

Abstract
The aryl hydrocarbon receptor (AHR) is well known for its role in mediating the toxic and adaptive responses to xenobiotic compounds. Recent studies also indicate that AHR ligands are endogenously produced and may be essential for normal development. Previously, we showed that the endogenous enzyme, aspartate aminotransferase (AST), generates the AHR proagonist, indole-3-pyruvic acid (I3P), by deamination of its substrate L-tryptophan. We hypothesized that other enzymatic pathways capable of producing I3P may generate AHR agonists in vivo. We now demonstrate that the enzyme D-amino acid oxidase (DAAO) catalyzes the production of AHR agonists through the enzymatic conversion of D-tryptophan to I3P. Moreover, we provide evidence that the non-enzymatic oxidation and condensation of I3P is a critical step in the generation of receptor agonists by DAAO and AST. Products of this process include two novel agonists, 1,3-di(1H-indol-3-yl)propan-2-one and 1-(1H-indol-3-yl)-3-(3H-indol-3-ylidene) propan-2-one (characterized in the accompanying paper, Chowdhury et al., Chem. Res. Toxicol. tx-2009-000418), both of which can potently activate the AHR at concentrations in the nanomolar range. These results show that endogenous AHR activity can be modulated by I3P production from amino acid precursors through multiple enzymatic pathways, including those catalyzed by DAAO and AST.
Introduction
The aryl hydrocarbon receptor (AHR) is a basic-helix-loop-helix-PAS protein (bHLH-PAS) that mediates vertebrate responses to many polycyclic aromatic hydrocarbons and halogenated dioxins, such as benzo[a]pyrene and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), respectively (1, 2). In its inactive form, the AHR is associated with chaperone proteins in the cytosol. Upon ligand-binding, the receptor undergoes a structural transformation that leads to the presentation of the nuclear localization signal, resulting in the translocation of the AHR (3, 4). In the nucleus, the AHR heterodimerizes with a second bHLH-PAS protein, the aryl hydrocarbon nuclear translocator (ARNT) (5). The bHLH domains found in both the AHR and ARNT enable their interaction with cognate DNA binding sequences, known as dioxin responsive elements (DREs) that are present upstream of target genes (6). Genes up-regulated by the AHR include the cytochrome P450 monooxygenases, Cyp1a1, Cyp1a2, and Cyp1b1 (7). The enzymes encoded by these genes catalyze the biotransformation of their substrates, leading to the metabolism of many AHR ligands and their derivatives.
In addition to mediating a metabolic response to xenobiotic agonists, the AHR also plays an essential role in developmental and adult physiology (8, 9). Evidence of this includes the observation that mice harboring mutations at the Ahr locus exhibit phenotypes consisting of a reduced liver weight, a patent ductus venosus, cardiac hypertrophy, and altered ocular vascularization (8–12). Furthermore, the presence of a developmentally important endogenous ligand for the AHR is supported by a number of observations, including (i) TCDD-induced AHR activation in utero can rescue the patent ductus venosus in Ahr-hypomorphic pups (12), (ii) constitutive activation of the AHR is reduced by overexpression of the Cyp1a1 gene (13), and (iii) the AHR is highly expressed at a number of specific sites in the developing embryo (14, 15).
Our previous efforts to understand endogenous activation of the AHR led to the elucidation of a biosynthetic pathway involving tryptophan-derived agonists. We demonstrated that in the presence of L-Trp, aspartate aminotransferase (AST) upregulates AHR activity through the catalytic formation of I3P, a compound that non-enzymatically converts to an array of AHR-activating derivatives (16). Furthermore, we showed that these I3P derivatives can competitively inhibit CYP1A1-mediated EROD activity (16), signifying that they can recognize the substrate-binding site and are likely metabolized by AHR-regulated enzymes. In order to understand the response of the receptor to tryptophan metabolites, we searched for additional enzymatic processes involving I3P that can lead to AHR activation. Analogous to AST, DAAO catalyzes the oxidative deamination of D-amino acids to produce achiral α-keto acids (17). We hypothesized, therefore, that DAAO reactions that produce I3P will generate AHR-activating compounds. In this work, we show that the enzymatic reaction of D-tryptophan (D-Trp) with DAAO leads to AHR activity through the generation of potent receptor agonists derived from I3P. The chemical structures of these agonists are described in an accompanying publication (Chowdhury et al, Chem Res Tox, tx-2009-000418).
Materials and Methods
Chemicals
Amino acids were dissolved in phosphate-buffered saline (PBS) and sterile-filtered with 0.2 micron nitrocellulose membrane filters. Dimethoxysulfoxide (DMSO) was the solvent for β-naphthoflavone (BNF) and dicumarol. Stock solutions of ethoxyresorufin were prepared in ethanol. Porcine D-amino acid oxidase and sodium benzoate were obtained from Sigma-Aldrich (St. Louis, MO) and resuspended in PBS. I3P and all other reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Cell lines and Growth Conditions
All cell lines were cultured in Dulbecco’s Modified Eagle Media with high glucose (Invitrogen Corp., Carlsbad, CA), and unless otherwise noted, were supplemented with 1% penicillin-streptomycin (w/v), 10% fetal bovine serum (v/v), 1 mM sodium pyruvate, 2 mM L-glutamine, and 100 μM MEM non-essential amino acids (Invitrogen Corp.). Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2 (v/v). The human hepatoma 101L cell line contains a stably-integrated luciferase reporter driven by a DRE-containing segment of the human CYP1A1 promoter (a gift of Dr. R. Tukey, University of California, San Diego) (18). The mouse hepatoma H1L6.1c3 cell line contains a similar DRE-driven reporter and was a gift from Dr. M. Denison (University of California, Davis).
Reporter Assays
High pressure liquid chromatography (HPLC) peaks were reconstituted in 100% CH3OH in the dark. Serial dilutions of each peak were used to treat cells at the indicated concentrations. The 101L or H1L6.1c3 cells were seeded in 96-well tissue culture plates and treated at ~60–70% confluency in media containing 1% fetal bovine serum (v/v). Eighteen hours after treatment, the cells were washed with PBS and lysed with Cell Culture Lysis Buffer (Promega, Madison, WI). Plates were incubated in the dark for 15 min prior to reagent injection (Luciferase Assay System, Promega) and luminescence quantitation on a Perkin Elmer MicroLumat Plus luminometer (Waltham, MA).
Cell Viability Assay
In order to control for variations in cell survival, viability assays were carried out in parallel with luciferase assays. Cells were treated as described above, and after 18 h, media was removed and replaced with complete phenol red-free media containing 10% FBS and 10% MTS reagent (CellTiter 96® AQueous One Cell Proliferation Assay, Promega). Between 30–60 min later, plates were read on a Molecular Devices SpectraMax Plus 384 spectrophotometer (Molecular Devices, Sunnyvale, CA) set at 490 nm. These cell viability data were divided into the relative luciferase units that were generated as described above.
Plasmids and Transient Transfection
A plasmid containing the human DAAO was constructed by amplification of the open reading frame from a full-length cDNA clone (Clone ID 5184628; Open Biosystems, Huntsville, AL) using the primers OL5997 (5’-CCGCCGCCATGCGTGTGGTGGTGATTGGAGC-3’) and OL5998 (5’-CCTCAGAGGTGGGATGGTGGCAT-3’). The amplification product was cloned by ligation into the pTARGET™ mammalian expression vector by the T/A method (Promega, Madison, WI). To assess AHR response to DAAO expression, this plasmid was co-transfected with a plasmid containing the SV40 early promoter driving the lacZ gene (pCH110) and a vector with a DRE-containing segment of the human CYP1A1 promoter driving the firefly luciferase gene (18). These plasmids were transfected into murine hepatoma Hepa1c1c7 cells using the FuGENE 6 transfection reagent (Roche Diagnostics, Indianapolis, IN). At ~12–18 h post-transfection, the cells were treated with 300 μM D-Trp or PBS. After an additional 24 h, luciferase activity was assessed as a fraction of β-galactosidase activity, to control for transfection efficiency (Promega, Madison, WI). Statistical significance for all experiments was determined using Student’s t test.
Ethoxyresorufin O-deethylase (EROD) Activity Assay
Cultured 101L cells were seeded in a 96-well plate at ~60% density and treated with 2 U/mL DAAO and 300 μM D-Trp. After 48 h, EROD activity was assayed by adapting a protocol described elsewhere (19). Briefly, the cells were washed with PBS and lysed by one freeze-thaw cycle. To each well, 150 μL of 50 mM HEPES buffer containing 40 μM dicumarol and 6.7 μM ethoxyresorufin were added. The samples were equilibrated at 30 °C for 15 min, followed by the addition of 50 μL of 0.5 mM NAPDH to initiate the reaction. Fluorescence at 530 nm excitation and 590 nm emission wavelengths was detected using an Fmax fluorometer (Molecular Devices, Sunnyvale, CA) and normalized to the number of cells.
Sample Preparation and Analysis of I3P Derivatives by HPLC
A solution of 500 μM I3P (prepared in 0.5 L PBS) was incubated at 37 °C for 24 h in the dark. To extract the reaction products, the solution was passed over UNIBOND™ C18 SPICE™ cartridges (Analtech, Inc., Newark, DE) and the clear flow-through fraction was discarded. The retained red product was eluted from the cartridges with 2 mL of ethanol and collected in microfuge tubes. The samples were dried under a gentle stream of nitrogen and stored at −30 °C until use. Each sample tube was resuspended with 500 μL of 50% CH3OH and spun for 5 min at 14,000 × g to remove particulates. Fractionation of the samples was performed using the 1100 Series Liquid Chromatography system (Agilent, Santa Clara, CA). The samples were injected onto a Microsorb-MV™ C18 column (250 × 4.6 mm × 100Å, 5 μm particle size) and resolved with a gradient of CH3OH and water containing 0.05% triethylamine (Varian, Inc., Palo Alto, CA). The chromatographic resolution of the samples proceeded with 5 min of 50% CH3OH, then a gradient that increased from 50–100% CH3OH over 25 minutes, followed by 5 minutes of 100% CH3OH. Absorbance at 280 nm was monitored and fractions were collected every 15 s. Assessment of AHR activity was achieved by applying 5 μL of each collected fraction to 100 μL of media containing 101L cells at ~60–70% confluence. Cells were incubated for ~20 h then washed and assayed for luciferase activity.
Results
D-Amino Acid Oxidase catalyzes the production of AHR agonists
DAAO catalyzes the dehydrogenation of non-polar and hydrophobic D-amino acids (20). To determine whether DAAO activity could lead to production of AHR agonists, we tested the response of 101L luciferase reporter cells to increasing concentrations of the enzyme. Porcine DAAO (dissolved in PBS) was added to cell culture medium and incubated for 20 h. As shown in Figure 1, the addition of DAAO caused a dose-dependent increase in luciferase activity, where a 3-fold increase was observed when cells were treated with 3 U/mL DAAO, the highest concentration tested (Figure 1).
Figure 1. DAAO activates the AHR.
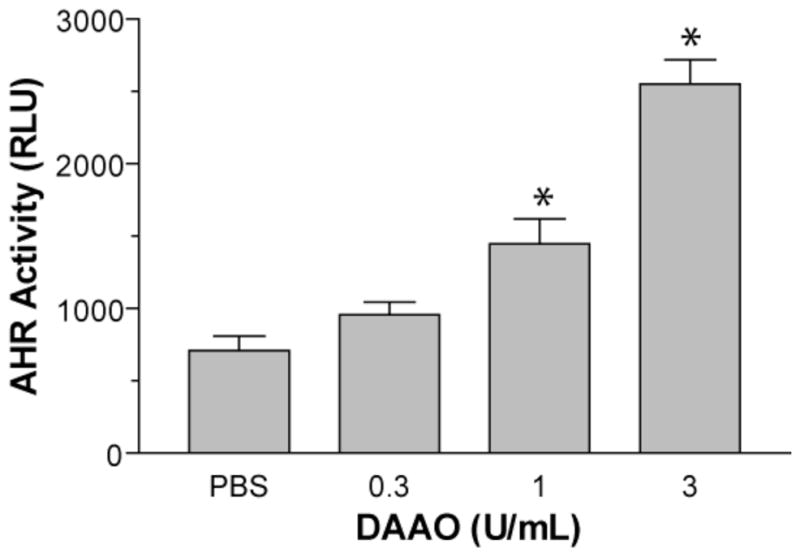
(A) 101L cells were treated with the indicated doses of DAAO or PBS for 18 h. Error bars: SD; (n = 3). *Denotes significant difference between treated and control samples, as determined by Student’s t test (p < 0.05).
D-Trp activates the AHR in the presence of DAAO
As agonists of the AHR are commonly small-molecule compounds (21), we next screened the 19 D-amino acids, including cystine (the oxidized dimeric form of cysteine), in order to identify the potential AHR agonist or agonist precursor. 101L cells were treated with 300 μM of each individual D-amino acid in the absence or presence of 2 U/mL DAAO. Following an overnight incubation, high luciferase activity was observed in the cell samples co-treated with DAAO and D-Trp or D-Tyr (Figure 2A). No activity was observed by the addition of either D-Trp or D-Tyr alone, indicating that activation of the AHR by the D-amino acids was dependent upon the enzyme. Furthermore, the L-enantiomers of Trp and Tyr did not activate the AHR in the presence of DAAO.
Figure 2. D-Trp activates the AHR in the presence of DAAO.


(A) Co-treatment of DAAO with D-Trp or D-Tyr activates DRE-driven luciferase activity. 101L cells were treated for ~20 h with 300 μM of each D-amino acid, L-Trp, L-Tyr, or PBS, plus 2 U/mL of DAAO or an equal volume of PBS. Gray bars: DAAO-treated. White bars: PBS-treated. Error bars: S.D; (n = 3). (B) The amino acids D-Trp and D-Tyr activate the AHR in a DAAO- and dose-dependent manner. 101L cells were exposed to increasing concentrations of D-Trp or D-Tyr, with or without 2 U/mL of DAAO. White bars: D-Trp. White hashed bars: D-Trp + DAAO. Black bars: D-Tyr. Gray bars: D-Tyr + DAAO. Error bars: S.D; (n = 3). (C) The reaction of DAAO with D-Trp induces EROD activity. 101L cells were treated with 300 μM D-Trp, 2 U/mL DAAO, or both for 48 h. Controls were treated with vehicle or 5 nM TCDD. Error bars: S.D; (n = 3).
To determine the relative potencies of D-Trp and D-Tyr for stimulation of AHR activity, dose response curves were generated by exposing 101L cells to concentrations of D-Trp and D-Tyr ranging from 1–500 μM (Figure 2B). When co-treated with DAAO, luciferase activity was observed beginning at 3 μM D-Trp or 10 μM D-Tyr. Maximal induction by each amino acid occurred at 300 μM, with D-Trp producing an 18-fold increase in luciferase activity (EC50 of 260 μM) and D-Tyr causing a 5-fold increase (EC50 of 93 μM). To understand the mechanism of AHR activation mediated by DAAO, we focused on D-Trp due to its greater potency.
To confirm the effect of D-Trp and DAAO on AHR activity, we next tested the induction of cytochromes P450 1A1 and 1A2, as measured by EROD assay. 101L cells were treated with 300 μM D-Trp, 2 U/mL of DAAO, or a combination of both reagents. After 48 h, the combined treatment of DAAO and D-Trp increased EROD activity 3.5-fold relative to the vehicle control, compared to the 15-fold induction by 5 nM TCDD (Figure 2C). Contrasting the effect on luciferase activity (Figure 1), incubating with DAAO alone did not increase EROD activity. This apparent lack of EROD induction by DAAO alone may be attributable to the reduced detection sensitivity of this enzyme activity assay.
Sodium benzoate inhibits DAAO-mediated AHR activity
Sodium benzoate is known to inhibit DAAO activity by impeding the interaction of the enzyme with the flavin prosthetic group (22–24). To confirm that the enzymatic function of DAAO is required for AHR activation by D-Trp and D-Tyr, we examined the effect of this inhibitor on the D-Trp-dependent activity. 101L cells were pre-treated for 45 min with 2 U/mL of DAAO and 20 mM sodium benzoate prior to the addition of 300 μM D-Trp. The pre-treatment of cells with sodium benzoate led to a 76% attenuation of luciferase activity induced by the combination of DAAO and D-Trp (Student’s t-test, p < 0.0001; Figure 3A). To control for any non-specific inhibitory affects of sodium benzoate, separate samples were also treated with 3 μM BNF in the presence or absence of sodium benzoate. While the BNF-induced activity was suppressed by 31% (p < 0.010), more extensive inhibition of D-Trp-induced activity was observed. These results indicate that AHR activation by D-Trp in the presence of DAAO was dependent upon the catalytic function of DAAO.
Figure 3. DAAO activity induces AHR activity.

(A) Sodium benzoate suppresses AHR activation by D-Trp/DAAO. 101L cells were pretreated for 45 min with 20 mM sodium benzoate (or an equivalent volume of PBS), then treated with 3 μM BNF or the combination of 2 U/mL DAAO and 300 μM D-Trp. White bars: PBS pre-treatment. Grey bars: sodium benzoate pre-treatment. Error bars: SD; (n = 4). Sodium benzoate greatly reduces the activity of DAAO/D-Trp (p < 0.0001) while affecting BNF to a lesser degree (p < 0.01). (B) Overexpression of DAAO activates the AHR. Mouse Hepa1c1c7 cells were transiently transfected with a DRE-driven luciferase expression plasmid, an SV40-lacZ vector, and either a plasmid encoding DAAO or the empty pTARGET vector. After 18 h, cells were incubated with 300 μM D-Trp or an equivalent volume of PBS for another 24 h. Luciferase activity was determined as a fraction of β-galactosidase activity to control for transfection efficiency. Overexpression of DAAO increased luciferase activity in both control- (p < 0.007) and D-Trp-treated samples (p < 0.003). White bars: PBS-treated. Grey bars: D-Trp-treated. Error bars: SD; (n = 3). Statistical analyses were determined using Student’s t-test.
Overexpression of DAAO activates the AHR
To confirm a role for DAAO in the regulation of AHR activity, we tested the effect of increased DAAO expression on the transcriptional activity of the receptor. The mouse hepatoma Hepa1c1c7 cell line was transiently co-transfected with plasmids that encode a DRE-driven luciferase gene, a lacZ reporter and the human DAAO gene. Cells mock-transfected with the pTARGET parent vector served as controls. The overexpression of DAAO led to a significant 2-fold increase in luciferase activity, both in the presence or absence of added D-Trp (Figure 3B). The addition of 300 μM D-Trp to the DAAO-transfected cells caused a slight enhancement of reporter activity (not significant at p 0.05; Figure 3B). These data indicate that the enzymatic activity of DAAO can lead to AHR activation.
Isolation of AHR agonists generated from I3P
Because D-Trp alone does not activate the AHR, the receptor activity must derive from I3P, the reaction product of DAAO with D-Trp. Indeed, we have previously shown that I3P is an AHR agonist precursor, whereby an aqueous solution of I3P incubated overnight at 37 °C was shown to produce numerous AHR-activating compounds (16). To purify and structurally clarify the agonists, we fractionated the products of an I3P solution. The resolution of the I3P reaction by reverse-phase HPLC revealed a complex group of derivatives, indicated by numerous 280 nm absorbance peaks ((16); Figure 4A). Furthermore, when 101L reporter cells were treated with eluate collected from the HPLC fractionation, several fractions that corresponded with chromatographic peaks (indicated by arrowheads) activated the AHR (Figure 4B).
Figure 4. The oxidation of I3P produces AHR agonists.

(A) Chromatogram of HPLC-fractionated I3P derivatives. A 500 μM solution of I3P was incubated for 24 h at 37 °C, protected from light. The reaction products were extracted and fractionated using HPLC as described under “Materials and Methods”. Arrowheads indicate the 280 nm absorbance of fractions that produced luciferase activity. (B) Luciferase activity of HPLC fractions. Five microliters of the elution fractions were added to 100 μL of culture media containing 101L reporter cells at 60–70% density. After 20 h of incubation, cells were washed and harvested for analysis of luciferase activity.
Using high resolution mass spectrometry (HRMS), NMV, UV and fluorescence spectroscopy, and subsequent analysis for reporter activation, two compounds were identified as agonists: 1,3-di(1H-indol-3-yl)propan-2-one (compound 1), and 1-(1H-indol-3-yl)-3-(3H-indol-3-ylidene) propan-2-one (compound 2) (Chowdhury et al, 2009 and Figure 6). Activation of the AHR by these molecules, as well as activation by the known AHR agonist, indirubin, was quantitated in both 101L (human) and H1L6.1c3 (mouse) cell lines and normalized to cell viability as determined by the MTS assay. Although we were not able to determine valid EC50 values due to effect saturation (compound 1) and cytotoxicity (compounds 1 and 2), both compounds appear to possess potencies comparable to that of indirubin. All three compounds proved to be more potent agonists in H1L6.1c3 cells relative to 101L cells, presumably because the mouse AHR has a greater affinity for ligand than does the human AHR.
Figure 6. Activation of the AHR by I3P derivatives: dose-response curves.

Compounds 1 and 2 were isolated by HPLC and reconstituted in CH3OH in the dark. 101L and H1L6.1c3 cells were then treated with each compound at increasing concentrations for 18 h, after which time luciferase assays were performed in parallel with MTS cell viability assays. Data are expressed as AHR activity (RLU) normalized to cell viability (MTS). Cytotoxicity was observed after treatment of H1L6.1c3 cells with compound 1 at the highest concentration tested. Likewise, toxicity was observed after treatment with either cell line with compound 2 at the highest dose. The potent AHR agonist indirubin was included for comparison. * Indicates lowest concentrations of significant activation.
Discussion
Numerous xenobiotics activate the AHR, but the physiologically relevant endogenous agonist(s) of the receptor remain elusive. We recently demonstrated that the mammalian enzyme AST could stimulate AHR activity by the catalytic deamination of L-Trp to I3P, a metabolite that readily converts to an array of AHR-activating compounds. To further understand the response of the receptor to tryptophan metabolites, we searched for additional biological sources of I3P and analyzed the activity of two isolated derivatives.
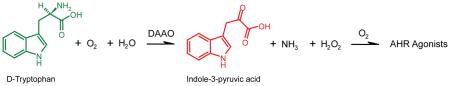
The enzyme DAAO was an obvious candidate as a generator of endogenous AHR agonists due to the similarity of its enzymatic process to the reaction of AST with Trp. We have previously shown that I3P, formed from the AST-mediated reaction of L-Trp with α-ketoglutaric acid (Figure 5A), is an AHR proagonist (16). While the reaction products α-ketoglutaric acid and L-glutamate do not activate the AHR, the “aging” or time-dependent incubation of an I3P solution was shown to increasingly activate the AHR (16). Although the D-enantiomer substrates of DAAO are less common than the L-enantiomers associated with AST, their biochemical reactions result in similar products, most notably I3P, which does not have a chiral center. Because DAAO catalyzes the deamination of D-amino acids to form the corresponding α-keto acids (17), the stimulation of AHR activity by DAAO/D-Trp is presumably also due to I3P (Figure 5B). In this study, we show that (i) the addition of DAAO to cell culture produces robust AHR activity as measured by a DRE-driven luciferase reporter and EROD assays, and (ii) this activity occurs through the enzymatic reaction with either D-Trp or D-Tyr. Of note, the lack of activity from L-Trp or L-Tyr is consistent with the substrate specificity of DAAO for D-amino acids. The receptor activation observed by the addition of DAAO alone to cell culture (Figure 1) or by the overexpression of DAAO in cells (Figure 3B) indicates that trace levels of D-enantiomer substrates exist in the cell culture system. As the DMEM media is supplemented with L-amino acids, this could be a source of D-amino acids. It is known that commercial preparations of common L-amino acids contain trace to percent level contamination of D-enantiomers (25). Additionally, D-amino acids may have arisen from racemase activity in serum.
Figure 5. Schematic of enzyme-mediated generation of AHR proagonist.

(A) AST catalyzes reaction of L-Trp with α-ketoglutaric acid to form I3P and L-glutamic acid. (B) D-Amino acid oxidase catalyzes deamination of D-Trp in the presence of molecular oxygen and water, to form I3P, ammonia, and hydrogen peroxide.
Our observation that an aqueous I3P solution can spontaneously yield numerous AHR-activating compounds led us to isolate and identify the chemical structures of select derivatives. The structural elucidation of two compounds using HRMS and NMR, as described in a corollary study (Chowdhury et al, 2009), indicates that these compounds may have formed from the dimerization of two I3P molecules. Capable of inducing DRE-driven reporters at concentrations in the nanomolar range, compounds 1 and 2 appear to possess potencies comparable to that of indirubin. The identification of these novel compounds thus demonstrates that I3P is a natural precursor of potent AHR agonists and supports the idea that the receptor mediates the metabolic response to both exogenous and endogenous agonists.
Our observations also provide insight on the indeterminate biological role of DAAO. Found in numerous species including yeast, bacteria, and mammals, DAAO is an enzyme with uncertain physiological relevance (26). DAAO is a flavoprotein that binds and deaminates D-amino acids, producing α-keto acids, hydrogen peroxide, and ammonia in the process (17). Despite the abundant expression of DAAO in the brain, liver, and kidneys of animals (27), the biological function of DAAO remains puzzling, because until recently, D-amino acids had not been found in high quantities in eukaryotes. In recent studies, D-Ser, -Asp, -Ala, -Leu and -Pro have been detected in the mammalian brain and central nervous system (28–31). Interestingly, the relatively high endogenous levels of D-serine along with its potent activation of the N-methyl-D-Asp receptor (NMDR) have led to the postulation that D-serine is an endogenous ligand of NMDR (32, 33). The discovery of a mammalian serine racemase (which catalyzes the conversion of L- to D-Ser), coupled with the detection of co-localized serine racemase and DAAO expression in brain tissue, in turn gave rise to the hypothesis that DAAO might play a role in regulating the endogenous level of D-Ser (34, 35).
DAAO is also hypothesized to play a role in metabolizing D-amino acids in mammals. Evidence of a metabolic role was gathered from observations of rodent models that lack normal DAAO activity. Unlike wildtype controls, rats deficient in DAAO expression do not experience nephrotoxicity when treated with high levels of D-serine, indicating that DAAO functions in the metabolism of renal D-amino acids (36). Furthermore, mice carrying a nonfunctional DAAO enzyme exhibit higher levels of D-amino acids in their tissues and urine than do wildtype mice (37, 38).
These current studies with DAAO were initiated after our earlier observation that I3P provided an in vivo route to AHR agonists (16). We postulated that I3P might come from sources in addition to the activity of AST on L-Trp. This study provided the additional biochemical evidence to support the generation of I3P from endogenous enzymatic activities. The L-Trp enantiomer is an essential amino acid that is primarily used for the production of protein and for the synthesis of kynurenine (39). Based on the estimate that D-Trp constitutes 0.2% of the physiological concentration of tryptophan (L- plus D-), which in human serum ranges from 50–71 μM (25, 40, 41), one might predict that very little I3P could be generated from the activity of DAAO in vivo. Although the serum concentrations of D-Trp under normal physiology are low, changes in D-amino acid levels have been observed under a variety of physiological states and cellular conditions. In this regard, high levels of D-amino acids in tissues are often associated with kidney failure, Alzheimer’s disease, and ageing (42–44). Moreover, while the cellular regulation of D-Trp is not well known, D-amino acids levels in normal serum and urine are known to be five- and ten-fold higher, respectively, than in the absence of DAAO (37). Based on these observations, we postulate that the collective production of I3P from DAAO and AST, as well as the production of related α-keto acids from tyrosine and phenylalanine could play a role in the endogenous production of AHR agonists in vivo under specific pathological conditions or cellular contexts.
Diet may also influence the level of endogenous AHR activation mediated by DAAO. Research from the last three decades show that certain methods of food fermentation or processing can result in the racemization of L-amino acids to D-enantiomers (45). Several strains of bacteria commonly found in fermentation produce high levels of D-Ala and D-Asp in their cell wall, as well as lower but significant amounts of other amino acids, including D-Tyr (45). In addition to the enzyme-catalyzed formation of D-amino acids associated with microorganisms, racemization occurring in food can also be catalyzed by extreme pH and/or heating (46–49). With heating at 95 °C, the level of D-Tyr in soybean protein was shown to increase by 30% (% of L + D) (50). Similarly, the alkali treatment (0.1 N NaOH, 75 °C, 3 h) of eight food proteins caused an increase of their D-Tyr composition to 15–35% (45). These multiple mechanisms of racemization in turn are underscored by the detection of D-amino acids in a broad range of food products that include wine and beer, coffee, bread, eggs, fruits, vegetables, meats and fermented dairy products such as cheese and yogurt. With processed foods comprising a third of the Western diet (45), they represent a potential source of AHR agonists derived from D-Trp and D-Tyr.
Compounds derived from three-substituted indoles comprise a growing class of AHR agonists. The endogenous metabolites indole acetic acid and tryptamine have both been reported to bind and activate the AHR in cell culture at concentrations in the micromolar range (51). A number of years ago, our own laboratory demonstrated the production of AHR agonists of the indolocarbazole class from the interaction of stomach acid on the plant metabolite indole-3-carbinol (52, 53). In addition, the potent AHR agonists 6-formylindolo[3,2-b]carbazole (FICZ) and 6,12-diinformylindolo[3,2-b]carbazole (dFICZ) have been identified from the UV-irradiation of tryptophan (54, 55). In general, indolocarbazoles are high affinity agonists of the AHR with EC50 in the picomolar range, which is comparable to that of TCDD (53, 56). Based on the observation that UV-B exposure causes an increase in CYP1A1 and CYP1B1 expression in human skin (57), high-potency AHR agonists derived from Trp have been suggested to occur endogenously (58).
In combination with earlier results from our lab, we have identified a novel mechanism of AHR agonist synthesis. We have shown that the endogenous enzymes DAAO and AST are capable of activating the AHR through chemical reactions with the D- and L-enantiomers, respectively, of Trp and Tyr. Moreover, we have shown that these pathways activate the AHR via I3P oligomerization to high-potency agonists. Although the physiological relevance of these compounds on endogenous AHR biology is as yet unknown, these results identify the DAAO/Trp/I3P pathway as a potential source of potent, intrinsically produced AHR ligands.
Acknowledgments
This work was supported by the National Institutes of Health Grants R37 ES005703, T32 CA009135, T32 ES007015 and P30 CA014520 (C.A.B.) and R37 CA090426, P30 ES000267 (F.P.G). One of the authors, C.A.B., has served as a scientific advisor to Dow Chemical Company in the area of dioxin toxicology. We thank Edward Glover for assistance with HPLC.
Abbreviations
- AHR
aryl hydrocarbon receptor
- DAAO
D-amino acid oxidase
- AST
aspartate aminotransferase
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- ARNT
aryl hydrocarbon nuclear translocator
- DREs
dioxin responsive elements
- PBS
phosphate-buffered saline
- BNF
β-naphthoflavone
- EROD
ethoxyresorufin O-deethylase
- HRMS
high resolution mass spectrometry
- I3P
indole-3-pyruvic acid
References
- 1.Sawyer T, Safe S. PCB isomers and congeners: induction of aryl hydrocarbon hydroxylase and ethoxyresorufin O-deethylase enzyme activities in rat hepatoma cells. Toxicol Lett. 1982;13:87–93. doi: 10.1016/0378-4274(82)90142-4. [DOI] [PubMed] [Google Scholar]
- 2.Poland A, Knutson J, Glover E. Studies on the mechanism of action of halogenated aromatic hydrocarbons. Clin Physiol Biochem. 1985;3:147–154. [PubMed] [Google Scholar]
- 3.Ikuta T, Eguchi H, Tachibana T, Yoneda Y, Kawajiri K. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J Biol Chem. 1998;273:2895–2904. doi: 10.1074/jbc.273.5.2895. [DOI] [PubMed] [Google Scholar]
- 4.Kazlauskas A, Sundstrom S, Poellinger L, Pongratz I. The hsp90 chaperone complex regulates intracellular localization of the dioxin receptor. Mol Cell Biol. 2001;21:2594–2607. doi: 10.1128/MCB.21.7.2594-2607.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 6.Whitlock JP., Jr Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- 7.Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci U S A. 1996;93:6731–6736. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor [see comments] Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- 10.Lahvis G, Lindell S, Thomas R, McCuskey R, Murphy C, Glover E, Bentz M, Southard J, Bradfield C. Portosystemic shunts and persistent fetal vascular structures in Ah-receptor deficient mice. Proc Natl Acad Sci U S A. 2000;97:10442–10447. doi: 10.1073/pnas.190256997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandez-Salguero PM, Ward JM, Sundberg JP, Gonzalez FJ. Lesions of aryl-hydrocarbon receptor-deficient mice. Veterinary Pathology. 1997;34:605–614. doi: 10.1177/030098589703400609. [DOI] [PubMed] [Google Scholar]
- 12.Walisser JA, Bunger MK, Glover E, Bradfield CA. Gestational exposure of Ahr and Arnt hypomorphs to dioxin rescues vascular development. Proc Natl Acad Sci U S A. 2004;101:16677–16682. doi: 10.1073/pnas.0404379101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang CY, Puga A. Constitutive activation of the aromatic hydrocarbon receptor. Mol Cell Biol. 1998;18:525–535. doi: 10.1128/mcb.18.1.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abbott BD, Birnbaum LS, Perdew GH. Developmental expression of two members of a new class of transcription factors: I. Expression of aryl hydrocarbon receptor in the C57BL/6N mouse embryo. Dev Dyn. 1995;204:133–143. doi: 10.1002/aja.1002040204. [DOI] [PubMed] [Google Scholar]
- 15.Jain S, Maltepe E, Lu MM, Simon C, Bradfield CA. Expression of ARNT, ARNT2, HIF1 alpha, HIF2 alpha, and Ah receptor mRNAs in the developing mouse. Mechanisms of Development. 1998;73:117–123. doi: 10.1016/s0925-4773(98)00038-0. [DOI] [PubMed] [Google Scholar]
- 16.Bittinger MA, Nguyen LP, Bradfield CA. Aspartate aminotransferase generates proagonists of the aryl hydrocarbon receptor. Mol Pharmacol. 2003;64:550–556. doi: 10.1124/mol.64.3.550. [DOI] [PubMed] [Google Scholar]
- 17.Berg CP. Physiology of the D-amino acids. Physiol Rev. 1953;33:145–189. doi: 10.1152/physrev.1953.33.2.145. [DOI] [PubMed] [Google Scholar]
- 18.Postlind H, Vu TP, Tukey RH, Quattrochi LC. Response of human CYP1-luciferase plasmids to 2,3,7,8-tetrachlorodibenzo-p-dioxin and polycyclic aromatic hydrocarbons. Toxicol Appl Pharmacol. 1993;118:255–262. doi: 10.1006/taap.1993.1031. [DOI] [PubMed] [Google Scholar]
- 19.Sanderson JT, Aarts JM, Brouwer A, Froese KL, Denison MS, Giesy JP. Comparison of Ah receptor-mediated luciferase and ethoxyresorufin-O-deethylase induction in H4IIE cells: implications for their use as bioanalytical tools for the detection of polyhalogenated aromatic hydrocarbons. Toxicol Appl Pharmacol. 1996;137:316–325. doi: 10.1006/taap.1996.0086. [DOI] [PubMed] [Google Scholar]
- 20.Konno R, Uchiyama S, Yasumura Y. Intraspecies and interspecies variations in the substrate specificity of D-amino acid oxidase. Comp Biochem Physiol B. 1982;71:735–738. doi: 10.1016/0305-0491(82)90490-4. [DOI] [PubMed] [Google Scholar]
- 21.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 22.Van den Berghe-Snorek S, Stankovich MT. Thermodynamic control of D-amino acid oxidase by benzoate binding. J Biol Chem. 1985;260:3373–3379. [PubMed] [Google Scholar]
- 23.Shindo H, Maeda T. Studies on the metabolism of D- and L-isomers of 3,4-dihydroxyphenylalanine (DOPA). VI. Metabolism of D-DOPA in rat kidney. Chem Pharm Bull (Tokyo) 1974;22:1721–1731. doi: 10.1248/cpb.22.1721. [DOI] [PubMed] [Google Scholar]
- 24.Gadda G, Negri A, Pilone MS. Reaction of phenylglyoxal with arginine groups in D-amino-acid oxidase from Rhodotorula gracilis. J Biol Chem. 1994;269:17809–17814. [PubMed] [Google Scholar]
- 25.Armstrong DW, Gasper M, Lee SH, Zukowski J, Ercal N. D-amino acid levels in human physiological fluids. Chirality. 1993;5:375–378. doi: 10.1002/chir.530050519. [DOI] [PubMed] [Google Scholar]
- 26.Pilone MS. D-Amino acid oxidase: new findings. Cell Mol Life Sci. 2000;57:1732–1747. doi: 10.1007/PL00000655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konno R, Yasumura Y. Mouse mutant deficient in D-amino acid oxidase activity. Genetics. 1983;103:277–285. doi: 10.1093/genetics/103.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunlop DS, Neidle A, McHale D, Dunlop DM, Lajtha A. The presence of free D-aspartic acid in rodents and man. Biochem Biophys Res Commun. 1986;141:27–32. doi: 10.1016/s0006-291x(86)80329-1. [DOI] [PubMed] [Google Scholar]
- 29.Hashimoto A, Nishikawa T, Hayashi T, Fujii N, Harada K, Oka T, Takahashi K. The presence of free D-serine in rat brain. FEBS Lett. 1992;296:33–36. doi: 10.1016/0014-5793(92)80397-y. [DOI] [PubMed] [Google Scholar]
- 30.Hashimoto A, Nishikawa T, Oka T, Takahashi K, Hayashi T. Determination of free amino acid enantiomers in rat brain and serum by high-performance liquid chromatography after derivatization with N-tert.-butyloxycarbonyl-L-cysteine and o-phthaldialdehyde. J Chromatogr. 1992;582:41–48. doi: 10.1016/0378-4347(92)80300-f. [DOI] [PubMed] [Google Scholar]
- 31.Hamase K, Konno R, Morikawa A, Zaitsu K. Sensitive determination of D-amino acids in mammals and the effect of D-amino-acid oxidase activity on their amounts. Biol Pharm Bull. 2005;28:1578–1584. doi: 10.1248/bpb.28.1578. [DOI] [PubMed] [Google Scholar]
- 32.Schell MJ, Molliver ME, Snyder SH. D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci U S A. 1995;92:3948–3952. doi: 10.1073/pnas.92.9.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schell MJ, Brady RO, Jr, Molliver ME, Snyder SH. D-serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J Neurosci. 1997;17:1604–1615. doi: 10.1523/JNEUROSCI.17-05-01604.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolosker H, Sheth KN, Takahashi M, Mothet JP, Brady RO, Jr, Ferris CD, Snyder SH. Purification of serine racemase: biosynthesis of the neuromodulator D-serine. Proc Natl Acad Sci U S A. 1999;96:721–725. doi: 10.1073/pnas.96.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dememes D, Mothet JP, Nicolas MT. Cellular distribution of D-serine, serine racemase and D-amino acid oxidase in the rat vestibular sensory epithelia. Neuroscience. 2006;137:991–997. doi: 10.1016/j.neuroscience.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 36.Maekawa M, Okamura T, Kasai N, Hori Y, Summer KH, Konno R. D-amino-acid oxidase is involved in D-serine-induced nephrotoxicity. Chem Res Toxicol. 2005;18:1678–1682. doi: 10.1021/tx0500326. [DOI] [PubMed] [Google Scholar]
- 37.Nagata Y, Konno R, Yasumura Y, Akino T. Involvement of D-amino acid oxidase in elimination of free D-amino acids in mice. Biochem J. 1989;257:291–292. doi: 10.1042/bj2570291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hashimoto A, Nishikawa T, Konno R, Niwa A, Yasumura Y, Oka T, Takahashi K. Free D-serine, D-aspartate and D-alanine in central nervous system and serum in mutant mice lacking D-amino acid oxidase. Neurosci Lett. 1993;152:33–36. doi: 10.1016/0304-3940(93)90476-2. [DOI] [PubMed] [Google Scholar]
- 39.Stone TW. Kynurenic acid antagonists and kynurenine pathway inhibitors. Expert Opin Investig Drugs. 2001;10:633–645. doi: 10.1517/13543784.10.4.633. [DOI] [PubMed] [Google Scholar]
- 40.Tcherkas YV, Kartsova LA, Krasnova IN. Analysis of amino acids in human serum by isocratic reversed-phase high-performance liquid chromatography with electrochemical detection. J Chromatogr A. 2001;913:303–308. doi: 10.1016/s0021-9673(00)01206-1. [DOI] [PubMed] [Google Scholar]
- 41.Pitkanen HT, Oja SS, Kemppainen K, Seppa JM, Mero AA. Serum amino acid concentrations in aging men and women. Amino Acids. 2003;24:413–421. doi: 10.1007/s00726-002-0338-0. [DOI] [PubMed] [Google Scholar]
- 42.Young GA, Kendall S, Brownjohn AM. D-Amino acids in chronic renal failure and the effects of dialysis and urinary losses. Amino Acids. 1994;6:283–293. doi: 10.1007/BF00813748. [DOI] [PubMed] [Google Scholar]
- 43.Nagata Y, Akino T, Ohno K, Kataoka Y, Ueda T, Sakurai T, Shiroshita K, Yasuda T. Free D-amino acids in human plasma in relation to senescence and renal diseases. Clin Sci (Lond) 1987;73:105–108. doi: 10.1042/cs0730105. [DOI] [PubMed] [Google Scholar]
- 44.Fuchs SA, Berger R, Klomp LW, de Koning TJ. D-amino acids in the central nervous system in health and disease. Mol Genet Metab. 2005;85:168–180. doi: 10.1016/j.ymgme.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 45.Friedman M. Chemistry, nutrition, and microbiology of D-amino acids. J Agric Food Chem. 1999;47:3457–3479. doi: 10.1021/jf990080u. [DOI] [PubMed] [Google Scholar]
- 46.Friedman M, Gumbmann MR. The utilization and safety of isomeric sulfur-containing amino acids in mice. J Nutr. 1984;114:2301–2310. doi: 10.1093/jn/114.12.2301. [DOI] [PubMed] [Google Scholar]
- 47.Friedman M, Gumbmann MR. The nutritive value and safety of D-phenylalanine and D-tyrosine in mice. J Nutr. 1984;114:2089–2096. doi: 10.1093/jn/114.11.2089. [DOI] [PubMed] [Google Scholar]
- 48.Casado FJ, Sanchez AH, Rejano L, Montano A. D-amino acid formation in sterilized alkali-treated olives. J Agric Food Chem. 2007;55:3503–3507. doi: 10.1021/jf0701685. [DOI] [PubMed] [Google Scholar]
- 49.Hayase F, Kato H, Fujimaki M. Racemization of amino acid residues in proteins and poly (L-amino acids) during roasting. J Agric Food Chem. 1975;23:491–494. doi: 10.1021/jf60199a055. [DOI] [PubMed] [Google Scholar]
- 50.Friedman M, Liardon R. Racemization kinetics of amino acid residues in alkali-treated soybean protein. J Agric Food Chem. 1985;33:666–672. [Google Scholar]
- 51.Heath-Pagliuso S, Rogers WJ, Tullis K, Seidel SD, Cenijn PH, Brouwer A, Denison MS. Activation of the Ah receptor by tryptophan and tryptophan metabolites. Biochemistry. 1998;37:11508–11515. doi: 10.1021/bi980087p. [DOI] [PubMed] [Google Scholar]
- 52.Bradfield CA, Bjeldanes LF. Effect of dietary indole-3-carbinol on intestinal and hepatic monooxygenase, glutathione S-transferase and epoxide hydrolase activities in the rat. Fd Chem Toxic. 1984;22:977–982. doi: 10.1016/0278-6915(84)90147-9. [DOI] [PubMed] [Google Scholar]
- 53.Bjeldanes LF, Kim JY, Grose KR, Bartholemew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: Comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc Natl Acad Sci USA. 1991;88:9543–9547. doi: 10.1073/pnas.88.21.9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rannug A, Rannug U, Rosenkranz HS, Winqvist L, Westerholm R, Agurell E, Grafstrom AK. Certain photooxidized derivatives of tryptophan bind with very high affinity to the Ah receptor and are likely to be endogenous signal substances. J Biol Chem. 1987;262:15422–15427. [PubMed] [Google Scholar]
- 55.Rannug U, Rannug A, Sjoberg U, Li H, Westerholm R, Bergman J. Structure elucidation of two tryptophan-derived, high affinity Ah receptor ligands. Chem Biol. 1995;2:841–845. doi: 10.1016/1074-5521(95)90090-x. [DOI] [PubMed] [Google Scholar]
- 56.Oberg M, Bergander L, Hakansson H, Rannug U, Rannug A. Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol Sci. 2005;85:935–943. doi: 10.1093/toxsci/kfi154. [DOI] [PubMed] [Google Scholar]
- 57.Katiyar SK, Matsui MS, Mukhtar H. Ultraviolet-B exposure of human skin induces cytochromes P450 1A1 and 1B1. J Invest Dermatol. 2000;114:328–333. doi: 10.1046/j.1523-1747.2000.00876.x. [DOI] [PubMed] [Google Scholar]
- 58.Rannug A, Fritsche E. The aryl hydrocarbon receptor and light. Biol Chem. 2006;387:1149–1157. doi: 10.1515/BC.2006.143. [DOI] [PubMed] [Google Scholar]