ability to unwind G4 DNA based on a recent study showing that genes with altered expression are preferentially those with the potential to form G4 structures [120]. Very recently, it was also reported that WS and BS cells have preferentially altered expression of genes with PQS [121]. It will be of interest to analyze transcription initiation and elongation of G-rich sequence elements in a reconstituted system to evaluate the importance of G4 binding factors and helicases for transcription.
Mouse studies suggest that premature aging and chromosomal instability of Werner syndrome reflects a role of WRN at telomeres
Although biochemical experiments demonstrate that the WRN and BLM helicases can efficiently unwind G4 structures in vitro, biological studies have only begun to elucidate the importance of WRN or BLM unwinding G-rich sequence elements in vivo. Model systems such as the WRN knockout mouse and defined human cell lines have been helpful in showing that WRN plays an important role in the metabolism of a G-rich DNA structure, the telomere. Although a disease phenotype in WRN-deficient mice is absent, late-generation telomerase and WRN-deficient mice (mTerc−/− WRN−/−) have dysfunctional telomeres and exhibit many of the clinical symptoms of premature aging and the types of tumors (osteocarcinomas, soft tissue sarcomas) typically observed in WS patients [122]. mTerc−/− WRN−/− cells displayed replicative senescence, chromosomal instability, accelerated telomere shortening and elevated recombination rates between the telomeres of sister chromatids [123]. Normal telomere sister-chromatid exchange was restored by the expression of helicase-active WRN protein. WRN-deficient telomere dysfunctional cells escaped senescence by the ALT pathway, suggesting that the chromosomal instability and cancer of WS is a consequence of aberrant telomere sister-chromatid exchange that activates the ALT pathway (for a review, see [124]).
mTerc−/− BLM−/− later-generation mice also showed an acceleration of phenotypes characteristic of later-generation Terc mutants and showed some characteristics such as bone loss reminiscent of BS [125]. mTerc−/− WRN−/− BLM−/− triple mutants showed even more severe phenotypes suggesting a synergistic relationship between the WRN and BLM helicases in telomere maintenance [125]. Overall, the mouse studies provided evidence that the complex pleiotropic phenotypes of WRN or BLM deficiency in mice relate to telomere shortening.
WRN confers telomeric stability by its helicase action on the G-rich lagging strand
The first evidence to suggest a role of human WRN in the maintenance of telomeres was that the cells cultured from WS patients senesce prematurely and display elevated telomere shortening rates [126,127]. Partial localization of WRN to telomeres and its interaction with telomere-associated factors further suggested a function of WRN at telomeres [128]. Cells deficient in the WRN protein display deletion of telomeres from sister chromatids [129]. Curiously, only telomeres that underwent lagging strand synthesis exhibited sister chromatid deletions, and the telomere loss was prevented by WRN helicase activity. Expression of telomerase activity could circumvent the lagging strand telomere deletions. From these elegant studies, the Karlseder laboratory concluded that cells lacking WRN helicase activity are defective in telomere lagging strand synthesis. This work is very significant in the field because it provided a signature form of genomic instability that characterized WS, a model disease for understanding aging. Demonstration that WRN-deficient cells were characterized by a telomere defect in the G-rich strand suggested that the ability of WRN to resolve G-quadruplex structures may be relevant to the peculiar form of telomeric DNA instability characteristic of the disease.
The Karlseder laboratory went on to further dissect the cause of genomic instability in telomeres of WRN-deficient cells by showing that replication-associated telomere loss is responsible for the chromosome fusions in WS fibroblasts [130]. Telomere extension by telomerase could prevent the accumulation of new chromosomal aberrations in WRN-deficient cells [130]. Altogether, the characterization of a telomere lagging strand defect in WS and the requirement of WRN helicase activity to prevent telomere deletion from sister chromatids point toward a specialized role of WRN to unwind an alternate DNA structure that forms in the G-rich strand of the telomere end. The ability of WRN to preferentially unwind G-quadruplex structures may be important for replication and telomere stability.
The stochastic loss of telomere length in WS cell lines may directly contribute to their premature senescence. The observation that the premature senescence and genomic instability prevalent in WS cells can be overcome by forced telomerase expression further suggests a role of WRN in telomere maintenance [131]. WRN and BLM interactions with shelterin proteins [132–134] and other telomere-associated factors [38] are consistent with a model that these helicases act directly on telomere-associated DNA structures. Based on the cellular evidence that WRN is necessary for efficient replication of G-rich telomeric DNA, and telomere lagging strand synthesis is defective in cells lacking WRN helicase activity, it is reasonable to propose that WRN action on telomeric G4 DNA structures is important for telomere maintenance.
Telomere-associated proteins and fragile-telomere phenotype
Using a procedure called proteomics of isolated chromatin segments, Dejardin and Kingston purified human telomeric chromatin and identified the majority of known telomere-associated proteins as well as novel factors [135]. Interestingly, the BLM and FANCJ helicases, but not WRN, were found specifically bound to ALT telomeres. Given the evidence that WRN is found at telomeres of ALT cells [132], it is conceivable that the WRN interaction is transient and was disrupted by the purification procedure. The FANCJ localization at telomeres of ALT, but not telomerase-positive cells, was verified by co-immunostaining with TRF2 [135].
Recent evidence implicates the shelterin protein TRF1 in suppression of telomere fragility as well as degenerative pathologies and cancer in mice [136,137]. Telomeres resemble aphidicolin-induced fragile sites that require TRF1 for stability and their efficient replication. TRF1 promotes efficient replication of TTAGGG repeats and prevents their stalling. To explore the hypothesis that TRF1 might alleviate telomere replication problems by recruiting G4 DNA helicases, various helicase-deficient mouse cells were examined for spontaneous fragile telomeres [137]. Both BLM and RTEL, but not WRN, were required to repress the fragile-telomere phenotype. Future work in this area will likely address the importance of TRF1–helicase interactions to prevent fork stalling and fragility at telomeres in mammalian cells.
Other G4 DNA helicases with roles in nucleic acid metabolism
DHX36
A novel G4 DNA resolvase activity from human placental tissue was isolated ∼ 13 years ago [138]. The G4 resolvase was able to catalytically unwind four-stranded G4 DNA to single-stranded DNA in an ATP- and Mg2+-dependent manner, but failed to unwind duplex or Hoogsteen-bonded triplex DNA. More recently, the same group purified this major non-RecQ, NTP-dependent G4 DNA-resolving enzyme from human cell lysates using G4 DNA affinity chromatography [139]. The protein was identified as the DEXH box helicase product of gene DHX36 (also known as RHAU) by mass spectrometry. The gene encoding DHX36 was cloned and the protein overexpressed in Escherichia coli from which it was purified to apparent homogeneity. The recombinant DHX36 protein displayed robust, highly specific G4 DNA-resolving activity. This enzyme preferred G4 DNA structures over conventional duplex molecules, and its NTPase activity and Mg2+ dependence characteristics were consistent with the properties of the native protein. Immunodepletion experiments using human cell lysates demonstrated that DHX36 protein was responsible for the majority of the detectable G4 DNA resolvase activity.
G4 Resolvase 1 (G4R1) encoded by DHX36 was further characterized and shown to bind and unwind both DNA and RNA quadruplexes with high affinity in vitro. RNA interference experiments demonstrated that G4R1 is the major source of G4 DNA and G4 RNA unwinding activity in HeLa cell lysates [140]. G4R1/RHAU is an RNA helicase associated with the AU-rich sequence of mRNAs that accelerates their degradation [141]. Upon transcription arrest, G4R1/RHAU was observed to colocalize in nucleolar caps and become associated with the RNA-processing helicases p68 and p72 [142]. Taken together, it is proposed that G4R1/RHAU may have an important role in RNA processing in mammalian cells, presumably mediated by its interaction with mRNA transcripts and robust G4 RNA-resolving activity.
It is notable that DHX36 shares sequence homology within the helicase core domain with C. elegans DOG-1 which is important for G-rich tract stability [99,101] and mouse Rtel that is important for maintaining telomere length [102]. Based on the biochemical studies of DHX36 (G4R1) from the Akman and Vaughn laboratories, it will be important to determine the biological consequences of DHX36 deficiency since the protein represents the major G4 DNA- and G4 RNA-resolving activity in human cells.
Dna2
Dna2 is a multifunctional enzyme with 5′–3′ DNA helicase, 3′ and 5′ exo/endonuclease, single-strand annealing and strand exchange activities [143]. Biochemical and genetic evidence (largely from yeast) suggest that Dna2 plays an important role in conjunction with Flap endonuclease 1 (FEN1) for Okazaki fragment maturation during cellular DNA replication [144,145] and in DNA repair [146]. Yeast Dna2 is localized to telomeres in both the G1 and G2 phases of the cell cycle [147]. Genetic evidence in yeast suggests that Dna2 is involved in the generation of G-rich overhangs, suggesting a role of Dna2 in processing of telomeric ends [148].
Recently, Masuda-Sasa et al. found that human and yeast Dna2 unwind G4 DNA structures in an ATP-dependent manner in vitro [149]. It was proposed that Dna2 resolves G4 DNA on G-rich flaps through its helicase and/or nuclease activity in conjunction with RPA, to prepare a substrate for FEN1. Dna2 may act upon G4 structures that form in ribosomal DNA because yeast Dna2 has been shown to be important for its stability [149]. Alternatively, the localization of Dna2 at telomeres and genetic evidence that Dna2 plays a role in telomere maintenance suggests that the enzyme may process telomeric G4 structures [147]. Although genetic and biochemical evidence demonstrates that Dna2 is important for DNA replication, a human disease genetically linked to mutation in Dna2 is yet to be identified, possibly because it is an essential gene.
Pif1
Pif1 is an evolutionarily conserved helicase involved in the maintenance of both nuclear and mitochondrial genomes [150]. Using yeast as a model organism, Ribeyre et al. determined that Pif1 prevents genomic instability of a G-quadruplex forming human minisatellite CEB1 sequence that was inserted into the genome of S. cerevisiae [151]. CEB1 repeats were found to be unstable in pif1 deletion cells only if the synthetic CEB1 minisatellites were able to form G4 structures in vitro. Mutations in the yeast helicase genes sgs1, dna2 or rrm3 did not destabilize the CEB1 locus. Consistent with the in vivo results, purified recombinant yeast Pif1 efficiently unwound G4 structures derived from the synthetic CEB1 arrays in vitro. It was proposed that the defective mitochondrial DNA maintenance of a pif1 mutant reflects defective processing of G4 structures that are prone to undergo recombination at the sites of breaks or at the G4 structures themselves. Nuclear Pif1 is implicated in telomere metabolism and was previously shown to inhibit telomerase [152]. Ribeyre et al. propose that Pif1 may also unwind G4 structures in telomeric DNA to insure its proper architecture [151]. The action of Pif1 on G4 DNA may also play a role in the replication or repair of nuclear DNA outside telomeres, based on its genetic interaction with Dna2 [153] and its importance in maintaining the replication fork barrier in G-rich ribosomal DNA that can also potentially form G-quadruplexes [149].
The specificity of Pif1 in conferring CEB1 stability suggests that other yeast G4 helicases may recognize G4 structures distinct from the human CEB1 G4 structure acted upon by Pif1. Applying the knowledge gained from the yeast Pif1 studies to mammalian systems will be a high priority to determine how human Pif1 is important for genomic stability through its action on alternate DNA structures such as G4. Human G4 DNA helicases genetically linked to human disease may act upon specific DNA structures that arise in distinct pathways of nucleic acid metabolism. It is interesting to note the number of different helicases that can process G4 structures (Table 1). Perhaps it relates to the fact that G4 species represent a large family of structures, or that the particular subgenomic location or phase of the cell cycle in which G4 structures form or persist require different activities.
Conclusions
The evidence that G-rich sequences can form G4 DNA in vitro and that G-rich sequences influence genomic stability raises challenges to researchers in the field. More elegant methods to detect G-quadruplexes would be extremely helpful to assess their abundance, enabling researchers to study the effects of genetic mutations that are linked to human disease on the prevalence of G4 nucleic acids. Genetic and cell biological systems to carefully evaluate the metabolism of G-rich DNA sequences should be further developed. Small molecules that specifically bind distinct G4 DNA structures will continue to be useful not only as tools to probe G4 dynamics but also potentially as anti-cancer drugs that target the regulation of oncogene expression, telomere maintenance and ultimately the nucleic acid structures and pathways in cancerous cells. Clear demonstration that G-quadruplexes exist in vivo is a high priority for the field. If this is accomplished, then researchers can focus on understanding how G4 nucleic acid structures are important in human disease.
Acknowledgements
This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging and the Fanconi Anemia Research Fund (RMB).
Abbreviations
- ALT
alternative lengthening of telomeres
- BS
Bloom’s syndrome
- FA
Fanconi anemia
- FXS
Fragile X syndrome
- G4
G-quadruplex
- ICL
interstrand cross-linking
- PQS
potential quadruplex sequence
- RPA
replication protein A
- RQC
RecQ C-terminal
- TEBP
telomere end-binding proteins
- TMS
telomestatin
- WS
Werner syndrome
References
- 1.Lipps HJ, Rhodes D. G-quadruplex structures: in vivo evidence and function. Trends Cell Biol. 2009;19:414–422. doi: 10.1016/j.tcb.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Huppert JL. Hunting G-quadruplexes. Biochimie. 2008;90:1140–1148. doi: 10.1016/j.biochi.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 3.Maizels N. Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nat Struct Mol Biol. 2006;13:1055–1059. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 4.Burge S, Parkinson GN, Hazel P, Todd AK, Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin Y, Hurley LH. Structures, folding patterns, and functions of intramolecular DNA G-quadruplexes found in eukaryotic promoter regions. Biochimie. 2008;90:1149–1171. doi: 10.1016/j.biochi.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brooks TA, Kendrick S, Hurley L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. FEBS J. 2010 doi: 10.1111/j.1742-4658.2010.07759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huppert JL. Structure, location and interactions of G-quadruplexes. FEBS J. 2010 doi: 10.1111/j.1742-4658.2010.07758.x. [DOI] [PubMed] [Google Scholar]
- 8.Maiti S. Human telomeric G-quadruplex. FEBS J. 2010;277:1097. doi: 10.1111/j.1742-4658.2009.07468.x. [DOI] [PubMed] [Google Scholar]
- 9.De CA, Lacroix L, Douarre C, Temime-Smaali N, Trentesaux C, Riou JF, Mergny JL. Targeting telomeres and telomerase. Biochimie. 2008;90:131–155. doi: 10.1016/j.biochi.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 10.Monchaud D, Teulade-Fichou MP. A hitchhiker’s guide to G-quadruplex ligands. Org Biomol Chem. 2008;6:627–636. doi: 10.1039/b714772b. [DOI] [PubMed] [Google Scholar]
- 11.Sharma S, Doherty KM, Brosh RM., Jr DNA helicases as targets for anti-cancer drugs. Curr Med Chem Anticancer Agents. 2005;5:183–199. doi: 10.2174/1568011053765985. [DOI] [PubMed] [Google Scholar]
- 12.Neidle S. The structures of quadruplex nucleic acids and their drug complexes. Curr Opin Struct Biol. 2009;19:239–250. doi: 10.1016/j.sbi.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Neidle S, Read MA. G-quadruplexes as therapeutic targets. Biopolymers. 2000;56:195–208. doi: 10.1002/1097-0282(2000)56:3<195::AID-BIP10009>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Eddy J, Maizels N. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006;34:3887–3896. doi: 10.1093/nar/gkl529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huppert JL, Balasubramanian S. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005;33:2908–2916. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eddy J, Maizels N. Selection for the G4 DNA motif at the 5′ end of human genes. Mol Carcinog. 2009;48:319–325. doi: 10.1002/mc.20496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verma A, Yadav VK, Basundra R, Kumar A, Chowdhury S. Evidence of genome-wide G4 DNA-mediated gene expression in human cancer cells. Nucleic Acids Res. 2009;37:4194–4204. doi: 10.1093/nar/gkn1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duquette ML, Huber MD, Maizels N. G-rich proto-oncogenes are targeted for genomic instability in B-cell lymphomas. Cancer Res. 2007;67:2586–2594. doi: 10.1158/0008-5472.CAN-06-2419. [DOI] [PubMed] [Google Scholar]
- 19.Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, Vuong B, Wang J, Phan RT, Datta A, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- 20.Maizels N. Immunoglobulin gene diversification. Annu Rev Genet. 2005;39:23–46. doi: 10.1146/annurev.genet.39.073003.110544. [DOI] [PubMed] [Google Scholar]
- 21.Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004;18:1618–1629. doi: 10.1101/gad.1200804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy D, Lieber MR. G clustering is important for the initiation of transcription-induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter. Mol Cell Biol. 2009;29:3124–3133. doi: 10.1128/MCB.00139-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Prospero NA, Fischbeck KH. Therapeutics development for triplet repeat expansion diseases. Nat Rev Genet. 2005;6:756–767. doi: 10.1038/nrg1690. [DOI] [PubMed] [Google Scholar]
- 24.Crawford DC, Acuna JM, Sherman SL. FMR1 and the fragile X syndrome: human genome epidemiology review. Genet Med. 2001;3:359–371. doi: 10.1097/00125817-200109000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fry M, Loeb LA. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc Natl Acad Sci USA. 1994;91:4950–4954. doi: 10.1073/pnas.91.11.4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kettani A, Kumar RA, Patel DJ. Solution structure of a DNA quadruplex containing the fragile X syndrome triplet repeat. J Mol Biol. 1995;254:638–656. doi: 10.1006/jmbi.1995.0644. [DOI] [PubMed] [Google Scholar]
- 27.Usdin K, Woodford KJ. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995;23:4202–4209. doi: 10.1093/nar/23.20.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fry M, Loeb LA. Human werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n. J Biol Chem. 1999;274:12797–12802. doi: 10.1074/jbc.274.18.12797. [DOI] [PubMed] [Google Scholar]
- 29.Wu Y, Shin-ya K, Brosh RM., Jr FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008;28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X, Mariappan SV, Catasti P, Ratliff R, Moyzis RK, Laayoun A, Smith SS, Bradbury EM, Gupta G. Hairpins are formed by the single DNA strands of the fragile X triplet repeats: structure and biological implications. Proc Natl Acad Sci USA. 1995;92:5199–5203. doi: 10.1073/pnas.92.11.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fojtik P, Kejnovska I, Vorlickova M. The guanine-rich fragile X chromosome repeats are reluctant to form tetraplexes. Nucleic Acids Res. 2004;32:298–306. doi: 10.1093/nar/gkh179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wells RD. Mutation spectra in fragile X syndrome induced by deletions of CGG*CCG repeats. J Biol Chem. 2009;284:7407–7411. doi: 10.1074/jbc.R800024200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 34.Fernando H, Sewitz S, Darot J, Tavare S, Huppert JL, Balasubramanian S. Genome-wide analysis of a G-quadruplex-specific single-chain antibody that regulates gene expression. Nucleic Acids Res. 2009;37:6716–6722. doi: 10.1093/nar/gkp740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McEachern MJ, Krauskopf A, Blackburn EH. Telomeres and their control. Annu Rev Genet. 2000;34:331–358. doi: 10.1146/annurev.genet.34.1.331. [DOI] [PubMed] [Google Scholar]
- 36.Palm W, de LT. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 37.de LT. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 38.Bhattacharyya S, Sandy A, Groden J. Unwinding protein complexes in ALTernative telomere maintenance. J Cell Biochem. 2009;109:7–15. doi: 10.1002/jcb.22388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henson JD, Neumann AA, Yeager TR, Reddel RR. Alternative lengthening of telomeres in mammalian cells. Oncogene. 2002;21:598–610. doi: 10.1038/sj.onc.1205058. [DOI] [PubMed] [Google Scholar]
- 40.Lansdorp PM. Telomeres and disease. EMBO J. 2009;28:2532–2540. doi: 10.1038/emboj.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, DePinho RA. Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell. 1999;97:515–525. doi: 10.1016/s0092-8674(00)80761-8. [DOI] [PubMed] [Google Scholar]
- 42.von FG, Hartmann D, Song Z, Rudolph KL. Role of telomere dysfunction in aging and its detection by biomarkers. J Mol Med. 2009;87:1165–1171. doi: 10.1007/s00109-009-0509-5. [DOI] [PubMed] [Google Scholar]
- 43.Varley H, Pickett HA, Foxon JL, Reddel RR, Royle NJ. Molecular characterization of inter-telomere and intra-telomere mutations in human ALT cells. Nat Genet. 2002;30:301–305. doi: 10.1038/ng834. [DOI] [PubMed] [Google Scholar]
- 44.Armanios M. Syndromes of telomere shortening. Annu Rev Genomics Hum Genet. 2009;10:45–61. doi: 10.1146/annurev-genom-082908-150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calado RT, Young NS. Telomere maintenance and human bone marrow failure. Blood. 2008;111:4446–4455. doi: 10.1182/blood-2007-08-019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 47.Yamaguchi H, Baerlocher GM, Lansdorp PM, Chanock SJ, Nunez O, Sloand E, Young NS. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 2003;102:916–918. doi: 10.1182/blood-2003-01-0335. [DOI] [PubMed] [Google Scholar]
- 48.Walne AJ, Vulliamy T, Marrone A, Beswick R, Kirwan M, Masunari Y, Al-Qurashi FH, Aljurf M, Dokal I. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet. 2007;16:1619–1629. doi: 10.1093/hmg/ddm111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vulliamy T, Beswick R, Kirwan M, Marrone A, Digweed M, Walne A, Dokal I. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci USA. 2008;105:8073–8078. doi: 10.1073/pnas.0800042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 2008;82:501–509. doi: 10.1016/j.ajhg.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008;112:3594–3600. doi: 10.1182/blood-2008-05-153445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y, Patel DJ. Solution structure of the human telomeric repeat dAG3(T2AG3)3 G-tetraplex. Structure. 1993;1:263–282. doi: 10.1016/0969-2126(93)90015-9. [DOI] [PubMed] [Google Scholar]
- 53.Parkinson GN, Lee MP, Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 54.Phan AT, Kuryavyi V, Luu KN, Patel DJ. Structure of two intramolecular G-quadruplexes formed by natural human telomere sequences in K+ solution. Nucleic Acids Res. 2007;35:6517–6525. doi: 10.1093/nar/gkm706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dai J, Carver M, Punchihewa C, Jones RA, Yang D. Structure of the Hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res. 2007;35:4927–4940. doi: 10.1093/nar/gkm522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu Y, Ishizuka T, Kurabayashi K, Komiyama M. Consecutive formation of G-quadruplexes in human telomeric-overhang DNA: a protective capping structure for telomere ends. Angew Chem Int Ed Engl. 2009;48:7833–7836. doi: 10.1002/anie.200903858. [DOI] [PubMed] [Google Scholar]
- 57.Prescott DM. The DNA of ciliated protozoa. Microbiol Rev. 1994;58:233–267. doi: 10.1128/mr.58.2.233-267.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, Patel DJ. Solution structure of the Tetrahymena telomeric repeat d(T2G4)4 G-tetraplex. Structure. 1994;2:1141–1156. doi: 10.1016/s0969-2126(94)00117-0. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, Patel DJ. Solution structure of the Oxytricha telomeric repeat dG4(T4G4)3 G-tetraplex. J Mol Biol. 1995;251:76–94. doi: 10.1006/jmbi.1995.0417. [DOI] [PubMed] [Google Scholar]
- 60.Schaffitzel C, Berger I, Postberg J, Hanes J, Lipps HJ, Pluckthun A. In vitro generated antibodies specific for telomeric guanine-quadruplex DNA react with Stylonychia lemnae macronuclei. Proc Natl Acad Sci USA. 2001;98:8572–8577. doi: 10.1073/pnas.141229498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paeschke K, Simonsson T, Postberg J, Rhodes D, Lipps HJ. Telomere end-binding proteins control the formation of G-quadruplex DNA structures in vivo. Nat Struct Mol Biol. 2005;12:847–854. doi: 10.1038/nsmb982. [DOI] [PubMed] [Google Scholar]
- 62.Zhang ML, Tong XJ, Fu XH, Zhou BO, Wang J, Liao XH, Li QJ, Shen N, Ding J, Zhou JQ. Yeast telomerase subunit Est1p has guanine quadruplex-promoting activity that is required for telomere elongation. Nat Struct Mol Biol. 2010;17:202–209. doi: 10.1038/nsmb.1760. [DOI] [PubMed] [Google Scholar]
- 63.Tang J, Kan ZY, Yao Y, Wang Q, Hao YH, Tan Z. G-quadruplex preferentially forms at the very 3′ end of vertebrate telomeric DNA. Nucleic Acids Res. 2008;36:1200–1208. doi: 10.1093/nar/gkm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maizels N. Genomic stability: FANCJ-dependent G4 DNA repair. Curr Biol. 2008;18:R613–R614. doi: 10.1016/j.cub.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oganesian L, Graham ME, Robinson PJ, Bryan TM. Telomerase recognizes G-quadruplex and linear DNA as distinct substrates. Biochemistry. 2007;46:11279–11290. doi: 10.1021/bi700993q. [DOI] [PubMed] [Google Scholar]
- 66.Temime-Smaali N, Guittat L, Sidibe A, Shin-ya K, Trentesaux C, Riou JF. The G-quadruplex ligand telomestatin impairs binding of topoisomerase IIIalpha to G-quadruplex-forming oligonucleotides and uncaps telomeres in ALT cells. PLoS One. 2009;4:e6919. doi: 10.1371/journal.pone.0006919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gomez D, O’Donohue MF, Wenner T, Douarre C, Macadre J, Koebel P, Giraud-Panis MJ, Kaplan H, Kolkes A, Shin-ya K, et al. The G-quadruplex ligand telomestatin inhibits POT1 binding to telomeric sequences in vitro and induces GFP-POT1 dissociation from telomeres in human cells. Cancer Res. 2006;66:6908–6912. doi: 10.1158/0008-5472.CAN-06-1581. [DOI] [PubMed] [Google Scholar]
- 68.Tahara H, Shin-ya K, Seimiya H, Yamada H, Tsuruo T, Ide T. G-quadruplex stabilization by telomestatin induces TRF2 protein dissociation from telomeres and anaphase bridge formation accompanied by loss of the 3′ telomeric osverhang in cancer cells. Oncogene. 2006;25:1955–1966. doi: 10.1038/sj.onc.1209217. [DOI] [PubMed] [Google Scholar]
- 69.Gomez D, Wenner T, Brassart B, Douarre C, O’Donohue MF, El KV, Shin-ya K, Morjani H, Trentesaux C, Riou JF. Telomestatin-induced telomere uncapping is modulated by POT1 through G-overhang extension in HT1080 human tumor cells. J Biol Chem. 2006;281:38721–38729. doi: 10.1074/jbc.M605828200. [DOI] [PubMed] [Google Scholar]
- 70.Tauchi T, Shin-ya K, Sashida G, Sumi M, Okabe S, Ohyashiki JH, Ohyashiki K. Telomerase inhibition with a novel G-quadruplex-interactive agent, telomestatin: in vitro and in vivo studies in acute leukemia. Oncogene. 2006;25:5719–5725. doi: 10.1038/sj.onc.1209577. [DOI] [PubMed] [Google Scholar]
- 71.Cogoi S, Xodo LE. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006;34:2536–2549. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cogoi S, Paramasivam M, Spolaore B, Xodo LE. Structural polymorphism within a regulatory element of the human KRAS promoter: formation of G4 DNA recognized by nuclear proteins. Nucleic Acids Res. 2008;36:3765–3780. doi: 10.1093/nar/gkn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hanakahi LA, Sun H, Maizels N. High affinity interactions of nucleolin with G-G-paired rDNA. J Biol Chem. 1999;274:15908–15912. doi: 10.1074/jbc.274.22.15908. [DOI] [PubMed] [Google Scholar]
- 74.Dempsey LA, Sun H, Hanakahi LA, Maizels N. G4 DNA binding by LR1 and its subunits, nucleolin and hnRNP D. A role for G-G pairing in immunoglobulin switch recombination. J Biol Chem. 1999;274:1066–1071. doi: 10.1074/jbc.274.2.1066. [DOI] [PubMed] [Google Scholar]
- 75.Gonzalez V, Guo K, Hurley L, Sun D. Identification and characterization of nucleolin as a c-myc G-quadruplex-binding protein. J Biol Chem. 2009;284:23622–23635. doi: 10.1074/jbc.M109.018028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vora KA, Tumas-Brundage KM, Lentz VM, Cranston A, Fishel R, Manser T. Severe attenuation of the B cell immune response in Msh2-deficient mice. J Exp Med. 1999;189:471–482. doi: 10.1084/jem.189.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Z, Scherer SJ, Ronai D, Iglesias-Ussel MD, Peled JU, Bardwell PD, Zhuang M, Lee K, Martin A, Edelmann W, et al. Examination of Msh6- and Msh3-deficient mice in class switching reveals overlapping and distinct roles of MutS homologues in antibody diversification. J Exp Med. 2004;200:47–59. doi: 10.1084/jem.20040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martomo SA, Yang WW, Gearhart PJ. A role for Msh6 but not Msh3 in somatic hypermutation and class switch recombination. J Exp Med. 2004;200:61–68. doi: 10.1084/jem.20040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Larson ED, Duquette ML, Cummings WJ, Streiff RJ, Maizels N. MutSalpha binds to and promotes synapsis of transcriptionally activated immunoglobulin switch regions. Curr Biol. 2005;15:470–474. doi: 10.1016/j.cub.2004.12.077. [DOI] [PubMed] [Google Scholar]
- 80.Salas TR, Petruseva I, Lavrik O, Bourdoncle A, Mergny JL, Favre A, Saintome C. Human replication protein A unfolds telomeric G-quadruplexes. Nucleic Acids Res. 2006;34:4857–4865. doi: 10.1093/nar/gkl564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zaug AJ, Podell ER, Cech TR. Human POT1 disrupts telomeric G-quadruplexes allowing telomerase extension in vitro. Proc Natl Acad Sci USA. 2005;102:10864–10869. doi: 10.1073/pnas.0504744102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.De Winter JP, Joenje H. The genetic and molecular basis of Fanconi anemia. Mutat Res. 2009;668:11–19. doi: 10.1016/j.mrfmmm.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 84.Thompson LH, Hinz JM. Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights. Mutat Res. 2009;668:54–72. doi: 10.1016/j.mrfmmm.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Levitus M, Waisfisz Q, Godthelp BC, de VY, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 86.Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 87.Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 88.Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 89.Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet. 2005;37:953–957. doi: 10.1038/ng1627. [DOI] [PubMed] [Google Scholar]
- 90.Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, Livingston DM. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci USA. 2004;101:2357–2362. doi: 10.1073/pnas.0308717101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 92.Frank B, Hemminki K, Meindl A, Wappenschmidt B, Sutter C, Kiechle M, Bugert P, Schmutzler RK, Bartram CR, Burwinkel B. BRIP1 (BACH1) variants and familial breast cancer risk: a case–control study. BMC Cancer. 2007;7:83. doi: 10.1186/1471-2407-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Karppinen SM, Vuosku J, Heikkinen K, Allinen M, Winqvist R. No evidence of involvement of germline BACH1 mutations in Finnish breast and ovarian cancer families. Eur J Cancer. 2003;39:366–371. doi: 10.1016/s0959-8049(02)00498-7. [DOI] [PubMed] [Google Scholar]
- 94.Lewis AG, Flanagan J, Marsh A, Pupo GM, Mann G, Spurdle AB, Lindeman GJ, Visvader JE, Brown MA, Chenevix-Trench G. Mutation analysis of FANCD2, BRIP1/BACH1, LMO4 and SFN in familial breast cancer. Breast Cancer Res. 2005;7:R1005–R1016. doi: 10.1186/bcr1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rutter JL, Smith AM, Davila MR, Sigurdson AJ, Giusti RM, Pineda MA, Doody MM, Tucker MA, Greene MH, Zhang J, et al. Mutational analysis of the BRCA1-interacting genes ZNF350/ZBRK1 and BRIP1/BACH1 among BRCA1 and BRCA2-negative probands from breast-ovarian cancer families and among early-onset breast cancer cases and reference individuals. Hum Mutat. 2003;22:121–128. doi: 10.1002/humu.10238. [DOI] [PubMed] [Google Scholar]
- 96.Sigurdson AJ, Hauptmann M, Chatterjee N, Alexander BH, Doody MM, Rutter JL, Struewing JP. Kin-cohort estimates for familial breast cancer risk in relation to variants in DNA base excision repair, BRCA1 interacting and growth factor genes. BMC Cancer. 2004;4:9. doi: 10.1186/1471-2407-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vahteristo P, Yliannala K, Tamminen A, Eerola H, Blomqvist C, Nevanlinna H. BACH1 Ser919Pro variant and breast cancer risk. BMC Cancer. 2006;6:19. doi: 10.1186/1471-2407-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu Y, Suhasini AN, Brosh RM., Jr Welcome the family of FANCJ-like helicases to the block of genome stability maintenance proteins. Cell Mol Life Sci. 2009;66:1209–1222. doi: 10.1007/s00018-008-8580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cheung I, Schertzer M, Rose A, Lansdorp PM. Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nat Genet. 2002;31:405–409. doi: 10.1038/ng928. [DOI] [PubMed] [Google Scholar]
- 100.Youds JL, Barber LJ, Ward JD, Collis SJ, O’Neil NJ, Boulton SJ, Rose AM. DOG-1 is the Caenorhabditis elegans BRIP1/FANCJ homologue and functions in interstrand cross-link repair. Mol Cell Biol. 2008;28:1470–1479. doi: 10.1128/MCB.01641-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kruisselbrink E, Guryev V, Brouwer K, Pontier DB, Cuppen E, Tijsterman M. Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr Biol. 2008;18:900–905. doi: 10.1016/j.cub.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 102.Ding H, Schertzer M, Wu X, Gertsenstein M, Selig S, Kammori M, Pourvali R, Poon S, Vulto I, Chavez E, et al. Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell. 2004;117:873–886. doi: 10.1016/j.cell.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 103.Barber LJ, Youds JL, Ward JD, McIlwraith MJ, O’Neil NJ, Petalcorin MI, Martin JS, Collis SJ, Cantor SB, Auclair M, et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell. 2008;135:261–271. doi: 10.1016/j.cell.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.London TB, Barber LJ, Mosedale G, Kelly GP, Balasubramanian S, Hickson ID, Boulton SJ, Hiom K. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J Biol Chem. 2008;283:36132–36139. doi: 10.1074/jbc.M808152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gupta R, Sharma S, Sommers JA, Kenny MK, Cantor SB, Brosh RM., Jr FANCJ (BACH1) helicase forms DNA damage inducible foci with replication protein A and interacts physically and functionally with the single-stranded DNA-binding protein. Blood. 2007;110:2390–2398. doi: 10.1182/blood-2006-11-057273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kumaraswamy E, Shiekhattar R. Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol Cell Biol. 2007;27:6733–6741. doi: 10.1128/MCB.00961-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brosh RM, Jr, Bohr VA. Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res. 2007;35:7527–7544. doi: 10.1093/nar/gkm1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 109.Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 1966;45:177–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- 110.Kudlow BA, Kennedy BK, Monnat RJ., Jr Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol. 2007;8:394–404. doi: 10.1038/nrm2161. [DOI] [PubMed] [Google Scholar]
- 111.Monnat RJ, Jr, Saintigny Y. Werner syndrome protein - unwinding function to explain disease. Sci Aging Knowledge Environ. 2004;2004:re3. doi: 10.1126/sageke.2004.13.re3. [DOI] [PubMed] [Google Scholar]
- 112.Chaganti RS, Schonberg S, German J. A manyfold increase in sister-chromatid exchanges in Bloom’s syndrome lymphocytes. Proc Natl Acad Sci USA. 1974;71:4508–4512. doi: 10.1073/pnas.71.11.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ellis NA, German J. Molecular genetics of Bloom’s syndrome. Hum Mol Genet. 1996;5:1457–1463. doi: 10.1093/hmg/5.supplement_1.1457. [DOI] [PubMed] [Google Scholar]
- 114.Sun H, Karow JK, Hickson ID, Maizels N.The Bloom’s syndrome helicase unwinds G4 DNA J Biol Chem 1998273s27587–27592. [DOI] [PubMed] [Google Scholar]
- 115.Mohaghegh P, Karow JK, Brosh JR, Jr, Bohr VA, Hickson ID. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huber MD, Duquette ML, Shiels JC, Maizels N. A conserved G4 DNA binding domain in RecQ family helicases. J Mol Biol. 2006;358:1071–1080. doi: 10.1016/j.jmb.2006.01.077. [DOI] [PubMed] [Google Scholar]
- 117.Popuri V, Bachrati CZ, Muzzolini L, Mosedale G, Costantini S, Giacomini E, Hickson ID, Vindigni A. The human RecQ helicases, BLM and RECQ1, display distinct DNA substrate specificities. J Biol Chem. 2008;283:17766–17776. doi: 10.1074/jbc.M709749200. [DOI] [PubMed] [Google Scholar]
- 118.von KC, Thoma NH, Czyzewski BK, Pavletich NP, Bohr VA. Werner syndrome protein contains three structure-specific DNA binding domains. J Biol Chem. 2003;278:52997–53006. doi: 10.1074/jbc.M308338200. [DOI] [PubMed] [Google Scholar]
- 119.Kamath-Loeb AS, Loeb LA, Johansson E, Burgers PM, Fry M. Interactions between the Werner syndrome helicase and DNA polymerase delta specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J Biol Chem. 2001;276:16439–16446. doi: 10.1074/jbc.M100253200. [DOI] [PubMed] [Google Scholar]
- 120.Hershman SG, Chen Q, Lee JY, Kozak ML, Yue P, Wang LS, Johnson FB. Genomic distribution and functional analyses of potential G-quadruplex-forming sequences in Saccharomyces cerevisiae. Nucleic Acids Res. 2008;36:144–156. doi: 10.1093/nar/gkm986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Johnson JE, Cao K, Ryvkin P, Wang LS, Johnson FB. Altered gene expression in the Werner and Bloom syndromes is associated with sequences having G-quadruplex forming potential. Nucleic Acids Res. 2010;38:1114–1122. doi: 10.1093/nar/gkp1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lombard DB, Beard C, Johnson B, Marciniak RA, Dausman J, Bronson R, Buhlmann JE, Lipman R, Curry R, Sharpe A, et al. Mutations in the WRN gene in mice accelerate mortality in a p53-null background. Mol Cell Biol. 2000;20:3286–3291. doi: 10.1128/mcb.20.9.3286-3291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Laud PR, Multani AS, Bailey SM, Wu L, Ma J, Kingsley C, Lebel M, Pathak S, DePinho RA, Chang S. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 2005;19:2560–2570. doi: 10.1101/gad.1321305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chang S. A mouse model of Werner syndrome: what can it tell us about aging and cancer? Int J Biochem Cell Biol. 2005;37:991–999. doi: 10.1016/j.biocel.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 125.Du X, Shen J, Kugan N, Furth EE, Lombard DB, Cheung C, Pak S, Luo G, Pignolo RJ, DePinho RA, et al. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol Cell Biol. 2004;24:8437–8446. doi: 10.1128/MCB.24.19.8437-8446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schulz VP, Zakian VA, Ogburn CE, McKay J, Jarzebowicz AA, Edland SD, Martin GM. Accelerated loss of telomeric repeats may not explain accelerated replicative decline of Werner syndrome cells. Hum Genet. 1996;97:750–754. doi: 10.1007/BF02346184. [DOI] [PubMed] [Google Scholar]
- 127.Faragher RG, Kill IR, Hunter JA, Pope FM, Tannock C, Shall S. The gene responsible for Werner syndrome may be a cell division ‘counting’ gene. Proc Natl Acad Sci USA. 1993;90:12030–12034. doi: 10.1073/pnas.90.24.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Multani AS, Chang S. WRN at telomeres: implications for aging and cancer. J Cell Sci. 2007;120:713–721. doi: 10.1242/jcs.03397. [DOI] [PubMed] [Google Scholar]
- 129.Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 130.Crabbe L, Jauch A, Naeger CM, Holtgreve-Grez H, Karlseder J. Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc Natl Acad Sci USA. 2007;104:2205–2210. doi: 10.1073/pnas.0609410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wyllie FS, Jones CJ, Skinner JW, Haughton MF, Wallis C, Wynford-Thomas D, Faragher RG, Kipling D. Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nat Genet. 2000;24:16–17. doi: 10.1038/71630. [DOI] [PubMed] [Google Scholar]
- 132.Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kolvraa S, May A, Seidman MM, Bohr VA. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell. 2004;14:763–774. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 133.Schawalder J, Paric E, Neff NF. Telomere and ribosomal DNA repeats are chromosomal targets of the bloom syndrome DNA helicase. BMC Cell Biol. 2003;4:15. doi: 10.1186/1471-2121-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lillard-Wetherell K, Machwe A, Langland GT, Combs KA, Behbehani GK, Schonberg SA, German J, Turchi JJ, Orren DK, Groden J. Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2. Hum Mol Genet. 2004;13:1919–1932. doi: 10.1093/hmg/ddh193. [DOI] [PubMed] [Google Scholar]
- 135.Dejardin J, Kingston RE. Purification of proteins associated with specific genomic loci. Cell. 2009;136:175–186. doi: 10.1016/j.cell.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Martinez P, Thanasoula M, Munoz P, Liao C, Tejera A, McNees C, Flores JM, Fernandez-Capetillo O, Tarsounas M, Blasco MA. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23:2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de LT. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Harrington C, Lan Y, Akman SA. The identification and characterization of a G4 DNA resolvase activity. J Biol Chem. 1997;272:24631–24636. doi: 10.1074/jbc.272.39.24631. [DOI] [PubMed] [Google Scholar]
- 139.Vaughn JP, Creacy SD, Routh ED, Joyner-Butt C, Jenkins GS, Pauli S, Nagamine Y, Akman SA. The DEXH protein product of the DHX36 gene is the major source of tetramolecular quadruplex G4 DNA resolving activity in HeLa cell lysates. J Biol Chem. 2005;280:38117–38120. doi: 10.1074/jbc.C500348200. [DOI] [PubMed] [Google Scholar]
- 140.Creacy SD, Routh ED, Iwamoto F, Nagamine Y, Akman SA, Vaughn JP. G4 resolvase 1 binds both DNA and RNA tetramolecular quadruplex with high affinity and is the major source of tetramolecular quadruplex G4 DNA and G4-RNA resolving activity in HeLa cell lysates. J Biol Chem. 2008;283:34626–34634. doi: 10.1074/jbc.M806277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tran H, Schilling M, Wirbelauer C, Hess D, Nagamine Y. Facilitation of mRNA deadenylation and decay by the exosome-bound, DExH protein RHAU. Mol Cell. 2004;13:101–111. doi: 10.1016/s1097-2765(03)00481-7. [DOI] [PubMed] [Google Scholar]
- 142.Iwamoto F, Stadler M, Chalupnikova K, Oakeley E, Nagamine Y. Transcription-dependent nucleolar cap localization and possible nuclear function of DExH RNA helicase RHAU. Exp Cell Res. 2008;314:1378–1391. doi: 10.1016/j.yexcr.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 143.Masuda-Sasa T, Imamura O, Campbell JL. Biochemical analysis of human Dna2. Nucleic Acids Res. 2006;34:1865–1875. doi: 10.1093/nar/gkl070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kao HI, Campbell JL, Bambara RA. Dna2p helicase/nuclease is a tracking protein, like FEN1, for flap cleavage during Okazaki fragment maturation. J Biol Chem. 2004;279:50840–50849. doi: 10.1074/jbc.M409231200. [DOI] [PubMed] [Google Scholar]
- 145.Kang HY, Choi E, Bae SH, Lee KH, Gim BS, Kim HD, Park C, MacNeill SA, Seo YS. Genetic analyses of Schizosaccharomyces pombe dna2(+) reveal that dna2 plays an essential role in Okazaki fragment metabolism. Genetics. 2000;155:1055–1067. doi: 10.1093/genetics/155.3.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Weitao T, Budd M, Hoopes LL, Campbell JL. Dna2 helicase/nuclease causes replicative fork stalling and double-strand breaks in the ribosomal DNA of Saccharomyces cerevisiae. J Biol Chem. 2003;278:22513–22522. doi: 10.1074/jbc.M301610200. [DOI] [PubMed] [Google Scholar]
- 147.Choe W, Budd M, Imamura O, Hoopes L, Campbell JL. Dynamic localization of an Okazaki fragment processing protein suggests a novel role in telomere replication. Mol Cell Biol. 2002;22:4202–4217. doi: 10.1128/MCB.22.12.4202-4217.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tomita K, Kibe T, Kang HY, Seo YS, Uritani M, Ushimaru T, Ueno M. Fission yeast Dna2 is required for generation of the telomeric single-strand overhang. Mol Cell Biol. 2004;24:9557–9567. doi: 10.1128/MCB.24.21.9557-9567.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Masuda-Sasa T, Polaczek P, Peng XP, Chen L, Campbell JL. Processing of G4 DNA by Dna2 helicase/nuclease and replication protein A (RPA) provides insights into the mechanism of Dna2/RPA substrate recognition. J Biol Chem. 2008;283:24359–24373. doi: 10.1074/jbc.M802244200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Boule JB, Zakian VA. Roles of Pif1-like helicases in the maintenance of genomic stability. Nucleic Acids Res. 2006;34:4147–4153. doi: 10.1093/nar/gkl561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ribeyre C, Lopes J, Boule JB, Piazza A, Guedin A, Zakian VA, Mergny JL, Nicolas A. The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet. 2009;5:e1000475. doi: 10.1371/journal.pgen.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang DH, Zhou B, Huang Y, Xu LX, Zhou JQ. The human Pif1 helicase, a potential Escherichia coli RecD homologue, inhibits telomerase activity. Nucleic Acids Res. 2006;34:1393–1404. doi: 10.1093/nar/gkl029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Budd ME, Reis CC, Smith S, Myung K, Campbell JL. Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase delta. Mol Cell Biol. 2006;26:2490–2500. doi: 10.1128/MCB.26.7.2490-2500.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer. 2009;9:644–654. doi: 10.1038/nrc2682. [DOI] [PubMed] [Google Scholar]
- 155.Bochman ML, Sabouri N, Zakian VA. Unwinding the functions of the Pif1 family helicases. DNA Repair (Amst) 2010;9:237–249. doi: 10.1016/j.dnarep.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cheng X, Dunaway S, Ivessa AS. The role of Pif1p, a DNA helicase in Saccharomyces cerevisiae, in maintaining mitochondrial DNA. Mitochondrion. 2007;7:211–222. doi: 10.1016/j.mito.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 157.Pike JE, Burgers PM, Campbell JL, Bambara RA. Pif1 helicase lengthens some Okazaki fragment flaps necessitating Dna2 nuclease/helicase action in the two-nuclease processing pathway. J Biol Chem. 2009;284:25170–25180. doi: 10.1074/jbc.M109.023325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Duxin JP, Dao B, Martinsson P, Rajala N, Guittat L, Campbell JL, Spelbrink JN, Stewart SA. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol Cell Biol. 2009;29:4274–4282. doi: 10.1128/MCB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zheng L, Zhou M, Guo Z, Lu H, Qian L, Dai H, Qiu J, Yakubovskaya E, Bogenhagen DF, Demple B, et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol Cell. 2008;32:325–336. doi: 10.1016/j.molcel.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kang YH, Lee CH, Seo YS. Dna2 on the road to Okazaki fragment processing and genome stability in eukaryotes. Crit Rev Biochem Mol Biol. 2010;45:71–96. doi: 10.3109/10409230903578593. [DOI] [PubMed] [Google Scholar]
- 161.Stewart JA, Campbell JL, Bambara RA. Significance of the dissociation of Dna2 by flap endonuclease 1 to Okazaki fragment processing in Saccharomyces cerevisiae. J Biol Chem. 2009;284:8283–8291. doi: 10.1074/jbc.M809189200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Huber MD, Lee DC, Maizels N. G4 DNA unwinding by BLM and Sgs1p: substrate specificity and substrate-specific inhibition. Nucleic Acids Res. 2002;30:3954–3961. doi: 10.1093/nar/gkf530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ashton TM, Hickson ID. Yeast as a model system to study RecQ helicase function. DNA Repair (Amst) 2010;9:303–314. doi: 10.1016/j.dnarep.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 164.Arimondo PB, Riou JF, Mergny JL, Tazi J, Sun JS, Garestier T, Helene C. Interaction of human DNA topoisomerase I with G-quartet structures. Nucleic Acids Res. 2000;28:4832–4838. doi: 10.1093/nar/28.24.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sun H, Yabuki A, Maizels N. A human nuclease specific for G4 DNA. Proc Natl Acad Sci USA. 2001;98:12444–12449. doi: 10.1073/pnas.231479198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang QS, Manche L, Xu RM, Krainer AR. hnRNP A1 associates with telomere ends and stimulates telomerase activity. RNA. 2006;12:1116–1128. doi: 10.1261/rna.58806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nakagama H, Higuchi K, Tanaka E, Tsuchiya N, Nakashima K, Katahira M, Fukuda H. Molecular mechanisms for maintenance of G-rich short tandem repeats capable of adopting G4 DNA structures. Mutat Res. 2006;598:120–131. doi: 10.1016/j.mrfmmm.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 168.Paramasivam M, Membrino A, Cogoi S, Fukuda H, Nakagama H, Xodo LE. Protein hnRNP A1 and its derivative Up1 unfold quadruplex DNA in the human KRAS promoter: implications for transcription. Nucleic Acids Res. 2009;37:2841–2853. doi: 10.1093/nar/gkp138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Fukuda H, Katahira M, Tsuchiya N, Enokizono Y, Sugimura T, Nagao M, Nakagama H. Unfolding of quadruplex structure in the G-rich strand of the minisatellite repeat by the binding protein UP1. Proc Natl Acad Sci USA. 2002;99:12685–12690. doi: 10.1073/pnas.152456899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Enokizono Y, Konishi Y, Nagata K, Ouhashi K, Uesugi S, Ishikawa F, Katahira M. Structure of hnRNP D complexed with single-stranded telomere DNA and unfolding of the quadruplex by heterogeneous nuclear ribonucleoprotein D. J Biol Chem. 2005;280:18862–18870. doi: 10.1074/jbc.M411822200. [DOI] [PubMed] [Google Scholar]
- 171.Xiao J, McGown LB. Mass spectrometric determination of ILPR G-quadruplex binding sites in insulin and IGF-2. J Am Soc Mass Spectrom. 2009;20:1974–1982. doi: 10.1016/j.jasms.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang Y, Zhang H, Ligon LA, McGown LB. Association of insulin-like growth factor 2 with the insulin-linked polymorphic region in cultured fetal thymus cells. Biochemistry. 2009;48:8189–8194. doi: 10.1021/bi900958x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Salas TR, Petruseva I, Lavrik O, Bourdoncle A, Mergny JL, Favre A, Saintome C. Human replication protein A unfolds telomeric G-quadruplexes. Nucleic Acids Res. 2006;34:4857–4865. doi: 10.1093/nar/gkl564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wu Y, Rawtani N, Thazhathveetil AK, Kenny MK, Seidman MM, Brosh RM., Jr Human replication protein A melts a DNA triple helix structure in a potent and specific manner. Biochemistry. 2008;47:5068–5077. doi: 10.1021/bi702102d. [DOI] [PMC free article] [PubMed] [Google Scholar]