

Abstract
Haspin is a serine/threonine kinase required for completion of normal mitosis that is highly expressed during cell proliferation, including in a number of neoplasms. Consequently, it has emerged as a potential therapeutic target in oncology. A high throughput screen of approximately 140,000 compounds identified an acridine analog as a potent haspin kinase inhibitor. Profiling against a panel of 270 kinases revealed that the compound also exhibited potent inhibitory activity for DYRK2, another serine/threonine kinase. An optimization study of the acridine series revealed that the structure-activity relationship (SAR) of the acridine series for haspin and DYRK2 inhibition had many similarities. However, several structural differences were noted that allowed generation of a potent haspin kinase inhibitor (33, IC50 < 60 nM) with 180-fold selectivity over DYRK2. In addition, a moderately potent DYRK2 inhibitor (41, IC50 < 400 nM) with a 5.4-fold selectivity over haspin was also identified.
Haspin (Haploid Germ Cell-Specific Nuclear Protein Kinase), also know as Gsg2 (Germ Cell Specific Gene-2),1 is a serine/threonine kinase expressed in a variety of tissues (e.g. testis, bone narrow, thymus and spleen) and in proliferating cells.2 It is also highly expressed in a number of neoplasms, including Burkitt’s lymphoma3 and diffuse large B cell lymphomas.4 Haspin’s kinase activity functions during mitosis, where it has been shown to phosphorylate histone H3 at Thr-3 (H3T3).5, 6 This phosphorylation begins in G2/early prophase, becomes maximal during prometaphase/metaphase and then diminishes during anaphase.5, 7 Depletion of haspin by RNA interference significantly reduces H3 Thr-3 phosphorylation in cells and prevents normal completion of mitosis.5, 8, 9
Human haspin, consisting of 798-amino acids, contains a C-terminal kinase domain that is a divergent member of the eukaryotic protein kinase (ePK) superfamily.10, 11 Sequence comparisons and recent crystal structures reveal that haspin contains a number of structural differences from other ePKs, particularly in the C-terminal lobe of the kinase domain. For example, the highly conserved DFG motif involved in ATP binding and the APE motif involved in stabilizing the activation loop in many ePKs are altered. Haspin adopts a constitutively active conformation and conserved serine, threonine and tyrosine residues are absent from its activation loop.10, 11
DYRKs (Dual-specificity Tyrosine-regulated Kinases) belong to the CMGC family of ePKs and contain a conserved kinase domain and adjacent N-terminal DYRK homology box. This group of kinases can be further divided into class 1 kinases (DYRK1A and 1B) that have an N-terminal nuclear localization signal and a C-terminal PEST region and class 2 kinases (DYRK2, 3 and 4), which lack these motifs and are predominantly cytosolic. Although DYRKs phosphorylate substrates on serine or threonine residues, their activity depends upon autophosphorylation of an essential activation loop tyrosine during synthesis.12 DYRKs appear to contribute to regulation of an array of signaling pathways, including NFAT signaling in the brain and immune system, Hedgehog signaling, caspase activity during apoptosis, cell cycle progression and mitosis, and p53 activation in response to DNA damage.13–16
In order to study the role of haspin’s kinase activity in mitosis (and other cellular processes) and its potential role in cancer, we sought to identify and optimize inhibitors. Utilizing a recently developed time-resolved fluorescence resonance energy transfer (TR-FRET) high throughput screening (HTS) assay with histone H3 peptide as substrate and a europium-labeled phosphospecific monoclonal antibody for detecting phosphorylated substrate (H3T3ph),17 the acridine derivative 1 was discovered as a potent inhibitor (Figure 1; IC50 = 0.010 μM). Kinase profiling of 1 revealed potent DYRK2 inhibitory activity as well. Herein, we describe the structure-activity relationship (SAR) of the acridine series for both haspin and DYRK2 inhibition.
Figure 1.
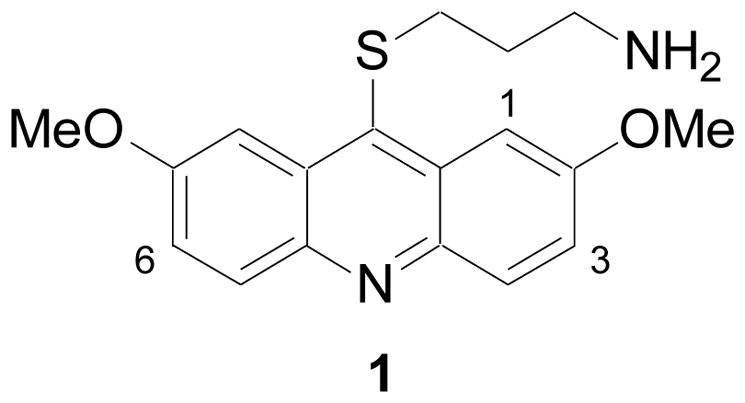
Haspin inhibitor identified by HTS. Also shown is the numbering system for acridines.
The synthesis of many of the acridine analogs was accomplished using the procedure outlined in Scheme 1. 2-Bromobenzoic acids 2 were coupled to anilines 3 using a copper-mediated procedure to give 4.18 Cyclization of 4 to 9-chloroacridines 5 was accomplished using phosphorus oxychloride. Treatment of 5 with P4S10 in the presence of DMPU gave 6. Alternatively, acid 4 was cyclized to ketone 7 in the presence of polyphosphoric acid (PPA), which was subsequently treated with Lawesson’s reagent with microwave (MW) heating at 110 °C to produce 6.19 The thioketone 6 could be alkylated with various amino-protected alkylbromides (BrCH2(CH2)nY; Y = NHBoc, NMeBoc, or NPhthalimide) in the presence of base (KOH) and the phase transfer catalyst tetrabutylammonium iodide (TBAI) in a mixture of toluene and water to give 8. Boc-protected analogs of 8 (Y = NHBoc or NMeBoc) upon treatment of 4N HCl in a mixture of 1,4-dioxane and methanol gave 9 (Z = NH2 or NHMe). Alternatively for analogs of 9 with Z = NH2, they could also be prepared directly from 6 via alkylation.
Scheme 1.

Reagents and conditions: (a) Cu, K2CO3, pentanol, 110 °C; (b) POCl3, 120 °C; (c) DMPU, P4S10, 95 °C; (d) PPA, 110 °C; (e) Lawesson’s reagent, THF, MW at 110 °C; (f) BrCH2(CH2)nY, KOH, TBAI (cat.), toluene, H2O, 110 °C; (g) BrCH2(CH2)nNH2•HBr, phenol, 80 °C; (h) 4N HCl in 1,4-dioxane, MeOH, rt.
Acridine analogs where the alkylamine groups were connected through an O or NH were prepared according to the procedure illustrated in Scheme 2. Ketone 10 was converted to 11 as previously described. Then a nucleophilic aromatic substitution with a mono-N-protected diamine followed by removal of the protecting group gave 12. Ketone 10 was also alkylated with N-Boc-protected 1-amino-3-bromopropane in the presence of KOH and phase-transfer catalyst (i.e. TBAI) followed by deprotection to give 13.
Scheme 2.

a) POCl3, 120 °C; (b) NH2(CH2)3NHBoc; (c) 4N HCl in 1,4-dioxane, MeOH, rt; (d) Br(CH2)3NHBoc, KOH, TBAI (cat.), toluene, H2O, 110 °C.
Acridine analogs where the alkylamine group was connected through a methylene were prepared using the method described in Scheme 3. Diphenylamine derivative 14 was condensed with acetic acid to give acridine analog 15.20 The methyl substituent in the 9-position was oxidized with selenium dioxide to give aldehyde 16.21 Addition of the anion of an N-Boc protected alkyne gave alcohol 17. Exposure of 17 to reducing conditions (Pd/C and Et3SiH) resulted in concomitant reduction of the alkyne and alcohol. Finally, removal of the protecting group on the amine yielded 18.
Scheme 3.

(a) InCl3, AcOH, MW at 230 °C; (b) SeO2, 1,4-dioxane, 80 °C; (c) HC≡CCH2NHBoc, n-BuLi, THF, −78 °C; (d) Pd/C, Et3SiH; (e) 4N HCl in 1,4-dioxane, MeOH, rt.
The synthesis of a tetrahydroacridine analog is outlined in Scheme 4. β-Ketoester 19 was allowed to react with 4-anisidine, 20, to produce 21. Cyclization of 21 in sulfuric acid gave ketone 22. This material was converted to the corresponding thioketone 23. Alkylation with 1-amino-3-bromopropane hydrobromide gave 24.
Scheme 4.

Reagents and conditions: (a) POCl3, cat. PTSA, 120 °C; (b) H2SO4, 120 °C; (c) DMPU, P4S10, 95 °C; (d) Br(CH2)3NH2 HBr, phenol, 80 °C.
Finally, a 2-phenylquinoline analog was synthesized accord to the procedure outlined in Scheme 5. The acetophenone derivative 25 was coupled with benzamide in the presence of a catalytic amount of CuI to give 26.22 Base-mediated cyclization of 26 gave 27. Conversion of this material to the thiol analog 28 was accomplished with Lawesson’s reagent. Then alkylation and deprotection as previously described for other analogs yielded 29.
Scheme 5.

(a) PhC(=O)NH2, 10 mol% CuI, (MeNHCH2)2, K2CO3, toluene, 170 °C; (b) NaOH, 1,4-dioxane, 110 °C; (c) Lawesson’s reagent, THF, MW at 150 °C; (d) Br(CH2)3NHBoc, KOH, TBAI (cat.), toluene, H2O, 110 °C; (e) 4N HCl in 1,4-dioxane, MeOH, rt.
Compound 1 was initially profiled for functional inhibitory activity against a panel of two hundred and seventy kinases at 10 μM. The results demonstrated that this compound was quite selective and only inhibited ten of the other kinases by ≥ 90% (Table S1 and Figure S1).23, 24 IC50 values were determined for these kinases (Table 1), with only five being potently inhibited (IC50 < 1 μM). DYRK2 was the most sensitive of these kinases (IC50 = 2 nM).23, 25
Table 1.
IC50 determinations for 1 against profile panel kinases with ≥ 90% inhibition at 10 μM.
| kinase | IC50 (μM) | kinase | IC50 (μM) |
|---|---|---|---|
| TRKB | 91 | ROS | 1.6 |
| CLK1 | 0.21 | HIPK1 | 1.4 |
| DYRK1A | 0.10 | HIPK2 | 1.3 |
| DYRK2 | 0.002 | PIM1 | 0.72 |
| DYRK3 | 0.019 | PIM2 | 56 |
For the SAR study, human haspin kinase inhibitory activity of the various compounds was evaluated using the same assay utilized for the HTS, except in the presence of varying test compound concentrations.17 DYRK2 kinase inhibitory activity was measured by 33P-incorporation into Woodtide peptide substrate in the presence of human DYRK2 containing an N-terminal GST-fusion protein and γ33P-ATP.23
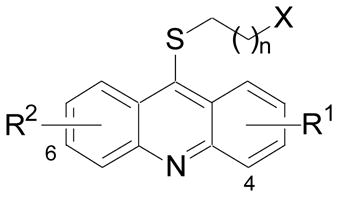
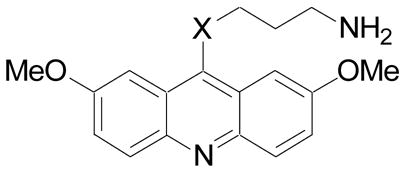
Only one of the methoxys in 1 appears necessary for potent haspin inhibition. When both of the methoxys were removed (30) inhibitory activity was dramatically reduced (Table 2). However, when only one of the methoxys was removed (31) or replaced with a methyl (32) or chlorine (33) potent activity (IC50 < 100 nM) was retained. The methoxy substituents of 1 could also be replaced with hydroxyls (34). Interestingly, transposition of the 7-methoxy to the 3-position (35) resulted in loss of activity. However, the 2-methoxy-3-chloro analog (36) was still quite active. The three aromatic rings that comprise the acridine also appeared necessary. Both 24 and 29 lacked haspin inhibitory activity. Next, the tether length between the thioether at the 9-position of the acridine and the primary amine was examined. Truncation (37) resulted in reduced activity, while addition of another methylene unit (38) had only a minimal impact on potency. Next, the contribution of the amine was examined. A secondary amine (39) was equally potent and a tertiary amine (40) only resulted in a slight decrease in activity. However, incorporation of the nitrogen into a phthalimide (41 and 42) resulted in a significant loss of activity. Finally, the thioether was examined (Table 3). Replacement of the thioether with an amine (12) or ether (13) was detrimental. But replacement of the sulfur with a methylene (18) retained potent inhibitory activity.
Table 2.
IC50 determinations for haspin and DYRK2 kinase inhibition.23
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | X | R1 | R2 | n | IC50 (μM) |
|
| haspin | DYRK2 | |||||
| 1 | NH2 | 2-OMe | 7-OMe | 2 | 0.01024 | 0.048 |
| 30 | NH2 | H | H | 2 | 9.8 | 7.7 |
| 31 | NH2 | 2-OMe | H | 2 | 0.017 | 0.19 |
| 32 | NH2 | 2-OMe | 7-Me | 2 | 0.070 | 0.43 |
| 33 | NH2 | 2-OMe | 7-Cl | 2 | 0.055 | 9.9 |
| 34 | NH2 | 2-OH | 7-OH | 2 | 0.044 | 0.17 |
| 35 | NH2 | 2-OMe, 3-OMe | H | 2 | ~ 7 | > 10 |
| 36 | NH2 | 2-OMe, 3-Cl | H | 2 | 0.079 | 9.4 |
| 37 | NH2 | 2-OMe | 7-OMe | 1 | 0.20 | 0.29 |
| 38 | NH2 | 2-OMe | 7-OMe | 3 | 0.040 | 0.036 |
| 39 | NHMe | 2-OMe | 7-OMe | 2 | 0.008 | 0.12 |
| 40 | NMe2 | 2-OMe | 7-OMe | 2 | 0.046 | 0.23 |
| 41 | Phth | 2-OMe | 7-OMe | 2 | 2.1 | 0.39 |
| 42 | Phth | 2-OMe | H | 2 | 8.8 | 1.3 |
Phth: phthalimide.
Table 3.
IC50 determinations for haspin and DYRK2 kinase inhibition.23
 | |||
|---|---|---|---|
| Cmpd | X | haspin IC50 (μM) | DYRK2 IC50 (μM) |
| 12 | NH | 1.7 | 6.5 |
| 13 | O | NA | NA |
| 18 | CH2 | 0.025 | 0.24 |
NA: Not active at 20 μM.
The SAR for DYRK2 inhibition had many similarities to that observed for haspin inhibition with some notable exceptions. Both methoxy groups appear to be necessary for DYRK2 inhibitory activity. For example, removal of both methoxy groups (30) was very detrimental, while removal of one methoxy (31) still resulted in a significant erosion of potency. Likewise, replacement of one methoxy with a methyl (32) or a chlorine atom (33) was not as tolerated compared to the results observed for haspin inhibition. Transposition of the 7-methoxy to the 3-position (35) also lead to loss in activity. Similarly, and unlike in the haspin SAR, removal of the 7-methoxy and addition of a 3-chloro group (36) was not tolerated for DYRK2 inhibition. The three aromatic rings that comprise the acridine also appear necessary for DYRK2 inhibition, with both 24 and 29 lacking activity. The effect of the tether length between the thioether at the 9-position of the acridine and the primary amine (37 and 38) was the same as previously observed for haspin inhibition. For DYRK2 inhibition the primary amine was better than secondary (39) or tertiary (40) amines. Interestingly, incorporation of the nitrogen into a phthalimide resulted in a less dramatic impact on DYRK2 inhibition compared to haspin and provided a moderately potent analog (41) that was 5.4-fold selective for DYRK2 verses haspin. Similar to the observations made with haspin inhibition, replacement of the thioether with an amine (12) or ether (13) was detrimental to DYRK2 inhibition, while replacement of the sulfur with a methylene (18) was tolerated, albeit with a 5-fold reduction in potency.
In conclusion, a SAR study of the acridine derivative 1, identified utilizing a recently developed HTS assay, was conducted for both haspin and DYRK2 inhibition. The study revealed that several structural features of 1, such as the three acridine aromatic rings, the presence of one or both methoxy groups, a three or four methylene tether between the thioether and the acridine, and a thioether or CH2 (but not an amine or ether) link to the acridine were necessary for both haspin and DYRK2 inhibition. However, several structural differences were noted that allowed generation of a potent haspin kinase inhibitor LDN-209929 (33, IC50 < 60 nM) with 180-fold selectivity verses DYRK2. In addition, a moderately potent DYRK2 inhibitor LDN-211848 (41, IC50 < 400 nM) with a 5.4-fold selectivity verses haspin was also identified. Additional optimization of the acridine series, potentially utilizing co-crystallization of these inhibitors with haspin and DYRK2, might provide more potent and selective inhibitors for both kinases. In addition, these inhibitors will serve as valuable molecular probes to study the cellular functions of these kinases and their potential roles in various diseases.
Supplementary Material
Acknowledgments
The authors thank Partners Healthcare for financial support. This work was also supported in part by NIH grant R01CA122608 to JMGH.
Footnotes
Supplementary data associated with this article can be found, in the online version, at ().
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Tanaka H, Yoshimura Y, Nozaki M, Yomogida K, Tsuchida J, Tosaka Y, Habu T, Nakanishi T, Okada M, Nojima H, Nishimune Y. J Biol Chem. 1999;274:17049. doi: 10.1074/jbc.274.24.17049. [DOI] [PubMed] [Google Scholar]; (b) Tanaka H, Yoshimura Y, Nishina Y, Nozaki M, Nojima H, Nishimune Y. FEBS Lett. 1994;355:4. doi: 10.1016/0014-5793(94)01155-9. [DOI] [PubMed] [Google Scholar]
- 2.Higgins JMG. Gene. 2001;267:55. doi: 10.1016/s0378-1119(01)00387-0. [DOI] [PubMed] [Google Scholar]
- 3.Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ, Greiner TC, Weisenburger DD, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Delabie J, Rimsza LM, Braziel RM, Grogan TM, Campo E, Jaffe ES, Dave BJ, Sanger W, Bast M, Vose JM, Armitage JO, Connors JM, Smeland EB, Kvoloy S, Holte H, Fisher RI, Miller TP, Montserrat E, Wilson WH, Bahl M, Zhao H, Yang L, Powell J, Simon R, Chan WC, Staudt LM. N Engl J Med. 2006;354:2431. doi: 10.1056/NEJMoa055759. [DOI] [PubMed] [Google Scholar]
- 4.Rosenwald A, Alizadeh AA, Widhopf G, Simon R, Davis RE, Yu X, Yang L, Pickeral OK, Rassenti LZ, Powell J, Botstein D, Byrd JC, Grever MR, Cheson BD, Chiorazzi N, Wilson WH, Kipps TJ, Brown PO, Staudt LM. J Exp Med. 2001;194:1639. doi: 10.1084/jem.194.11.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai J, Sultan S, Taylor SS, Higgins JMG. Genes Dev. 2005;19:472. doi: 10.1101/gad.1267105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higgins JMG. Chromosoma. 2010;119:137. doi: 10.1007/s00412-009-0250-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polioudaki H, Markaki Y, Kourmouli N, Dialynas G, Theodoropoulos PA, Singh PB, Georgatos SD. FEBS Lett. 2004;560:39. doi: 10.1016/S0014-5793(04)00060-2. [DOI] [PubMed] [Google Scholar]
- 8.Dai J, Sullivan BA, Higgins JMG. Dev Cell. 2006;11:741. doi: 10.1016/j.devcel.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 9.Dai J, Kateneva AV, Higgins JMG. J Cell Sci. 2009;122:4168. doi: 10.1242/jcs.054122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Higgins JMG. Cell Mol Life Sci. 2003;60:446. doi: 10.1007/s000180300038. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Higgins JMG. Protein Sci. 2001;10:1677. doi: 10.1110/ps.49901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Eswaran J, Patnaik D, Filippakopoulos P, Wang F, Stein RL, Murray JW, Higgins JMG, Knapp S. Proc Natl Acad Sci USA. 2009;106:20198. doi: 10.1073/pnas.0901989106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Villa F, Capasso P, Tortorici M, Forneris F, de Marco A, Mattevi A, Musacchio A. Proc Natl Acad Sci USA. 2009;106:20204. doi: 10.1073/pnas.0908485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lochhead PA, Sibbet G, Morrice N, Cleghon V. Cell. 2005;121:925. doi: 10.1016/j.cell.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida K. Biochem Pharmacol. 2008;76:1389. doi: 10.1016/j.bcp.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 14.Chih-Chien K, Klancer R, Singson A, Seydoux G. Cell. 2009;139:560. doi: 10.1016/j.cell.2009.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laguna A, Aranda S, Barallobre MJ, Barhoum R, Fernández E, Fotaki V, Delabar JM, de la Luna S, de la Villa P, Arbonés ML. Dev Cell. 2008;15:841. doi: 10.1016/j.devcel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 16.Varjosola M, Borklund M, Cheng F, Syvänen H, Kivioja T, Kilpinen S, Sun Z, Kallioniemi O, Stunnenberg HG, He WW, Ojala P, Taipale J. Cell. 2008;133:537. doi: 10.1016/j.cell.2008.02.047. [DOI] [PubMed] [Google Scholar]
- 17.Patnaik D, Xian J, Glicksman MA, Cuny GD, Stein RL, Higgins JMG. J Biomol Screen. 2008;13:1025. doi: 10.1177/1087057108326081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orda-Zgadzaj M, Abraham W. Synthesis. 2007;21:3345. [Google Scholar]
- 19.Cava MP, Levinson MI. Tetrahedron. 1985;41:5061. [Google Scholar]
- 20.Joseph J, Kuruvilla E, Achuthan AT, Ramaiah D, Schuster GB. Bioconjug Chem. 2004;15:1230. doi: 10.1021/bc0498222. [DOI] [PubMed] [Google Scholar]
- 21.Tasior M, Gryko DT. Heterocycles. 2007;71:2735. [Google Scholar]
- 22.Jones CP, Anderson KW, Buchwald SL. J Org Chem. 2007;72:7968. doi: 10.1021/jo701384n. [DOI] [PubMed] [Google Scholar]
- 23.See supplementary data for details.
- 24.Carna Biosciences (http://www.carnabio.com/) determined a haspin inhibition IC50 = 22 nM for 1. The assay used full-length human haspin fused at the N-terminus to GST and expressed using baculovirus, histone H3 peptide as substrate and [ATP] of 100 μM.
- 25.Determined by Carna Biosciences. Full-length human DYRK2 fused at the N-terminus to GST and expressed using baculovirus, DYRKtide-F as substrate and [ATP] of 10 μM were used.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.