

Abstract
Vertebrate photoreceptors have a modified cilium composed of a basal body, axoneme and outer segment. The outer segment includes stacked membrane discs, containing opsin and the signal transduction apparatus mediating phototransduction. In photoreceptors, two distinct classes of vesicles are trafficked. Synaptic vesicles are transported down the axon to the synapse, whereas opsin-containing vesicles are transported to the outer segment. The continuous replacement of the outer segments imposes a significant biosynthetic and trafficking burden on the photoreceptors. Here, we show that Ahi1, a gene that when mutated results in the neurodevelopmental disorder, Joubert syndrome (JBTS), is required for photoreceptor sensory cilia formation and the development of photoreceptor outer segments. In mice with a targeted deletion of Ahi1, photoreceptors undergo early degeneration. Whereas synaptic proteins are correctly trafficked, photoreceptor outer segment proteins fail to be transported appropriately or are significantly reduced in their expression levels (i.e., transducin and Rom1) in Ahi1−/− mice. We show that vesicular targeting defects in Ahi1−/− mice are cilium specific, and our evidence suggests that the defects are caused by a decrease in expression of the small GTPase Rab8a, a protein required for accurate polarized vesicular trafficking. Thus, our results suggest that Ahi1 plays a role in stabilizing the outer segment proteins, transducin and Rom1, and that Ahi1 is an important component of Rab8a-mediated vesicular trafficking in photoreceptors. The retinal degeneration observed in Ahi1−/− mice recapitulates aspects of the retinal phenotype observed in patients with JBTS and suggests the importance of Ahi1 in photoreceptor function.
Introduction
The primary cilium is a microtubule-based organelle found on the surface of most quiescent cells (Pan and Snell, 2007; Pugacheva et al., 2007). During ciliogenesis, the mother centriole is delivered to the apical cell surface via attachment to a Golgi-derived centriolar vesicle (Sorokin, 1962). Accessory structures are recruited that ultimately form the basal body, the immature axoneme projects from the mother centriole, and vesicular fusion events produce the ensheathment of the elongating axonemal shaft. The centriolar vesicle then fuses with the plasma membrane (Gilula and Satir, 1972; Satir and Christensen, 2007). Subsequent elongation and maintenance of the cilium are regulated by intraflagellar transport (Rosenbaum and Witman, 2002; Scholey and Anderson, 2006; Scholey, 2008).
The rod and cone photoreceptor cells of the retina are examples of cells that use specialized sensory cilia for sensation, in this case, light (Adams et al., 2007; Liu et al., 2007). Failure of cilium formation and/or function has recently been implicated in many diseases; such diseases are referred to as ciliopathies (Badano et al., 2006). An important example of ciliopathies is inherited retinal degeneration, in which the outer segments of the rod and cone photoreceptor cells are affected. The connection here is that the light sensitive outer segments are specialized sensory cilia. Like other cilia, these photoreceptor sensory cilia contain an axoneme, which begins at the basal bodies, and passes through a transition zone (also called the “connecting cilium”) and into the outer segment (Beisson and Wright, 2003; Liu et al., 2007).
Joubert syndrome (JBTS) is considered to be one such ciliopathy (Badano et al., 2006; Hildebrandt and Zhou, 2007). JBTS is an autosomal recessive disorder classically characterized by brainstem malformations (i.e., the “molar tooth” sign) and cerebellar vermis aplasia/hypoplasia, but also retinal dystrophy (Parisi et al., 2007; den Hollander et al., 2008; Zaki et al., 2008). Multiple genes are known to cause JBTS, with 10–15% of all cases of JBTS attributed to mutations in the Abelson-helper integration site-1 (AHI1) gene (Dixon-Salazar et al., 2004; Ferland et al., 2004; Parisi et al., 2006). AHI1 encodes a cytoplasmic multidomain protein that is thought to serve as a scaffolding protein (Jiang et al., 2002). Only recently has the function of Ahi1 begun to be elucidated in a recent report that demonstrated the requirement of Ahi1 for the formation of primary nonmotile cilia (Hsiao et al., 2009).
Here we show that in Ahi1−/− mice on embryonic day 18.5 (E18.5), basal bodies are found to be normally distributed in the outer retina, and immature axonemes are beginning to form. Postnatal Ahi1−/− mice fail to develop complete photoreceptor sensory cilia, and they exhibit degeneration of photoreceptor cells. Proteins that are trafficked through the transition zone to the outer segments are mistargeted in Ahi1−/− mice, with many of the aberrantly targeted proteins being seen instead in the photoreceptor cell bodies. Our data demonstrate that the absence of Ahi1 results in a dramatic failure of photoreceptor sensory cilia formation, and emphasize that inherited retinal degenerative disorders are a subclass of the ciliopathies.
Materials and Methods
Animals.
Generation of Ahi1 knock-out mice on a C57BL/6J genetic background (B6-Ahi1+/−) has been described previously (Hsiao et al., 2009). However, since Ahi1−/− mice on a C57BL/6J (B6) background die at birth, we performed backcrossing of our B6-Ahi1+/− mice onto multiple, independent inbred lines; the result was that one inbred line, BALB/cJ, was found to be capable of overcoming the postnatal lethality phenotype. The BALB/cJ-Ahi1+/− animals were then repeatedly backcrossed to BALB/cJ mice. To obtain tissue for analysis, we examined animals from Ahi1+/− X Ahi1+/− crosses, with wild-type littermates serving as controls. Mice were maintained on a normal 12 h light/dark cycle (6:00 A.M. to 6:00 P.M.) with unlimited access to food and water. All mouse procedures were performed under approval from the Institutional Animal Care and Use Committees of the Albany Medical College, Rensselaer Polytechnic Institute, and the Wadsworth Center (New York State Department of Health), in accordance with The National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Histological analysis.
Ahi1−/−, Ahi1+/−, and Ahi1+/+ mice at various ages were overdosed with avertin or sodium pentobarbital, and then perfused transcardially with PBS, pH 7.4, at room temperature, followed by cold 4% paraformaldehyde (PFA) made in PBS. Eyes were removed and postfixed in 4% PFA, followed by cryoprotection in a 30% sucrose solution made in PBS. Enucleated mouse eyes were sectioned at a thickness of 10–20 μm on a Microm cryostat (Richard-Allan Scientific), and were then mounted on positively charged glass slides (Superfrost Plus; Fisher Scientific). Sections were allowed to air dry for 30 min before being stored at −80°C.
To obtain E18.5 pups, we overdosed pregnant females with avertin or sodium pentobarbital, and removed the embryos and placed them into ice-cold 0.1 m PBS. Embryos were perfused under a Zeiss dissecting microscope and processed as described above.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining, using the In Situ cell death detection kit (Roche Diagnostics), was performed on retinas from Ahi1−/−, Ahi1+/−, and Ahi1+/+ mice at various ages to establish when the retinal degeneration was occurring in the photoreceptor layer. Briefly, 4% PFA was applied to slides containing retinal sections that had been perfused previously as described above. The slides were then washed for 30 min in PBS, followed by incubation in 0.1% Triton X-100 and 0.1% sodium citrate, for 2 min on ice. The slides were rinsed briefly in PBS before being incubated in the TUNEL reaction mixture (prepared according to the manufacturer's instructions) at 37°C for 1 h in the dark. Slides were rinsed in PBS and coverslipped with Fluoromount-G; they were analyzed on a Zeiss AxioImager-Z1 microscope and imaged with an AxioCam MRm camera.
Immunostaining and visualization.
Ahi1−/−, Ahi1+/−, and Ahi1+/+ mice at various ages were overdosed with avertin or sodium pentobarbital and perfused as described above. The only method differences were as follows: (1) for immunostaining, mice were perfused with either cold 1% or 4% PFA; (2) the eyes were enucleated and, after removal of the cornea and lens, were fixed for an additional 2 to 3 h at 4°C (Liu et al., 2004). The eyecups were then transferred to a 30% sucrose solution in PBS, incubated overnight at 4°C, and embedded in optimal cutting temperature freezing medium for cryosectioning. Eyes were sectioned at a thickness of 10–20 μm. Sections were blocked in 10% fetal bovine serum (FBS) in PBS with 0.04% Triton X-100 (PBS-TX) for 1 h, before application of primary antibodies. Primary antibodies were applied overnight at 4°C in 1% FBS. Secondary antibodies were applied for 2 h at room temperature in 1% FBS. All antibody solutions were prepared in PBS-TX. Vectashield mounting medium with 4′,6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories) was applied after staining. Slides were visualized using either (1) a Zeiss AxioImager-Z1 microscope and a Zeiss Apotome, with imaging by an AxioCam MRm camera and AxioVision software release 4.5 (Zeiss), or (2) a Zeiss LSM 510 Meta confocal microscope, with image processing using the Zeiss Meta 510 software. All images were processed in Adobe Photoshop CS2 (version 9.0.2; Adobe Systems). Contrast and brightness of images were adjusted through linear level adjustments, as needed, to optimize the intensity range of the images.
Western blot analysis.
Retinas were dissected from postnatal day 12 (PN12) eyes (Ahi1−/−, n = 3; Ahi1+/+, n = 4) and homogenized in RIPA buffer (50 mm Tris, pH 8, 150 mm NaCl, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, 1 mm DTT, 1 mm phenylmethylsulfonyl fluoride, and a 1× protease inhibitor cocktail; Roche Applied Science). After incubation on ice for 30 min, the homogenized retina was centrifuged at 10,000 × g for 30 min at 4°C. Protein concentrations of the supernatant for each retina sample were determined with the Advanced Protein Assay Reagent kit (Cytoskeleton). The protein lysates (5 μg) were resolved on an SDS/PAGE gel and transferred onto polyvinylidene fluoride membrane (Millipore). The membrane was blocked in 5% skim milk/TBSTx (100 mm Tris, pH 7.4, 150 mm NaCl, and 0.01% Triton X-100) for 1 h at room temperature. The membrane was then incubated with primary antibody (loading controls consisted of using chicken anti-βIII tubulin antibodies; 1:1000; Millipore) diluted in blocking solution at 4°C overnight. Primary antibodies [transducin (1:5000) and Rom1 (1:1000)] were detected with either the SuperSignal West Femto Maximum Sensitivity Substrate Chemiluminescence kit (Pierce) or with fluorescent secondary antibodies (Alexa Fluor 488; Invitrogen). Signals were analyzed with a Syngene G:Box iChemi XT imaging system and the GeneTools analysis software (Synoptics). To quantify and compare the signal intensities of each sample, the detected signals were unsaturated and in the linear range of detection.
Antibodies.
The following primary antibodies were used for immunostaining at the indicated final dilutions: Ahi1 (rabbit IgG; 1:1000) (Doering et al., 2008); rhodopsin (mouse IgG, clone 4D2, 1:1000, kindly provided by Dr. R. S. Molday, University of British Columbia, Vancouver, Canada; and mouse IgG, clone 1D4, 1:1000, Sigma) (Molday and MacKenzie, 1983); synaptotagmin (mouse IgG2a, 1:100; Calbiochem) (Matthew et al., 1981); anti-γ-tubulin (mouse IgG1, 1:300; Sigma) (Hsiao et al., 2009); Rab8a (mouse IgG2b, 1:50; BD Biosciences) (Hsiao et al., 2009); α-transducin (rabbit IgG, 1:1000; Santa Cruz Biotechnology) (Chen et al., 2007); Rom1 (mouse IgG, 1D5, 1:40; kindly provided by Dr. R. S. Molday) (Bascom et al., 1992); Rp1 (chicken IgY, 1:2000) (Liu et al., 2002); Rpgrip1 (rabbit IgG, 1:5000; kindly provided by Dr. Tiansen Li, Massachussetts Eye and Ear Infirmary, Boston, MA) (Hong et al., 2001); and anti-cyclic nucleotide gated channel α 1 (mouse IgG, 1D1, 1:10; kindly provided by Dr. R. S. Molday) (Cook et al., 1989). Detection of the primary antibodies was determined via the appropriate fluorophore-labeled secondary antibodies [Invitrogen (Alexa Fluor) or Jackson ImmunoResearch].
Light and electron microscopy.
Retinas from Ahi1−/− mice and Ahi1+/+ littermate control mice at PN12 were prepared for light and electron microscopy as described previously (Liu et al., 2003, 2009). Briefly, animals were killed and perfused with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 m PBS. Eyes were enucleated, and the corneas were removed. Eyecups were then fixed in the same fixative for an additional 4 h at 4°C. Retinas were trimmed into 2 mm pieces, and the pieces were postfixed in 1% OsO4, dehydrated through a graded ethanol series followed by propylene oxide infiltration, and embedded in Epon (EMbed812; Electron Microscopy Sciences). Semithin (0.5 μm) sections were cut and stained with alkaline toluidine blue for light microscopy. Ultrathin sections (60 nm) were cut and stained with 2% uranyl acetate and lead citrate, and imaged in an FEI Tecnai transmission electron microscope.
Results
Ahi1 expression in the retina
We performed localization studies of Ahi1 in the retina of the postnatal mouse. In the wild-type (Ahi1+/+) mouse, Ahi1 immunolabeling was observed in the outer nuclear layer photoreceptors (Fig. 1A). More specifically, Ahi1 protein was found in the transition zone of the photoreceptor sensory cilium, colocalizing with the transition zone marker, Rpgrip1 (Fig. 1C, top row). To determine whether Ahi1 localizes to the axoneme, we compared the immunostaining patterns of Ahi1 and the axonemal marker Rp1. Ahi1 immunostaining was observed proximal to Rp1 staining, indicating that Ahi1 is present at the proximal end of the axoneme (Fig. 1C, bottom row). Ahi1 immunolabeling was also observed in the inner retina of the wild-type mouse (Fig. 1A). More specifically, Ahi1 immunostaining was found to occur at the edge of horizontal, bipolar, and ganglion cell bodies. These immunostaining patterns were not observed in mice with a targeted deletion of Ahi1 (Ahi1−/−) (Fig. 1B). Moreover, no Ahi1 protein was detected in dissected Ahi1−/− retina by Western blotting (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.
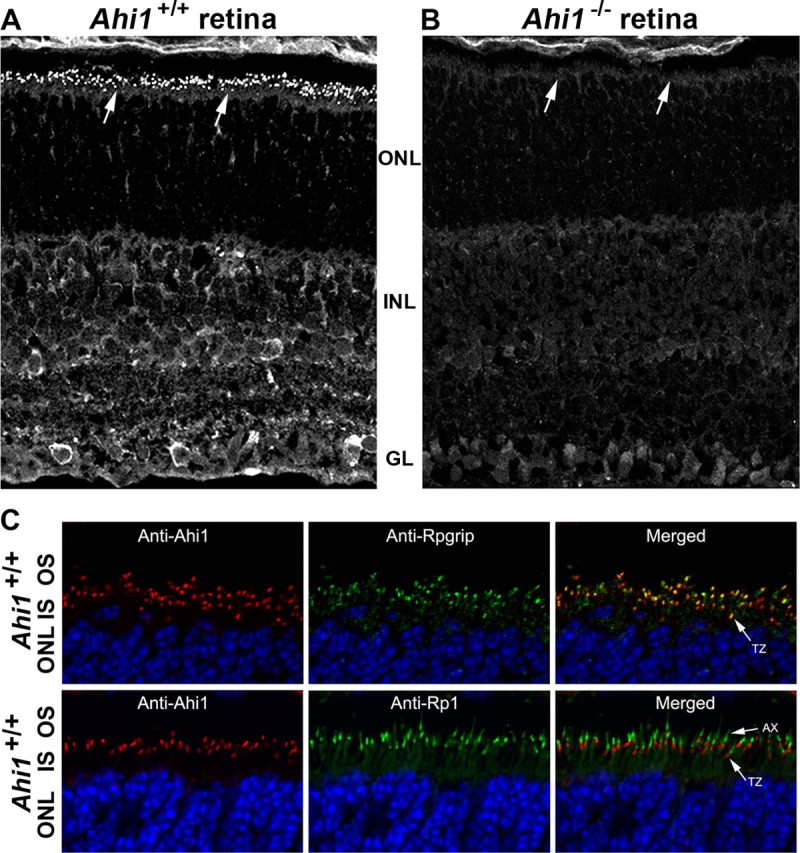
Ahi1 localization in the transition zone of the specialized cilium of the photoreceptors and in the inner retina. A, Immunostaining of Ahi1 in wild-type (Ahi1+/+) PN12 retinas. Arrows indicate targeting of Ahi1 to the ciliary transition zone. B, Lack of Ahi1 immunostaining in retina from age-matched mice that have a targeted deletion of Ahi1 (Ahi1−/−). Arrows indicate that lack of targeting of Ahi1 to the ciliary transition zone. C, PN12 wild-type (Ahi1+/+) mice with coimmunolabeling of Ahi1 (red) and the transition zone marker, Rpgrip (green in top row of images). Also labeled is the axoneme marker, Rp1 (green in bottom row of images). DNA is visualized with DAPI (blue). GL, Ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; AX, axoneme; IS, inner segments; OS, outer segments; TZ, transition zone.
Mice with targeted deletion of Ahi1 (Ahi1−/−) have early photoreceptor degeneration
Although individuals with JBTS have been reported to have retinal dystrophy (Parisi et al., 2006), little is known about the pathogenesis of this process in JBTS. Histological analysis of retinas from Ahi1−/− mice revealed complete absence of the photoreceptor outer segments in PN12 animals (Fig. 2A, compare layer at black arrow in Ahi1+/+ retina with the lack of this layer at the black arrow in Ahi1−/− retina). By PN24, significant loss of photoreceptor cells was observed in the outer nuclear layer (Fig. 2A). The photoreceptor nuclear layer showed progressive decline in thickness, and was absent in mice at PN200 (Fig. 2A). At PN22, apoptosis was apparent in the photoreceptor cells in retinas from Ahi1−/− mice, but not in retinas from Ahi1+/+ littermates (Fig. 2B).
Figure 2.
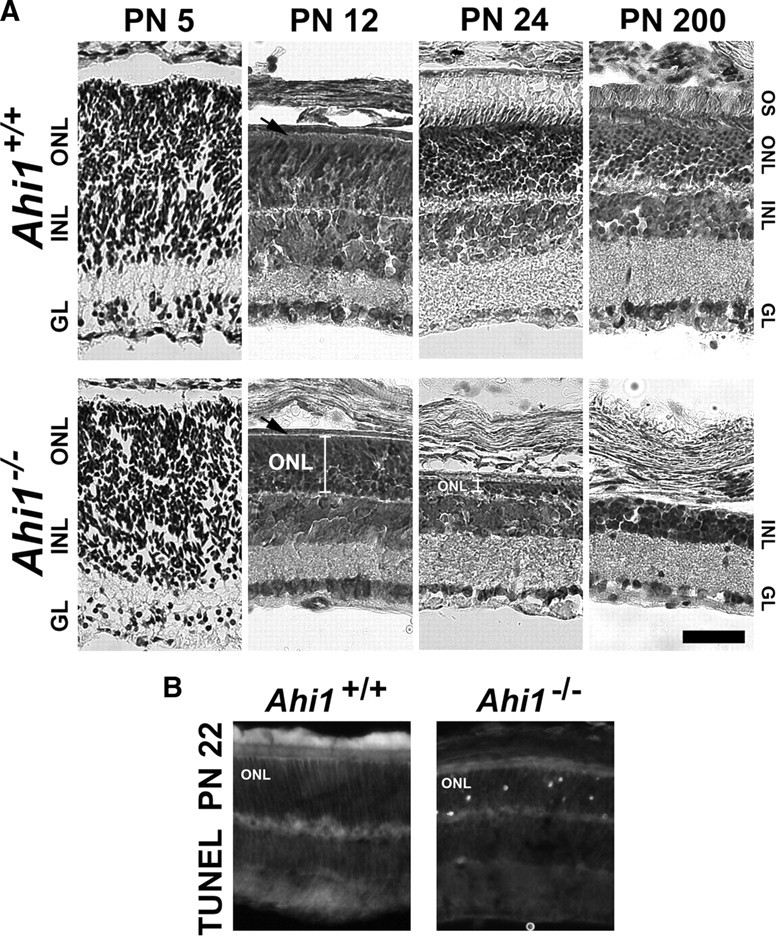
Retinal degeneration in the photoreceptors (outer nuclear layer) of mice with a targeted deletion of Ahi1. A, Cresyl violet staining of retinas from age-matched, wild-type mice (Ahi1+/+; top row) and mice with a targeted deletion of Ahi1 (Ahi1−/−; bottom row) at PN5, PN12, PN24, and PN200, demonstrating the progressive degeneration of the photoreceptor layer in Ahi1−/− retina, but not in Ahi1+/+ retina. The white brackets in the images from the Ahi1−/− retinas at PN12 and PN24 highlight the decrease in size of the photoreceptor layer. The black arrows are pointing to the outer segment layer in Ahi1+/+ retina and the absence of this layer in Ahi1−/− retina. Scale bar, 50 μm. B, TUNEL staining of apoptotic photoreceptor nuclei at PN22 in Ahi1−/− retinas (right), but not in Ahi1+/+ retinas (left). INL, Inner nuclear layer; IGL, ganglion cell layer; ONL, outer nuclear layer; OS, outer segments.
Studies have demonstrated that rod photoreceptor cells are born late in embryonic development in rodents, and further, that rhodopsin expression can be detected in late embryonic development in a small number of rod photoreceptors (Morrow et al., 1998; Rutherford et al., 2004). Therefore, to differentiate whether the absence of the photoreceptor layer in Ahi1−/− mice was attributable to an error in retinal developmental, or whether it was the result of a degenerative process, we examined the retinas of mice at earlier time points. Examination of immature photoreceptor cells in E18.5 pups, using rhodopsin antibodies, showed rhodopsin immunostaining in some cells both in Ahi1+/+ and in Ahi1−/− mice, consistent with the idea that early photoreceptor cell development is not disrupted in Ahi1−/− mice (Fig. 3A). Similarly, no major defects were observed in the developing Ahi1−/− retinas at PN5 (Fig. 2A), suggesting that the retinal degeneration occurs after cell differentiation. In addition, the appearance of rhodopsin-containing immature outer segments suggests that, at least initially, immature outer segments could form in the Ahi1−/− retina (Fig. 3A, white arrows).
Figure 3.

Basal bodies in retinal photoreceptors and early photoreceptor cell development were not abnormal in E18.5 Ahi1−/− mice. A, Rhodopsin-positive photoreceptor cells (red) in the outer retina extend processes that terminate in broadened segments in retinas from day E18.5 Ahi1+/+ and Ahi1−/− mice, characteristic of developing connecting cilia. Arrows are pointing to immature rhodopsin-positive outer membranes. DNA is visualized with DAPI (blue). B, Day E18.5 mouse retinas from Ahi1+/+ and Ahi1−/− mice were immunostained for the ciliary basal body (centriole) marker, γ-tubulin (green). Scale bars, 20 μm. BB, Basal bodies; ONL, outer nuclear layer.
Normal basal body localization in embryonic Ahi1−/− mice
To determine whether primary cilium formation is initiated in the absence of Ahi1, we examined the retinas of Ahi1−/− and Ahi1+/+ pups at day E18.5. Basal bodies (visualized via antibodies to the basal body marker, γ-tubulin) were found to be properly targeted in the apical region of the photoreceptor neuroblasts in Ahi1−/− retinas (Fig. 3B). Since γ -tubulin was distributed at the outer edge of the outer nuclear layer in both Ahi1−/− and Ahi1+/+ pups, we concluded that basal body mispositioning is not the cause of the impaired development of the photoreceptor outer segments.
Absence of photoreceptor outer segments in Ahi1−/− mice before degeneration
We examined further whether photoreceptor sensory cilia were present in the PN12 retina. Whereas outer segments were discernible in light micrographs of epoxy-embedded retinal sections from Ahi1+/+ mice, no outer segments were observed in sectioned Ahi1−/− retinas (Fig. 4A,B). These results were confirmed at the ultrastructural level. Outer segments in the Ahi1+/+ retina appeared, at PN12, as straight, parallel cylinders projecting from inner segments toward the retinal pigment epithelium (Fig. 4C). In contrast, outer segments were completely absent in PN12 Ahi1−/− mice (Fig. 4D). No disc structures can be seen in the space between the inner segments and the retinal pigment epithelium (Fig. 4D). We further examined the structure of the photoreceptor sensory cilium components in Ahi1+/+ and Ahi1−/− retinas. The stacks of nascent discs were organized and were oriented perpendicular to the long axis of the axoneme that extended from the basal body and transition zone in Ahi1+/+ mice (Fig. 4E). In the Ahi1−/− mice, the basal body and transition zone showed structures comparable to that in the Ahi1+/+ retina (Fig. 4F). Strikingly, the axoneme was normal in appearance and extended into the space between the inner segment and the retinal pigment epithelium (Fig. 4F). However, no disc membranes were seen along the axoneme (Fig. 4F). Instead, the axoneme was surrounded, at its distal end, by disorganized membranous material (Fig. 4F).
Figure 4.
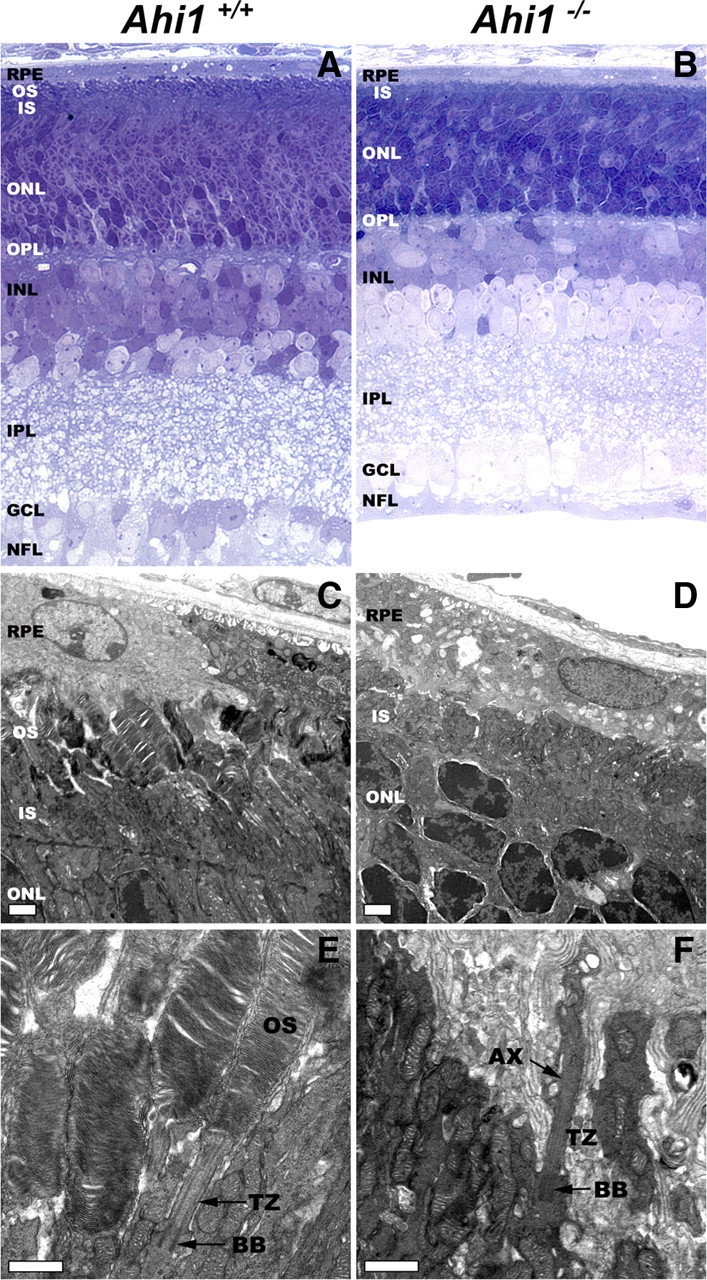
Ahi1−/− mice fail to develop photoreceptor outer segments. A, B, Light micrographs of epoxy-embedded sections of central retinas from PN12 Ahi1+/+ (A) and Ahi1−/− (B) mice. The outer segments did not develop in retinas from Ahi1−/− mice. The thickness of the outer nuclear layer was reduced, compared with retinas from Ahi1+/+ littermate controls. C, D, Electron microscopic images showing the ultrastructure of photoreceptors from PN12 Ahi1+/+ (C) and Ahi1−/− (D) mice. Whereas the outer segments in the Ahi1+/+ retina had a normal appearance, the outer segments in the retinas from Ahi1−/− mice were completely absent. Scale bars, 2 μm. E, F, Details of the photoreceptor sensory cilium components, in retinas from PN12 Ahi1+/+ (E) and Ahi1−/− (F) mice. The stacks of nascent discs, the axoneme, basal body, and transition zone had a normal appearance in Ahi1+/+ mice. However, in the Ahi1−/− mice, although the axoneme, basal body, and transition zone appeared normal, no disc membranes were observed along the axoneme; instead, the axoneme was surrounded by disorganized membranous material at its distal end. Scale bars, 1 μm. AX, Axoneme; BB, basal body; INL, inner nuclear layer; IPL, inner plexiform layer; IS, inner segments; GCL, ganglion cell layer; NFL, nerve fiber layer; ONL, outer nuclear layer; OPL, outer plexiform layer; OS, outer segments; RPE, retinal pigment epithelium; TZ, transition zone.
Altered photoreceptor outer segment protein targeting and levels in Ahi1−/− mice
Ahi1+/+ mice exhibited a high level of rhodopsin expression that was localized specifically to the outer segments by PN11 (Fig. 5A,B). The outer segments were seen to be more pronounced at the PN22 and PN42 time points (Fig. 5A). Although Ahi1−/− retinas also exhibited strong rhodopsin expression, rhodopsin was not present in the outermost layers of the outer nuclear layer (outer segments). Instead, it was aberrantly distributed to the inner side of the outer nuclear layer (cell bodies), as well as in the inner segments of the photoreceptors (Fig. 5A,B). Photoreceptor degeneration, as indicated by a thinning of the outer nuclear layer, was observed in retinas from Ahi1−/− mice by PN22, and was nearly complete by PN42; at the latter time point, only a few rhodopsin-positive cells remained, and the outer nuclear layer was only approximately one cell thick (Fig. 5A, high-magnification image).
Figure 5.
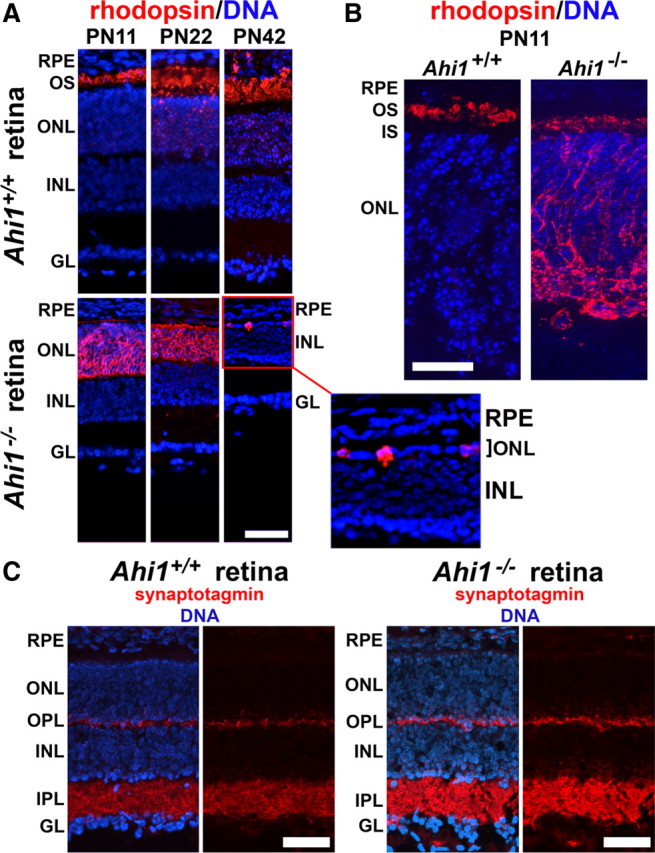
Photoreceptor outer segments fail to mature in retinas from Ahi1−/− mice. A, Rhodopsin (red) is sparsely observed throughout the outer nuclear layer, but is highly present in photoreceptor outer segments in Ahi1+/+ mice (top row). The outer segments are shown elongating from PN11 to PN42. In the Ahi1−/− retina (bottom row), a thin line of rhodopsin immunostaining (red) can be seen along the photoreceptor inner segments; however, most of the rhodopsin has been mistargeted to the outer nuclear layer (ONL). Additionally, the ONL is thinner than in the wild type at PN22, and only a few rhodopsin-positive cells remain by PN42 (see the high-magnification image of the ONL in the Ahi1−/− retina). DNA is visualized with DAPI (blue). Scale bar, 50 μm. B, Higher magnification of rhodopsin immunostaining (red) in PN11 mouse retinas from Ahi1+/+ and Ahi1−/− mice. DNA is visualized with DAPI (blue). Scale bar, 20 μm. C, The synaptic vesicle marker synaptotagmin is not mistargeted in retinas from Ahi1−/− mice. PN11 retinas from Ahi1+/+ and Ahi1−/− mice were immunostained for synaptotagmin (red). Synaptotagmin staining is restricted primarily to the inner and outer plexiform layers in both Ahi1+/+ and Ahi1−/− mice. DNA is visualized with DAPI (blue). Scale bars, 50 μm. INL, Inner nuclear layer; IPL, inner plexiform layer; GL, ganglion cell layer; OPL, outer plexiform layer; OS, outer segments; RPE, retinal pigment epithelium.
To determine the fate of other outer segment proteins in Ahi1−/− retinas, we performed localizations of several outer segment proteins. In Ahi1+/+ retinas, the outer segment proteins Cnga1, Rom1, and transducin were found in their normal locations in the outer segments (Figs. 6C, 7A,C). However, as seen with rhodopsin localization in Ahi1−/− retinas, Cnga1 and Rom1 were mistargeted to the inner segments and cell bodies of photoreceptor cells (Figs. 6B,D, 7B). Interestingly, the levels of Rom1 and transducin were notably lower in Ahi1−/− retinas than in Ahi1+/+ retinas, both immunohistochemically (Fig. 7, A vs B, C vs D) and by Western blotting (Fig. 7E–H).
Figure 6.

Abnormal location of photoreceptor outer segment proteins in retinas from Ahi1−/− mice. A–D, Frozen sections of retinas from Ahi1+/+ and Ahi1−/− mice were labeled with antibodies to the photoreceptor outer segment proteins: rhodopsin (A, B) and Cnga1 (C, D). The outer segment proteins rhodopsin and Cnga1 were mistargeted to the inner segments and cell bodies of photoreceptor cells in Ahi1−/− retinas (B, D), compared to their outer segment location in the Ahi1+/+ control retinas (A, C). INL, Inner nuclear layer; IS, inner segments; ONL, outer nuclear layer; OS, outer segments.
Figure 7.
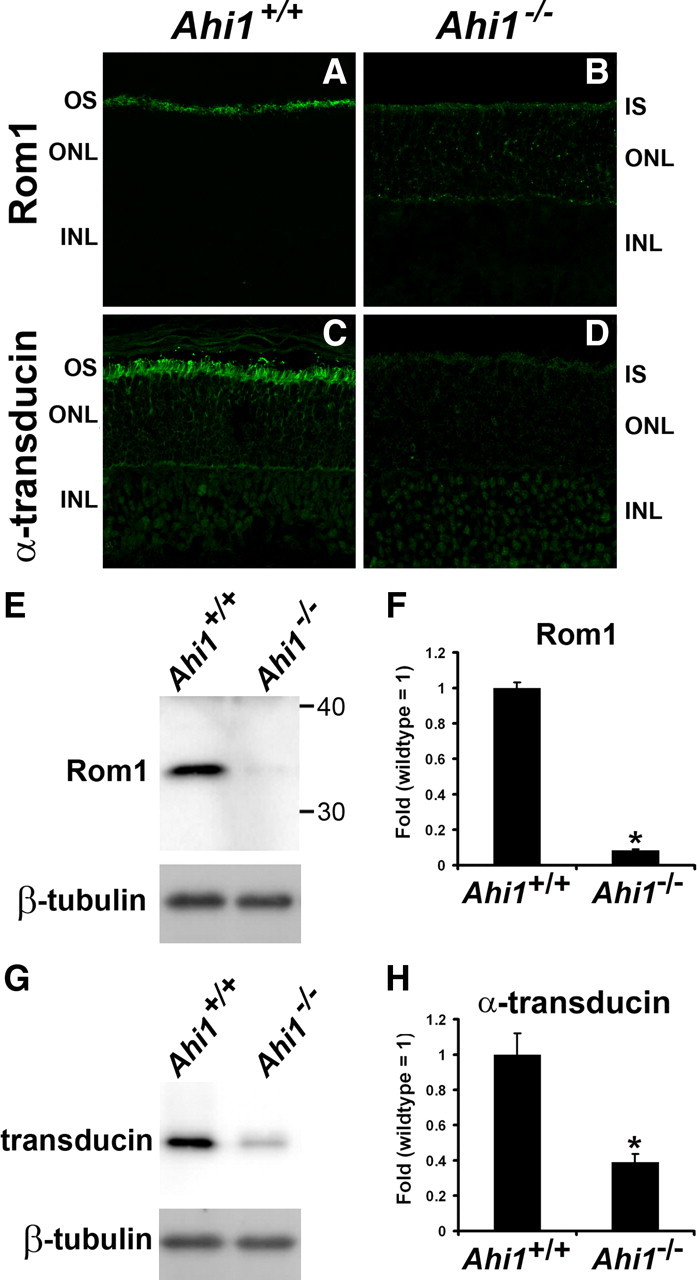
Decreased levels of photoreceptor outer segment proteins in retinas from Ahi1−/− mice. A–D, Frozen sections of retinas from Ahi1+/+ and Ahi1−/− mice were labeled with antibodies to other photoreceptor outer segment proteins: Rom1 (A, B), and α-transducin (C, D). The outer segment protein Rom1 was mistargeted to the inner segments and cell bodies of photoreceptor cells in Ahi1−/− retinas (B) compared to their outer segment location in the Ahi1+/+ control retinas (A). Also, the levels of Rom1 and α-transducin were significantly reduced in Ahi1−/− retinas (A vs B, C vs D, respectively). E–H, These reductions in Rom1 and α-transducin levels in the photoreceptors were confirmed by Western blotting. The protein levels of endogenous Rom1 and α-transducin from dissected retinas from Ahi1+/+ and Ahi1−/− mice were analyzed by Western blotting (representative blots, E, G) and graphically displayed (F, H; n ≥ 3/genotype). The level of β-tubulin represents the loading control. The error bars represent SEM. Asterisks denote significance from Ahi1+/+ (Rom1, t6 = 23.55, p < 0.0001; α-transducin, t4 = 4.78, p < 0.001). INL, Inner nuclear layer; IS, inner segments; ONL, outer nuclear layer; OS, outer segments.
Nonciliary vesicular transport is not impaired in the retinas of Ahi1−/− mice
To determine whether the mistargeting of the outer segment proteins that we observed was attributable to a general defect in vesicular trafficking, we investigated the localization of a second class of vesicles that are targeted to a distinct cellular domain, the synaptic vesicles. For this, we determined whether these vesicles were being correctly targeted to the axon terminals. We performed a localization of the synaptic vesicle marker synaptotagmin in the retina, at a time point before the onset of retinal degeneration in Ahi1−/− mice. Synaptotagmin immunostaining of Ahi1+/+ retinas was strong in the plexiform (synaptic) layers of the retina, at PN11, consistent with the known distribution pattern of presynaptic markers in the retina (Fig. 5C, left). Ahi1−/− retinas showed an identical synaptotagmin pattern at PN11 (Fig. 5C, right). This result indicates that Ahi1 is not required for the transport of synaptic vesicles to the cell periphery, nor is it required for targeting of the presynaptic vesicles to the axon and presynapse. Apparently, Ahi1 is not required for synaptic vesicle trafficking or targeting, but it is required for specific targeting of vesicles for outer segment formation.
The polarized vesicular trafficking protein, Rab8a, is reduced in the photoreceptors of Ahi1−/− mice
We examined Rab8a expression and distribution in retinas from day E18.5 Ahi1−/− pups. Rab8a was found to localize to photoreceptor neuroblasts in both Ahi1+/+ and Ahi1−/− retinas (Fig. 8A,B). However, Rab8a levels were reduced in the photoreceptors from Ahi1−/− mice (Fig. 8A,B). This result and previous work from our lab indicates that Rab8a is destabilized in the absence of Ahi1 (Hsiao et al., 2009).
Figure 8.
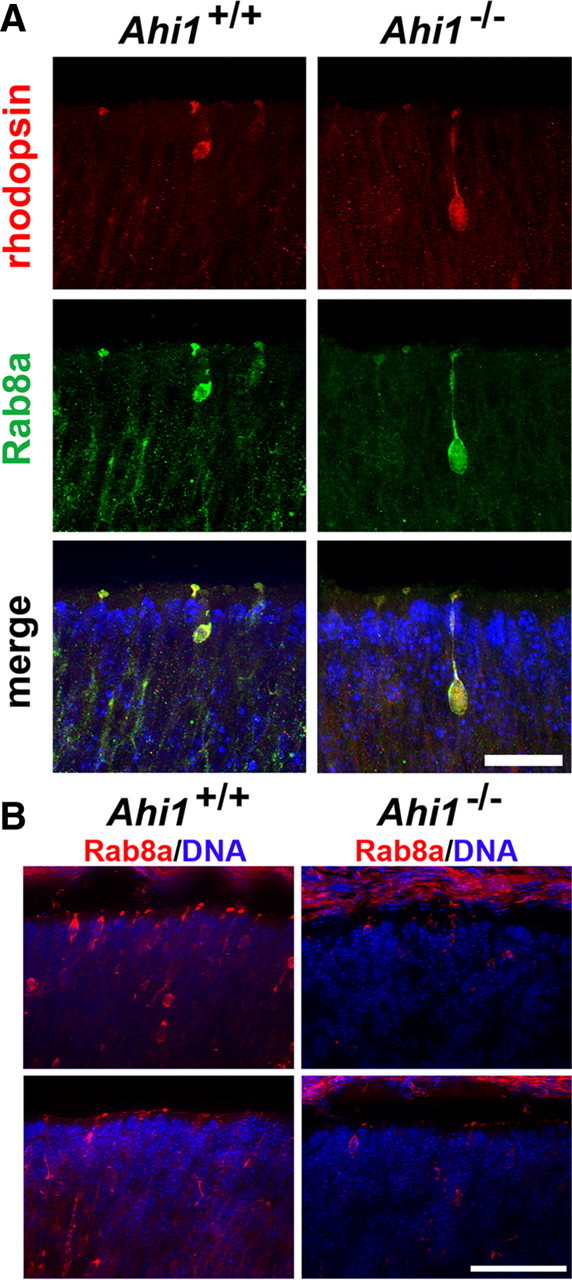
Reduced Rab8a expression in retinal photoreceptors from day E18.5 Ahi1−/− mice. A, Retinas from Ahi1+/+ and Ahi1−/− mice were immunostained for the membrane trafficking protein, Rab8a (green), and were colabeled with rhodopsin (red). Rab8a and rhodopsin colocalized in immature photoreceptor cells, in both Ahi1+/+ and Ahi1−/− mice. The same exposure settings were used for the Ahi1+/+ and Ahi1−/− retinas. DNA is visualized with DAPI (blue). Scale bar, 20 μm. B, Lower-magnification images of Rab8a (red) immunostaining in retinal photoreceptors from Ahi1+/+ and Ahi1−/− mice. Much weaker Rab8a immunostaining was observed in Ahi1−/− photoreceptors (right) than in Ahi1+/+ photoreceptors (left). The same exposure settings were used for the Ahi1+/+ and Ahi1−/− retinas. DNA is visualized with DAPI (blue). Scale bar, 50 μm.
Discussion
The data that we have presented demonstrate that Ahi1 is required for photoreceptor outer segment development, and that the lack of Ahi1 results in early photoreceptor degeneration. The failure of outer segment development in Ahi1−/− mice is complete, and may be the result of a ciliary trafficking defect, as evidenced by mistargeting of several outer segment proteins to the inner segments and cell bodies of photoreceptor cells in Ahi1−/− mice. The findings that basal bodies are positioned correctly, that axonemes begin to form, and that Rab8a levels are notably decreased in the photoreceptor cells of the Ahi1−/− mice are all consistent with the concept of such a ciliary trafficking defect. In contrast, nonciliary trafficking appears to be normal in the retinas of Ahi1−/− mice, as indicated by the normal distribution seen for synaptotagmin. The requirement of Ahi1 for photoreceptor outer segment formation is consistent with, and provides insight into, the retinal degeneration phenotype in patients with JBTS.
Basal bodies, which are a clear marker of cellular polarity and are also required for cilium formation, were found to be in their proper apical position in the photoreceptors of Ahi1−/− mice. This suggests that Ahi1 is not required for the positioning of the basal body to the photoreceptor membrane, or for cell polarity. Interestingly, at late embryonic time points, rhodopsin-containing extensions from the photoreceptors were observed that morphologically resembled immature cilia. Such features could indicate that in the retina, Ahi1 is not required for initiation of cilium formation, but rather for ciliary growth and maturity, and/or outer segment maintenance.
The role just proposed for Ahi1 in outer segment growth/maturity could arise from the interaction of Ahi1 with Rab8a. Knockdown of Ahi1 in ciliated cells has been shown to result in significant decreases in Rab8a expression (Hsiao et al., 2009). We confirm here that loss of Ahi1 results in a reduction in Rab8a in photoreceptors. This result implicates the involvement of Ahi1 in stabilization of Rab8a levels in photoreceptor cells (Hsiao et al., 2009). It is likely that the reduction in Rab8a, induced by loss of Ahi1, plays a role in the failure of the process involved in outer segment development and maintenance. Moreover, since the loss of Rab8a results in ciliary defects (Nachury et al., 2007; Hsiao et al., 2009), our present results implicate the involvement of both Rab8a and Ahi1 as critical mediators of the growth and maturation of photoreceptor outer segments.
It is important to note that the outer segments of rod and cone photoreceptor cells are specialized sensory cilia. Membrane-bound vesicles carrying newly synthesized outer segment proteins are transported from the post-Golgi membranes and fuse with the plasma membrane near the transition zone, through which the proteins are delivered to the outer segments (Young, 1968; Deretic, 2006). Rab8a is sited near the base of the transition zone in photoreceptor cells and is thought to be necessary for proper rhodopsin-containing vesicle docking and transport from the post-Golgi to the plasma membrane of the transition zone (Deretic et al., 1995). This idea is supported by studies in which alterations in Rab8a function induced accumulation of rhodopsin along the outer edge of the rod inner segments, with no transport of rhodopsin occurring through the transition zone into the outer segments (Deretic et al., 1995; Deretic, 1997; Moritz et al., 2001). Such a loss of rhodopsin transport ultimately leads to photoreceptor cell degeneration (Deretic et al., 1995; Deretic, 1997; Moritz et al., 2001). The abnormal pattern of rhodopsin distribution in photoreceptors from Ahi1−/− mice, followed by loss of the outer nuclear layer, strongly resembles the Rab8a phenotype in Xenopus (Moritz et al., 2001). Moreover, we have shown recently that knockdown of Ahi1 results in impairments of trafficking from the Golgi to the plasma membrane (Hsiao et al., 2009), and that Ahi1 is involved (likely through its effects on Rab8a) in vesicular trafficking in inner medullary collecting duct (IMCD3) cells. Nevertheless, general polarized vesicle transport is unlikely to be affected, given that synaptotagmin was seen to localize to the plexiform layers without any extraneous mistargeting to the outer nuclear layer in Ahi1−/− mice. Moreover, our synaptotagmin result indicates that Ahi1 is not required for the transport of synaptic vesicles to the axon and presynapse. As a whole, these results suggest that polarized vesicular trafficking is affected in ciliary, but not synaptically, targeted vesicles. Together, our results demonstrate that trafficking of proteins/vesicles to the outer segments is altered in the absence of Ahi1, suggesting that disruptions of this trafficking pathway are responsible for the failure of proper rhodopsin transport, and/or the growth and maintenance of the photoreceptor outer segments.
The absence of Ahi1 in photoreceptor cells results in a significant decrease in levels of Rab8a, as well as in decreases in levels of some outer segment proteins (i.e., transducin and Rom1), although not all of them (i.e., rhodopsin and Cnga1). These findings suggest either an involvement of Ahi1 in the stabilization of proteins in photoreceptors, or else an effect of Ahi1 on the transcription of the genes that encode these proteins. Future studies will be aimed at elucidating which of these two roles Ahi1 is actually playing in the cell.
The retinal degeneration observed in our Ahi1−/− mice recapitulates aspects of the retinal abnormalities that are observed in patients with JBTS (Parisi et al., 2006, 2007; Utsch et al., 2006), and demonstrates the importance of Ahi1 in retinal function. Recently, Cep290, another JBTS-related protein, was found to associate with Rab8a, and to be required for Rab8a targeting to the centrosome and to the basal body of the primary cilium (Kim et al., 2008; Tsang et al., 2008); such a distribution is similar to our result for Ahi1 (Hsiao et al., 2009). Both Ahi1−/− mice and Cep290-deficient mice show early postnatal photoreceptor degeneration (Chang et al., 2006). Interestingly, Cep290-deficient mice, in contrast to our Ahi1−/− mice, do appear to form outer segments (Chang et al., 2006), suggesting that photoreceptor degeneration that occurs in Ahi1−/− and Cep290-deficient mice is possibly mediated by two distinct pathways. In support of this, it is becoming increasingly clear that photoreceptor degeneration can occur through the presence of active opsin in the photoreceptor cell body (Alfinito and Townes-Anderson, 2002; Lee and Flannery, 2007; Chinchore et al., 2009). Given the common retinal degeneration phenotype between our Ahi1−/− mice and Cep290-deficient mice, this suggests the importance of polarized trafficking of rhodopsin for maintaining photoreceptor health; both Ahi1−/− and Cep290-deficient mice have abnormal rhodopsin expression in the photoreceptor cell body. It further suggests the possibility that the activity of Rab8a, in controlling rhodopsin localization, is regulated together by Ahi1 and Cep290 in the photoreceptor sensory cilia. However, since Cep290-deficient mice appear to form outer segments, this suggests a unique role of Ahi1 in the formation of the photoreceptor sensory cilia, possibly through a role in the trafficking of membrane constituents necessary for the formation of outer segments (Hsiao et al., 2009).
Many ciliopathies are characterized by loss of vision, and many other systemic disorders that cause retinal degeneration can have a ciliopathic component (Adams et al., 2007). In only a few of these have the intercellular transport defects been characterized. In Bardet-Biedl syndrome, opsin is mistargeted to the outer nuclear layer; however, outer segments are present, are only slightly stunted, and contain opsin (Nishimura et al., 2004; Abd-El-Barr et al., 2007). In Usher syndrome, there is, depending on which gene is mutated, either an impairment or else a complete block of opsin transport to the outer segment, with opsin accumulation seen in the connecting cilium. The onset of degradation in this case is seemingly a function of the severity of the transport defect (Liu et al., 1998, 1999). In both of the above disorders, rhodopsin is either able to traffic to the cilium or else it collects in or at the base of the cilium. The phenotype that we have seen in our Ahi1−/− mice is distinct from the above pattern in that a loss of outer segments occurs with little accumulation of rhodopsin at the distal ends of the inner segments. Rhodopsin-bearing vesicles may initially be docking and starting to accumulate; however, they are cleared through an endocytotic mechanism that randomly distributes rhodopsin across the cell. We have shown previously that targeting of endocytotic vesicles is impaired in Ahi1-knockdown cells (Hsiao et al., 2009). Another explanation could be mistargeting of rhodopsin-containing post-Golgi vesicles, causing most vesicles to shuttle indiscriminately, thereby preventing accumulation in the inner segments and also causing the high level of rhodopsin accumulation seen at the inner side of the photoreceptors. Such mistargeting could be attributable to loss of the interaction between Ahi1 and Rab8a, which we have shown previously to exist (Hsiao et al., 2009).
We have shown that Ahi1, a gene that causes Joubert syndrome when mutated, is required for the maintenance of photoreceptor outer segments, through the proper preservation of polarized vesicular trafficking to the cilia, and when absent results in retinal degeneration. This is consistent with findings from other mouse models of photoreceptor degeneration that show failure of outer segment formation. A classic example is the homozygous retinal degeneration slow mice, which have a mutation in the peripherin 2 (Prph2) gene, which encodes the peripherin 2 protein that is thought to be required for outer segment disc formation (Molday, 1998). Mutations in PRPH2 cause retinitis pigmentosa and several dominant macular dystrophies in humans (Farrar et al., 1991; Kajiwara et al., 1991; Nichols et al., 1993; Wells et al., 1993). Last, our current findings not only suggest a role for Ahi1 in Rab8a-mediated vesicle trafficking (Hsiao et al., 2009), but also provide clues toward a better understanding of the pathogenesis of ciliopathies.
Footnotes
This work was supported in part by National Institutes of Health Grants EY12910 to E.A.P. and MH71801 to R.J.F., the Kirby Foundation (E.A.P.), the Foundation Fighting Blindness (E.A.P.), Research to Prevent Blindness (E.A.P.), the Rosanne Silbermann Foundation (E.A.P.), and March of Dimes Foundation Grant 5-FY09-29 to R.J.F. We thank Dr. Adriana Verschoor (Wadsworth Center) for critical reading and input on our manuscript as well as the Wadsworth Center for allowing us to maintain our animal colonies there since 2005.
References
- Abd-El-Barr MM, Sykoudis K, Andrabi S, Eichers ER, Pennesi ME, Tan PL, Wilson JH, Katsanis N, Lupski JR, Wu SM. Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet-Biedl syndrome. Vision Res. 2007;47:3394–3407. doi: 10.1016/j.visres.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams NA, Awadein A, Toma HS. The retinal ciliopathies. Ophthalmic Genet. 2007;28:113–125. doi: 10.1080/13816810701537424. [DOI] [PubMed] [Google Scholar]
- Alfinito PD, Townes-Anderson E. Activation of mislocalized opsin kills rod cells: a novel mechanism for rod cell death in retinal disease. Proc Natl Acad Sci U S A. 2002;99:5655–5660. doi: 10.1073/pnas.072557799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annual review of genomics and human genetics. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- Bascom RA, Manara S, Collins L, Molday RS, Kalnins VI, McInnes RR. Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identifies a disk rim protein family implicated in human retinopathies. Neuron. 1992;8:1171–1184. doi: 10.1016/0896-6273(92)90137-3. [DOI] [PubMed] [Google Scholar]
- Beisson J, Wright M. Basal body/centriole assembly and continuity. Curr Opin Cell Biol. 2003;15:96–104. doi: 10.1016/s0955-0674(02)00017-0. [DOI] [PubMed] [Google Scholar]
- Chang B, Khanna H, Hawes N, Jimeno D, He S, Lillo C, Parapuram SK, Cheng H, Scott A, Hurd RE, Sayer JA, Otto EA, Attanasio M, O'Toole JF, Jin G, Shou C, Hildebrandt F, Williams DS, Heckenlively JR, Swaroop A. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet. 2006;15:1847–1857. doi: 10.1093/hmg/ddl107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Wu M, Sezate SA, McGinnis JF. Light threshold-controlled cone alpha-transducin translocation. Invest Ophthalmol Vis Sci. 2007;48:3350–3355. doi: 10.1167/iovs.07-0126. [DOI] [PubMed] [Google Scholar]
- Chinchore Y, Mitra A, Dolph PJ. Accumulation of rhodopsin in late endosomes triggers photoreceptor cell degeneration. PLoS Genet. 2009;5:e1000377. doi: 10.1371/journal.pgen.1000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook NJ, Molday LL, Reid D, Kaupp UB, Molday RS. The cGMP-gated channel of bovine rod photoreceptors is localized exclusively in the plasma membrane. J Biol Chem. 1989;264:6996–6999. [PubMed] [Google Scholar]
- den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Deretic D. Rab proteins and post-Golgi trafficking of rhodopsin in photoreceptor cells. Electrophoresis. 1997;18:2537–2541. doi: 10.1002/elps.1150181408. [DOI] [PubMed] [Google Scholar]
- Deretic D. A role for rhodopsin in a signal transduction cascade that regulates membrane trafficking and photoreceptor polarity. Vision Res. 2006;46:4427–4433. doi: 10.1016/j.visres.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Deretic D, Huber LA, Ransom N, Mancini M, Simons K, Papermaster DS. rab8 in retinal photoreceptors may participate in rhodopsin transport and in rod outer segment disk morphogenesis. J Cell Sci. 1995;108:215–224. doi: 10.1242/jcs.108.1.215. [DOI] [PubMed] [Google Scholar]
- Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, Gleeson JG. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004;75:979–987. doi: 10.1086/425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering JE, Kane K, Hsiao YC, Yao C, Shi B, Slowik AD, Dhagat B, Scott DD, Ault JG, Page-McCaw PS, Ferland RJ. Species differences in the expression of Ahi1, a protein implicated in the neurodevelopmental disorder Joubert syndrome, with preferential accumulation to stigmoid bodies. J Comp Neurol. 2008;511:238–256. doi: 10.1002/cne.21824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar GJ, Kenna P, Jordan SA, Kumar-Singh R, Humphries MM, Sharp EM, Sheils DM, Humphries P. A three-base-pair deletion in the peripherin-RDS gene in one form of retinitis pigmentosa. Nature. 1991;354:478–480. doi: 10.1038/354478a0. [DOI] [PubMed] [Google Scholar]
- Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, Shugart YY, Ruvolo M, Walsh CA. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet. 2004;36:1008–1013. doi: 10.1038/ng1419. [DOI] [PubMed] [Google Scholar]
- Gilula NB, Satir P. The ciliary necklace. A ciliary membrane specialization. J Cell Biol. 1972;53:494–509. doi: 10.1083/jcb.53.2.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol. 2007;18:1855–1871. doi: 10.1681/ASN.2006121344. [DOI] [PubMed] [Google Scholar]
- Hong DH, Yue G, Adamian M, Li T. Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J Biol Chem. 2001;276:12091–12099. doi: 10.1074/jbc.M009351200. [DOI] [PubMed] [Google Scholar]
- Hsiao YC, Tong ZJ, Westfall JE, Ault JG, Page-McCaw PS, Ferland RJ. Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum Mol Genet. 2009;18:3926–3941. doi: 10.1093/hmg/ddp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Hanna Z, Kaouass M, Girard L, Jolicoeur P. Ahi-1, a novel gene encoding a modular protein with WD40-repeat and SH3 domains, is targeted by the Ahi-1 and Mis-2 provirus integrations. J Virol. 2002;76:9046–9059. doi: 10.1128/JVI.76.18.9046-9059.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiwara K, Hahn LB, Mukai S, Travis GH, Berson EL, Dryja TP. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature. 1991;354:480–483. doi: 10.1038/354480a0. [DOI] [PubMed] [Google Scholar]
- Kim J, Krishnaswami SR, Gleeson JG. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet. 2008;17:3796–3805. doi: 10.1093/hmg/ddn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ES, Flannery JG. Transport of truncated rhodopsin and its effects on rod function and degeneration. Invest Ophthalmol Vis Sci. 2007;48:2868–2876. doi: 10.1167/iovs.06-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zhou J, Daiger SP, Farber DB, Heckenlively JR, Smith JE, Sullivan LS, Zuo J, Milam AH, Pierce EA. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest Ophthalmol Vis Sci. 2002;43:22–32. [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Lyubarsky A, Skalet JH, Pugh EN, Jr, Pierce EA. RP1 is required for the correct stacking of outer segment discs. Invest Ophthalmol Vis Sci. 2003;44:4171–4183. doi: 10.1167/iovs.03-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zuo J, Pierce EA. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J Neurosci. 2004;24:6427–6436. doi: 10.1523/JNEUROSCI.1335-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Tan G, Levenkova N, Li T, Pugh EN, Jr, Rux JJ, Speicher DW, Pierce EA. The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteomics. 2007;6:1299–1317. doi: 10.1074/mcp.M700054-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Saveliev A, Pierce EA. The severity of retinal degeneration in Rp1h gene-targeted mice is dependent on genetic background. Invest Ophthalmol Vis Sci. 2009;50:1566–1574. doi: 10.1167/iovs.08-2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ondek B, Williams DS. Mutant myosin VIIa causes defective melanosome distribution in the RPE of shaker-1 mice. Nat Genet. 1998;19:117–118. doi: 10.1038/470. [DOI] [PubMed] [Google Scholar]
- Liu X, Udovichenko IP, Brown SD, Steel KP, Williams DS. Myosin VIIa participates in opsin transport through the photoreceptor cilium. J Neurosci. 1999;19:6267–6274. doi: 10.1523/JNEUROSCI.19-15-06267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthew WD, Tsavaler L, Reichardt LF. Identification of a synaptic vesicle-specific membrane protein with a wide distribution in neuronal and neurosecretory tissue. J Cell Biol. 1981;91:257–269. doi: 10.1083/jcb.91.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday RS. Photoreceptor membrane proteins, phototransduction, and retinal degenerative diseases. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1998;39:2491–2513. [PubMed] [Google Scholar]
- Molday RS, MacKenzie D. Monoclonal antibodies to rhodopsin: characterization, cross-reactivity, and application as structural probes. Biochemistry (Mosc) 1983;22:653–660. doi: 10.1021/bi00272a020. [DOI] [PubMed] [Google Scholar]
- Moritz OL, Tam BM, Hurd LL, Peranen J, Deretic D, Papermaster DS. Mutant rab8 Impairs docking and fusion of rhodopsin-bearing post-Golgi membranes and causes cell death of transgenic Xenopus rods. Mol Biol Cell. 2001;12:2341–2351. doi: 10.1091/mbc.12.8.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow EM, Belliveau MJ, Cepko CL. Two phases of rod photoreceptor differentiation during rat retinal development. J Neurosci. 1998;18:3738–3748. doi: 10.1523/JNEUROSCI.18-10-03738.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- Nichols BE, Sheffield VC, Vandenburgh K, Drack AV, Kimura AE, Stone EM. Butterfly-shaped pigment dystrophy of the fovea caused by a point mutation in codon 167 of the RDS gene. Nat Genet. 1993;3:202–207. doi: 10.1038/ng0393-202. [DOI] [PubMed] [Google Scholar]
- Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, Andorf JL, Mykytyn K, Swiderski RE, Yang B, Carmi R, Stone EM, Sheffield VC. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A. 2004;101:16588–16593. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Snell W. The primary cilium: keeper of the key to cell division. Cell. 2007;129:1255–1257. doi: 10.1016/j.cell.2007.06.018. [DOI] [PubMed] [Google Scholar]
- Parisi MA, Doherty D, Eckert ML, Shaw DW, Ozyurek H, Aysun S, Giray O, Al Swaid A, Al Shahwan S, Dohayan N, Bakhsh E, Indridason OS, Dobyns WB, Bennett CL, Chance PF, Glass IA. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet. 2006;43:334–339. doi: 10.1136/jmg.2005.036608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300) Eur J Hum Genet. 2007;15:511–521. doi: 10.1038/sj.ejhg.5201648. [DOI] [PubMed] [Google Scholar]
- Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351–1363. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- Rutherford AD, Dhomen N, Smith HK, Sowden JC. Delayed expression of the Crx gene and photoreceptor development in the Chx10-deficient retina. Invest Ophthalmol Vis Sci. 2004;45:375–384. doi: 10.1167/iovs.03-0332. [DOI] [PubMed] [Google Scholar]
- Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- Scholey JM. Intraflagellar transport motors in cilia: moving along the cell's antenna. J Cell Biol. 2008;180:23–29. doi: 10.1083/jcb.200709133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholey JM, Anderson KV. Intraflagellar transport and cilium-based signaling. Cell. 2006;125:439–442. doi: 10.1016/j.cell.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Sorokin S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J Cell Biol. 1962;15:363–377. doi: 10.1083/jcb.15.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang WY, Bossard C, Khanna H, Peranen J, Swaroop A, Malhotra V, Dynlacht BD. CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev Cell. 2008;15:187–197. doi: 10.1016/j.devcel.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utsch B, Sayer JA, Attanasio M, Pereira RR, Eccles M, Hennies HC, Otto EA, Hildebrandt F. Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol. 2006;21:32–35. doi: 10.1007/s00467-005-2054-y. [DOI] [PubMed] [Google Scholar]
- Wells J, Wroblewski J, Keen J, Inglehearn C, Jubb C, Eckstein A, Jay M, Arden G, Bhattacharya S, Fitzke F, Bird A. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat Genet. 1993;3:213–218. doi: 10.1038/ng0393-213. [DOI] [PubMed] [Google Scholar]
- Young RW. Passage of newly formed protein through the connecting cilium of retina rods in the frog. J Ultrastruct Res. 1968;23:462–473. doi: 10.1016/s0022-5320(68)80111-x. [DOI] [PubMed] [Google Scholar]
- Zaki MS, Abdel-Aleem A, Abdel-Salam G, Marsh SE, Silhavy JL, Barkovich AJ, Ross ME, Saleem SN, Dobyns WB, Gleeson JG. The molar tooth sign: a new Joubert syndrome and related cerebellar disorders classification system tested in Egyptian families. Neurology. 2008;70:556–565. doi: 10.1212/01.wnl.0000277644.12087.fd. [DOI] [PubMed] [Google Scholar]