

Abstract
HIV infection remains a major global public health problem, in part because of the ability of the virus to elude antiretroviral therapies. Most conventional drugs were designed to directly target virus-encoded mechanisms. However, there is increasing appreciation that certain host-encoded molecules are comparably important for the viral life cycle and could therefore represent potential antiviral targets. Prominent among these is TSG101, a cytoplasmic molecule that is “hijacked” by HIV and used to facilitate viral budding and release. In our present report, we demonstrate thatTSG101 is uniquely exposed on the surface of HIV-infected cells and is available to antibody-based therapies. We also characterize the development of a monoclonal antibody, CB8-2, which reduces virus production from infected cells. These studies demonstrate the potential of TSG101-directed antibodies to combat HIV/AIDS.
Keywords: TSG101, HIV, monoclonal antibody, CB8-2, HIV/AIDS therapy
Introduction
Despite considerable efforts, HIV/AIDS remains a worldwide problem for the public health community. In part, the menace of the disease arises from a high mutation rate, which favors the development of drug resistant variants [1-4]. Compounding this, toxicities associated with conventional antiretroviral therapies often decrease patient compliance. As a result, HIV drugs are progressively less effective as resistant variants emerge and become more prevalent[5].
The standard approach for developing new antiretroviral therapies has been to target virus-encoded pathways. This strategy facilitated many successful therapies such as inhibitors of HIV-encoded reverse transcriptase or protease. A newer approach has focused on targeting host molecules that are necessary for HIV binding or entry [6-8]. This approach differs from the classic strategy in that host targets are invoked as potential targets for therapy. More recently, this concept has extended to determining host targets that are essential for other aspects of the viral life cycle [9, 10]. This new approach is based on the well-established fact that the viral machinery “hijacks” the host cell to facilitate many aspects of the viral life cycle [11-14]. In doing so, these host molecules are manipulated in a manner that differs remarkably from their normal function in non-infected cells. A prominent example of this concept is TSG101.
TSG101 is a cytoplasmic molecule that functions as a component of the vesicular protein sorting machinery. TSG101 function is associated with multivesicular bodies, where it regulates lysosomal protein degradation [15-17]. TSG101 also regulates the budding process of some enveloped viruses. Specifically, viruses that encode for proteins with amino acid sequences encoding for late domain motifs, most notably HIV and Ebola viruses, bind TSG101 and that this interaction is necessary for progeny virus release from the cell membrane. [17-24]. In the case of HIV, the late domain motif is part of the p6Gag protein, which binds TSG101 and facilitates the budding and release of mature virions [19,20,24].
In our present report, we demonstrate that TSG101 is exposed on the outer membrane of cells that have been infected with HIV. This change in subcellular localization is widely applicable to different HIV strains, including drug resistant strains, and SIV. We also demonstrate that TSG101 specific antibodies can reduce virus production in infected cells and that TSG101 cell surface exposure can serve as a focus for targeted intervention using monoclonal antibodies. These results suggest an opportunity for a novel therapeutic approach for combating HIV/AIDS that is broadly applicable to different viral strains.
Materials and methods
Antibodies
Polyclonal antibody 1299 was generated by immunizing a rabbit against a 19 amino acid peptide derived from the N-terminal region of TSG101 (TIKTGKHVDANGKIYLPYL).
Proteins
The expression vector pET-21b containing the ubiquitin E2 variant (UEV) domain of TSG101 was generously provided by Dr. M. Javad Aman from the United States Army Medical Research Institute for Infectious Diseases. The UEV domain was expressed as a C-terminal His-tag fusion protein in the E. coli strain BL21 and purified using immobilized metal affinity chroma-tography. The C-terminus of TSG101 was inserted into the expression vector pLLexp and expressed as a 5′ Fc-fusion protein in the 293T cell line. The fusion protein was purified with a protein A column. The full-length TGS101 protein was expressed as an N-terminal His-tag fusion protein in 293T or HeLa cells by Q-Biogene Inc. The fusion protein was purified under denaturing condition and refolded with Pierce's protein refolding kit following the manufacturer's instruction.
Monoclonal antibody selection and construction
Selection of antigen-specific scFv antibodies: Specific phage displayed scFv antibodies were affinity-selected by using TSG101 full-length protein, UEV fragment or C-terminal fragement absorbed to immunotubes [25]. For selections using the TSG101 C-terminal fragment (a human Fc fusion protein), the phage library was preincubated with 100 μg of irrelevant human IgG to deplete the library of human Fc binders. After binding of phage library to the immobilized antigen and wash of non-specific binders, the bound phage was eluted from each selection and used to infect E. coli TG1 cells. The rescue-selection-plating cycle was repeated twice, after which specific antigen binding by individual clones was confirmed by enzyme linked immu-nosorbent assays (ELISA). The DNA sequence of individual clones was determined for subsequent construction of full-length antibody (see below).
Affinity maturation of scFv CB8: To improve the affinity of scFv CB8 binding to the full-length TSG101, the DNA corresponding to the CB8 sequence was cloned into pYD2 vector for display on yeast surface [26] and mutated by error-prone PCR to introduce random changes [27]. After three rounds of flow cytometry sorting, individual scFv with increased binding affinity were selected and analyzed for cell surface recognition of TSG101 on HIV-infected cells as described below.
Cloning and expression of full-length CB8-2 human monoclonal antibody: In order to construct the full-length human monoclonal antibody CB8-2, the antibody backbone containing the IgG1 heavy chain and leader sequence, (obtained from plasmid pJL100K) the lambda light chain constant region (synthesized by Geneart, Burlin-game, CA) was cloned into plasmid pCEP4-neo-HuIgGLam, an EBNA-1 episomal antibody expression vector. The CB8-2 heavy and light chains were cloned in frame into pCEP4-neo-HuIgGLam yielding the CB8-2 antibody expression vector. CHO-S cells (Invitrogen, Carlsbad, CA), growing at a density of 1×106 cells/ml in 250 ml Freestyle CHO Expression Medium (Invitrogen, Carlsbad, CA) containing 8 mM L-glutamine, are transfected with the CB8-2 expression vector using Freestyle MAX reagent (Invitrogen, Carlsbad, CA) following the manufacturer's protocol. The antibody DNA (stock concentration is 1 ug/ul in H2O) and lipid mixes are made separately in OptiPRO SFM, immediately mixed together for a total volume of 10 ml, incubated for 10 minutes at room temperature, and then added to the cells. Transfected cells are incubated at 37°C with 8% CO2 with shaking at 135 rpm. Antibody production is allowed to proceed for 7 days before supernatant is harvested. IgG was purified from culture supernatant using protein A affinity chromatography.
Cell culture and virus infections
MT-4 cells were grown in RPMI 1640 medium containing 10% heat-inactivated (56°C, 30 min) FBS (HyClone, Long, UT) supplemented with 2 mM glutamine (Invitrogen, Carlsbad, CA), 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA). Peripheral blood samples were obtained by venipuncture in Vacutainer tubes containing sodium heparin from clinically healthy seronega-tive subjects. All participants signed an informed consent form before the study. Peripheral blood mononuclear cells (PBMC) were obtained by Ficoll-Hypaque (Sigma-Aldrich, St Louis, MO) gradient centrifugation. The cells were stimulated with PHA (3 μg/ml; Sigma-Aldrich, St Louis, MO) for 3 days in RPMI 1640 medium containing 10% heat-inactivated (56°C, 30 min) FBS (HyClone, Long, UT) supplemented with 2 mM glutamine (Invitrogen, Carlsbad, CA), 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA), 100 U/ml penicillin (BioWhittaker), and 100 U/ml IL-2 (Roche). CD4+ cells from the activated PBMC were subsequently obtained by positive selection using immunomagnetic (IM) beads bearing anti-CD4 antibodies (Miltenyi Biotec).
The following viruses or virus molecular clones were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: pNL4-3 from Dr. Malcolm Martin [28], HIV-1ME1 from Dr. Phalguni Gupta [29] and Protease-resistant HIV-1 (L10R/M46I/L63P/V82T/I84V) from Dr. Emilio Emini [30]. SIVmac251 was obtained from Advanced Biosciences Laboratories, Kensington, MD. Virus infections were performed with HIV-1 at varying amounts of virus as indicated. After 1 h, the cells were washed and then cultured in the RPMI 1640 growth medium containing 100 U/ml IL-2 (Roche). For PBMC experiments CB8-2 or isotype control antibody was added with the growth medium. To monitor virus release, half the culture supernatant was collected every two days and replaced with fresh culture medium containing half the amount of C-B8-2 or isotype control antibody originally added to the culture. Infection culture supernatants were then transferred to the TZM-bl indicator cell line for determination of infectious viral particles. Relative Luminescence Unit (RLU) measurement was obtained on TZM-bl cells after they were treated with Bright-Glo Luciferase Assay System (Promega, Madison, WI) 3 days post infection (dpi). Percent virus inhibition was calculated by comparing the amount of luciferase produced by antibody treated cells to untreated infected cells.
Antibody dependent cellular cytotoxicity (ADCC) assay
MT-4 cells were infected with HIV-1NL4-3 as described above and incubated at 37°C for 3 days. Natural killer (NK) cells purified from donor derived PBMCs using the Human NK Cell Isolation Kit, (Miltenyi, Auburn, CA) and C-B8-2 antibody were added to MT-4 cells in MEM media and incubated at 37°C for 4 hours. Levels of lactose dehydrogenase (LDH) released upon cell death were measured using Cytotox 96 – non-radioactive cytotoxicity assay (Promega, Madison, WI). Percent cell killing was calculated by comparing the amount of LDH released from the NK cell and antibody treated cells to control MT-4 cells well treated with lysis buffer (total lysis).
Flow cytometry
For flow cytometry analysis, 1×105 MT-4 cells were incubated on ice with 10 μg/ml TSG101 antibody for 30 min. Cells were washed three times with PBS (Invitrogen, Carlsbad, CA, San Diego, CA) containing 1% BSA (Sigma-Aldrich, St. Louis, MO) on ice and incubated with FITC conjugated goat anti-rabbit antibody (Becton Dickinson, San Jose, CA) or FITC conjugated goat anti-human antibody (Jackson Immu-nolabs, West Grove, PA) diluted in PBS-BSA for 30 min on ice. Following 3 washes in wash buffer the cells were fixed in PBS with 1% para-formaldehyde for TSG101 cell surface staining studies or used to determine viability. For viability assays, propidium iodide (PI; Becton Dickinson, San Jose, CA) or 7-amino-actinomycinD (7AAD; Guava Technologies, Hayward, CA) was added to unfixed cells according to manufacturer instructions. Cells stained with viability dyes were subjected to analysis within 15 minutes of the addition of 7AAD or PI. Fix and Perm Cell Permeabilization Reagents (Invitrogen, Carlsbad, CA) were used for detection of intra-cellular TSG101 following the manufacturer's instructions. Fluorescence data were acquired in an EasyCyte Flow Cytometer (Guava Technologies, Hayward, CA) and data was analyzed using FlowJo analysis software.
Tissue cross reactivity
Tissue cross reactivity studies were conducted at LifeSpan Biosciences (Seattle, WA). Briefly, FITC conjugated C-B8-2 antibody was tested on human formalin fixed, paraffin embedded tissues or infected cells (positive control). An anti-FITC antibody was used as a secondary followed by HRP-DAB (brown precipitate) detection. Tissues were also stained with positive control antibodies (CD31 and vimentin) to ensure that the tissue antigens were preserved and accessible for immunohistochemical analysis. A scale of 0-4 was used to evaluate the level of staining present in the tissues 0 - negative, 1- blush, 2-faint, 3-moderate, 4-strong.
Results
HIV-1 infection causes membrane exposure of TSG101
A previous study demonstrated that TSG101 redistributes with its cognate viral late domain protein (VP40) to a membrane-proximal site in cells expressing Ebola viral like proteins (VLPs) [31]. These findings, coupled with evidence that TSG101 plays an essential role in HIV budding [18-20], led us to ask if TSG101 might be exposed on the outer surface of the cell membrane. To address this question, MT4 human T lymphocytes were infected with HIV-1NL4-3 for 4 days. The cells were then labeled with a purified TSG101 polyclonal antibody at 4°C to prevent internalization and stained with fluoro-phore-conjugated goat anti-rabbit IgG. Flow cytometric analyses revealed that TSG101 rabbit polyclonal antibodies specifically bound the surface of HIV-infected cells while matched iso-type control antibodies did not (Figure 1A). This response was unique to infected cells since parallel studies with non-infected cells did not identify TSG101 on the cell surface.
Figure 1.

Flow cytometry analysis of TSG101 antibody binding to the surface of HIV infected cells. Panel A shows MT-4 cells infected with HIV-1NL4-3. Infected (green) and uninfected (red) cells were incubated with polyclonal antibody 1299 (pAb 1299), which is specific to the N-terminal region of TSG101. Membrane integrity of TGS101 positive cells was assessed using propidium iodide as shown in Panel B. The quadrant plot shows MT-4 cells 4 days post infection with HIV-1NL4-3. The lower right quadrant shows cells cell surface expression of TSG101 on cells that exclude PI. The upper right quadrant shows cells with compromised membrane integrity as evidenced by the uptake of PI. Panel C shows lack of binding of 1299 to uninfected MT-4 cells (Red) in the absence of permeabilization reagent, and positive binding when the cells were permeabilized (Green).
The studies above indicate that TSG101 is uniquely exposed on the surface of HIV-infected cells. To verify surface exposure, we considered that compromised membrane integrity might have caused TSG101 antibodies to gain access to antigen. In this scenario, the observed staining might have represented an artifact of dead or dying cells. To test this, TSG101 membrane exposure was evaluated in HIV-infected cells using two-color flow cytometry using TSG101 antibodies and propidium iodine (PI), a marker of membrane integrity. TSG101 surface exposure was verified on infected cells that retained membrane integrity (i.e., did not stain with PI) (Figure 1B). As expected, dead or dying cells (which stain positive for PI) also stained with TSG101 antibodies. As a further control, we determined that TSG101 antibodies were only able to stain uninfected cells if the cell membrane had been first stripped away using non-ionic detergents. For example, permeabilization of uninfected cells facilitated TSG101 surface exposure whereas intact, uninfected cells did not stain with TSG101 antibodies (Figure 1C). Altogether, these findings indicate that TSG101 is found on the surface of HIV-infected cells and prior to loss of cell membrane integrity.
Having confirmed that antibodies recognize TSG101 on the surface of intact, HIV-infected cells, we determined the time course of TSG101 surface exposure. MT-4 cells were infected with HIV at a low multiplicity of infection (MOI = 0.004) and monitored daily for TSG101 cell surface exposure. To verify the infection had been productive, a parallel assessment of virus production was performed using a standard TZM-bl cell reporter assay. Under these conditions, TSG101 was first detected on the cell surface three days after infection and progressively increased thereafter (Figure 2A). This timing indicated that detection of TSG101 surface exposure coincided with the initial detection and subsequent increase of virus particle release (Figure 2B). We also conducted similar studies using a higher initial multiplicity of infection (MOI = 1) to increase the population of infected cells at earlier time points(Figure 2C, D). Peak virus production was observed at 36 hours post infection and this coincided with optimal TSG101 cell surface exposure. These studies align known knowledge of TSG101's role in the viral life cycle with our current evidence for TSG101 surface exposure during virus infection.
Figure 2.

TSG101 exposure on the surface of infected cells is consistent with release of progeny virus. MT-4 cells were infected with HIV-1NL4-3 for the length of time indicated at either low or high MOI as indicated and stained with pAb1299 (Panels A and C) and evaluated using flow cytometry. Virus release was quantified by luciferase expression of TZM-bl cells exposed to cell culture supernatant from each time point (Panels Band D).
Selective recognition of HIV-infected cells with CB8-2 human monoclonal antibody
Since TSG101 exposure on the surface of cells was limited to infected cells, we asked whether this unique change might be exploited as an opportunity for therapeutic targeting of virus-infected cells using a monoclonal antibody. Monoclonal antibodies are renowned for their exquisite specificity and we considered that this could allow us to characterize TSG101 exposure on infected cells with more focused reagents. We developed TSG101 specific monoclonal antibodies using a non-immune human single chain variable fragment (scFv) library of phage was used to select TSG101-specific antibodies [32]. This library consisted of a phage encompassing approximately 109 different specificities [33, 34]. The antigens used for selection included the TSG101 UEV domain (amino acids 1-145), C-terminus of TSG101 (amino acids 218-390) and recombinant full-length TSG101. After two rounds of selection, the binding specificity of individual scFv candidates was determined using ELISA assays. Analysis of the clones revealed that 75% recognized full-length TSG101 whereas 20% were restricted to epitopes on the C-terminal region of TSG101, and 2% recognized epitopes within the UEV domain. After characterizing the DNA sequences to confirm independent clones, 40 candidates were prioritized based on their ability to recognize TSG101 on the surface of virus-infected cells. Of the 40 scFv that were tested for TSG101 surface recognition, 18 recognized TSG101 that was exposed on the surface of HIV-infected cells. The most promising candidate, C-B8 was selected for further optimization.
A complete full-length human IgG1 antibody was constructed based on the CB-8 scFV sequence. This full-length antibody was then optimized for enhanced affinity using site-directed mutagene-sis, resulting in the affinity-enhanced antibody, known hereafter as CB8-2. We confirmed that CB8-2 continued to selectively recognize TSG101 on the surface of cells infected with HIV or SIV (Figure 3A). In these studies CD4-positive T-cells were isolated from peripheral blood mononuclear cells (PBMC) and infected with HIV strains that differed in their tropism (CCR5 or CXCR4) and antiretroviral drug susceptibility. C-B82 also bound TSG101 on the surface of CD4+ T-cells (isolated from rhesus macaque PBMC) that had been infected with SIVmac251. These studies revealed that the appearance of TSG101 on the surface of infected cells is not restricted to the MT-4 cell line or to any particular HIV strain. As we observed with polyclonal TSG101 antibodies, CB8-2 antibodies stained the surface of cells that contain an intact cell membrane (as determined by exclusion of the vital dye, 7AAD; Figure 3B). Consistent with the findings presented above, the binding of TSG101 to uninfected cells was observed only when the cells were treated with permeabilization reagents to allow the antibody access to the inside of the cell (Figure 3C).
Figure 3.

Human monoclonal antibody CB8-2 recognizes TSG101 on the surface of cells infected with different HIV strains and SIV. Histograms in panel A show TSG101 exposure on the surface of primary CD4 T-cells infected with HIV-1NL4-3 (CXCR4), HIV-1ME1 (CCR5), HIV-1 L10R/M46I/L63P/V82T/I84V (CXCR4, protease resistant) and CD4+ T-cells isolated from rhesus macaques PBMC infected with SIVmac251. Infected (green) and uninfected (red) cells were incubated with antibody CB8-2 and a FITC-goat anti-human secondary antibody. The membrane integrity of cells positive for CB8-2 binding cells was assessed using 7AAD as shown in Panel B. The lower right quadrant shows cell surface expression of TSG101 on cells that exclude 7AAD. The upper right quadrant shows cells with compromised membrane integrity, as evidenced by the uptake of 7AAD. Panel C shows lack of binding of CB8-2 to MT-4 uninfected cells (Red) in the absence of permeabilization reagent, and positive binding when the cells were permeabilized (Green).
Preliminary safety screening and epitope identification
The ultimate goal of our work is the development of a potential new therapeutic to combat HIV/AIDS. This potential is based on the fact that TSG101 surface exposure is restricted to virus-infected cells. To facilitate potential future use in people, we addressed the importance of safety, which was particularly important since TSG101 is a highly-conserved, ubiquitously-expressed host molecule. Tissue cross-reactivity provides an accepted means to evaluate potential non-specific or undesired binding of antibodies to normal human tissues. Tissue samples obtained from human vital organs (brain, heart, kidney, liver, and lung) were subjected to immu-nohistochemical analyses with CB8-2. No CB8-2 immunoreactivity was observed with frozen specimens of heart or kidney. On the few cells of the brain, liver and lung that stained with CB8-2 (Figure 4A), this immunoreactivity was limited to faint staining of the cytoplasm and no membrane staining was observed (Figure 4B).
Figure 4.

Weak CB8-2 immunoreactivity with uninfected tissues. CB8-2 was used for immunohistochemical staining of frozen sections of human brain, heart, liver and lung as shown in Panel A. Panel B shows a table of the location and relative levels of staining in each tissue.
To determine the epitope recognized by CB8-2 on TSG101, we used matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF)-based mass spectrometry to analyze purified full length TSG101 that had been digested with trypsin. These studies revealed a series of three missing peaks in samples incubated with CB8-2 (Figure 5A) that corresponded to noncontiguous segments of approximately 9 aminoacids (Figure 5B). These results suggested that CB8-2 recognizes a conformational epitope.
Figure 5.

CB8-2 binds a conformational epitope on TSG101. MALDI-TOF analysis were undertaken to determine the binding site of CB8-2 on TSG101. Panel A shows mass spectrometry analysis of MALDI-TOF using protease digested TSG101 incubated with CB8-2 monoclonal antibodies. Note the absence of three peaks which correspond to the three segments of TSG101 shown in panel B. Panel B illustrates the amino acid sequence of human TSG101 with the three regions recognized by the antibody highlighted in yellow.
CB8-2 elimination of HIV-infected cells
To determine the efficacy of TSG101 antibodies in controlling HIV infection, normal donor derived human PBMCs were infected with HIV-1NL4-3 in the presence or absence of CB8-2. After incubation for 8 days, the amount of virus in the cell culture supernatant was measured using the TZM-bl reporter assay. Figure 6 shows the results obtained from the treatment of HIV-infected PBMC that had been treated with CB8-2 at day 8 post-infection. The antibody specifically decreased HIV-1 levels and in a dose dependent manner. These results conveyed an estimated EC50 for CB8-2 of less than 10 nM. Interestingly, studies performed using MT-4 cells infected with HIV-1NL4-3 in the absence of PBMCs showed no decrease in virus replication in spite of the fact that TSG101 is detectable on the surface of these cells. (Figure 7A and B). These findings indicated that the antibody alone is not sufficient to reduce virus production in cell culture and instead suggested that the mechanistic basis of CB8-2 antiviral activity may have resulted from immune clearance mechanisms such as antibody dependent functions that are mediated by natural killer (NK) cells or macrophages, which are present in PBMCs. To address the potential role of effector cell mechanisms, specifically antibody dependent cellular cytotoxicity (ADCC) in the anti-HIV activity of C-B8-2, we measured the amount of cytotoxicity observed in MT-4 cells infected with HIV-1NL4-3 in the presence of 5 ng/mL of C-B8-2 antibody and NK cells isolated from donor derived PBMCs. The results show that a ratio of 16:1 NK cells: infected M-T4 cells resulted 40% cytotoxicity when compared to the total lysis control in which a comparable number of cells had been treated with detergent (Figure 7C). Reducing the amount of NK cells also reduced the amount of killing observed. Incubation of uninfected MT-4 cells with NK cells did not result in cell killing even at higher antibody concentrations (up to 500 ng/mL) (data not shown). These findings provide strong evidence that the anti-HIV activity of C-B8-2 is a result of infected cell targeting and subsequent elimination by effector cell mechanisms.
Figure 6.
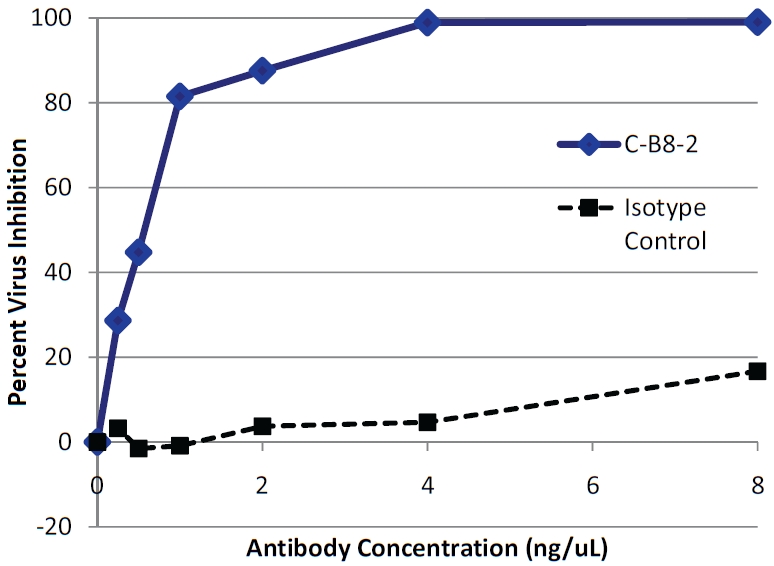
CB8-2 inhibits HIV replication in infected human PBMC. Donor derived human PBMCswere infected with HIV-1NL4-3 and incubated for 8 days respectively days in the presence or absence of a CB8-2 or an isotype control antibody. Culture supernatants were collected and virus output was determined by luciferase expression of TZM cells exposed to the culture supernatant. Results are represented as percent viral inhibition as compared to infected PBMC cells without antibody. The graph shows the level of inhibition of virus production in infected PBMCs by different concentrations of antibody.
Figure 7.

Antiviral Mechanism of CB8-2 Involves ADCC. Panel A MT-4 cells were infected HIV-1NL4-3 and incubated for 5 days in the presence or absence of C-B8-2 or an isotype control antibody. Supernatants were collected from the MT-4 cultures and virus production was determined by luciferase expression of TZM-bl cells exposed to the culture supernatants. Panel B. Flow cytometry analysis on the infected MT-4 cells used in the virus inhibition experiments in Panel A to demonstrate the level of TSG101 cell surface exposure. Panel C shows the results of a cytoxicity assay exposing HIV-1 infected MT-4 cells to C-B8-2 (5 ng/mL) antibody and varying amounts of NK cells. The levels of cyto-toxicity were determined by measuring LDH release as a result of cell death. Results are represented as percent killing as compared to a total cell lysis control to which lysis buffer was added.
Discussion
In the current study, we demonstrate HIV infection causes TSG101 to be exposed on the surface of HIV infected cells. Comparable findings were obtained using cell lines as well as acutely infected donor-derived primary CD4+ T-lymphocytes. (Figures 1 and 3). The timing of TSG101 exposure on the cell surface coincides with the budding and release of virus particles (Figure 2), consistent with the established role of TSG101 in HIV budding.
TSG101 membrane localization upon HIV infection has been previously noted. In two independent reports, TSG101 was shown to redistribute to a site near the cell membrane only in virus-infected cells and presumably as a direct result of virus infection [19, 31]. In a third study, TSG101 localized to the cell membrane regardless of viral infection and infection did not increase the amount of TSG101 at the cell surface. [35]. Our studies demonstrate the presence of TSG101 at the cell surface from a different perspective in that it is exposed on the outside of the cell. This exposure is unique to virus-infected cells and given the role of TSG101 in virus budding may result from interactions of TSG101 with p6Gag. An alternative means to attribute TSG101 surface exposure would be to invoke the mechanisms such as those that induce anionic phospholipid asymmetry at the plasma membrane following cell activation, malignant transformation and other cellular stress-ors [36] and have been shown to occur as a result of virus infection [37]. Further studies are necessary to confirm the mechanism of TSG101 membrane translocation.
Our findings do suggest that a large portion of TSG101 is exposed on the surface of infected cells. For instance, TSG101 surface exposure is observed with monoclonal and polyclonal antibodies that target different epitopes throughout TSG101 (Figure 1 and Figure 5). Such findings are consistent with the fact that TSG101 does not encode for a transmembrane domain and thus would not have regions that are specifically directed to the cell surface. Rather, we propose that TSG101 is transferred across the cell membrane during virus budding, presumably as a result of specific interactions with HIV p6Gag. The mechanistic basis of this change may not necessarily be unique to TSG101 and we cannot exclude that other components of the ESCRT complex that have been implicated in the HIV budding process are not similarly exposed on the surface of HIV-infected cells. Nevertheless, the major finding of this study is that regardless of the mechanism(s) that contribute to TSG101 surface exposure, the revelation of an epitope on the surface provides an opportunity for antibody-mediated elimination of the infected cell.
Our results also demonstrate that many different HIV-1 strains cause TSG101 to be exposed on the cell surface. Comparable findings were obtained with HIV-1 strains that differ in their tropism or drug sensitivity. This suggests a potential for broad-spectrum application to HIV-1 infections. Likewise, the availability of TSG101 on the surface of cells infected with SIVmac251 suggests that TSG101 surface exposure is comparably applicable beyond HIV-1 to other retrovirus infections. However, TSG101 surface exposure is not universally applicable to virus infection. For example, infection of host cells by non-enveloped viruses, such as Rotavirus, does not cause TSG101 to be exposed on the cell surface (data not shown).
CB8-2 antibody treatment reduced virus production but only in infected PBMCs but did not decrease HIV levels in studies with infected MT-4 cells (Figure 6 and 7A). Such findings imply that CB8-2 binding does not directly inhibit virus budding or release, but rather needs other cells present in the PBMCs in order to decrease virus release in cell culture. ADCC assays performed using purified human NK cells and HIV infected MT-4 cells showed dose dependent cell killing in the presence of antibody and increasing amounts of NK cells (Figure 7C). In light of the fact that PBMCs provide syngeneic target CD4+ cells and effector natural killer (NK) cells [38, 39] we interpret these results as suggesting that CB8-2 recruits effector cells to eliminate HIV-infected cells via mechanisms such as ADCC and consequently reduce the release and spread of mature, infectious virions.
To characterize the TSG101 epitope recognized by CB8-2, we performed a series of studies to assess tissue cross-reactivity studies as well as MALDI-TOF-based epitope identification (Figures 4 and 5). In the former case, a lack of tissue cross-reactivity minimized the potential for unwanted binding of TSG101 antibodies to unin-fected cells. Epitope identification eliminated epitopes that might vary within the human population. The benefits of monoclonal antibody therapy can be thwarted if genetic polymorphisms within the population limit the breadth of patients, who might benefit from the therapy. A search of the SNP databases (www.ncbi.nlm.gov) revealed no relevant polymorphisms in TSG101 that might obviate CB8-2 binding. Indeed, a study conducted of the TSG101 open reading frame from 50 healthy blood donors confirmed the lack of non-synonymous changes in this region [40]. Such findings favor the advancement of CB8-2 as a potential therapeutic candidate to target HIV in the wider human population.
Our present findings suggest TSG101 could provide an opportunity for passive immunother-apy using monoclonal antibodies. Such results might also be extended further to elicit active immunotherapy via TSG101-directed vaccines. The opportunity afforded by a TSG101 vaccine would include the ability to mount a sustained challenge to HIV infection. These benefits would have to be balanced against the potential for autoimmunity against TSG101 within non-infected host cells. Our present findings suggest that sufficient safety can be achieved by selective targeting of certain epitopes that are not exposed in normal cells. Indeed, the concept of targeting a human host molecule with a vaccine is not unprecedented as evidenced by the ongoing studies of an angiotensin II vaccine to manage hypertensive disease [41-43].
In summary, the surface exposure of TSG101 on infected cells could provide an important new tool to combat HIV/AIDS. Our present studies indicate that this unique change can facilitate monoclonal antibody-based elimination of infected cells via effector cell mechanisms such as ADCC. This approach appears to be broadly relevant to all variants of HIV and SIV tested to date, which raises an intriguing possibility that TSG101 antibodies, such as CB8-2, could provide a broad-spectrum means to control HIV infection.
References
- 1.Shafer RW, Schapiro JM. HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. Aids Reviews. 2008;10:67–84. [PMC free article] [PubMed] [Google Scholar]
- 2.Malta M, Strathdee SA, Magnanini MMF, Bastos FI. Adherence to antiretroviral therapy for human immunodeficiency virus/acquired immune deficiency syndrome among drug users: a systematic review. Addiction. 2008;103:1242–1257. doi: 10.1111/j.1360-0443.2008.02269.x. [DOI] [PubMed] [Google Scholar]
- 3.Ines SM, Moralejo L, Marcos M, Fuertes A, Luna G. Adherence to highly active antiretroviral therapy in HIV-infected inmates. Current Hiv Research. 2008;6:164–170. doi: 10.2174/157016208783885047. [DOI] [PubMed] [Google Scholar]
- 4.Bennett DE, Bertagnolio S, Sutherland D, Gilks CF. The World Health Organization's global strategy for prevention and assessment of HIV drug resistance. Antiviral Therapy. 2008;13:1–13. [PubMed] [Google Scholar]
- 5.Tang JW, Pillay D. Transmission of HIV-1 drug resistance. Journal of Clinical Virology. 2004;30:1–10. doi: 10.1016/j.jcv.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Hughes A, Nelson M. HIV entry: new insights and implications for patient management. Current Opinion in Infectious Diseases. 2009;22:35–42. doi: 10.1097/QCO.0b013e3283213093. [DOI] [PubMed] [Google Scholar]
- 7.Wu XS, Lee VC, Chevalier E, Hwang ST. Chemokine Receptors as Targets for Cancer Therapy. Current Pharmaceutical Design. 2009;15:742–757. doi: 10.2174/138161209787582165. [DOI] [PubMed] [Google Scholar]
- 8.Hunt JS, Romanelli F. Maraviroc, a CCR5 Coreceptor Antagonist That Blocks Entry of Human Immunodeficiency Virus Type 1. Pharmacotherapy. 2009;29:295–304. doi: 10.1592/phco.29.3.295. [DOI] [PubMed] [Google Scholar]
- 9.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 10.Konig R, Zhou YY, Elleder D, Diamond TL, Bonamy GMC, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calistri A, Salata C, Parolin C, Palu G. Role of multivesicular bodies and their components in the egress of enveloped RNA viruses. Reviews in Medical Virology. 2009;19:31–45. doi: 10.1002/rmv.588. [DOI] [PubMed] [Google Scholar]
- 12.Smith GA, Enquist LW. Break ins and break outs: Viral interactions with the cytoskeleton of mammalian cells. Annual Review of Cell and Developmental Biology. 2002;18:135–161. doi: 10.1146/annurev.cellbio.18.012502.105920. [DOI] [PubMed] [Google Scholar]
- 13.Dohner K, Sodeik B. The role of the cytoskeleton during viral infection. Membrane Trafficking in Viral Replication. 2004;285:67–108. doi: 10.1007/3-540-26764-6_3. [DOI] [PubMed] [Google Scholar]
- 14.Lehmann-Che J, Saib A. Early stages of HIV replication: How to hijack cellular functions for a successful infection. Aids Reviews. 2004;6:199–207. [PubMed] [Google Scholar]
- 15.Li L, Cohen SN. Tsg101: a novel tumor susceptibility gene isolated by controlled homozygous functional knockout of allelic loci in mammalian cells. Cell. 1996;85:319–329. doi: 10.1016/s0092-8674(00)81111-3. [DOI] [PubMed] [Google Scholar]
- 16.Carter CA. Tsg101: HIV-1's ticket to ride. Trends in Microbiology. 2002;10 doi: 10.1016/s0966-842x(02)02350-8. PII S0966-0842X (0902)02350-B. [DOI] [PubMed] [Google Scholar]
- 17.Martin-Serrano J, Zang T, Bieniasz PD. Role of ESCRT-I in retroviral budding. J Virol. 2003;77:4794–4804. doi: 10.1128/JVI.77.8.4794-4804.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demirov DG, Ono A, Orenstein JM, Freed EO. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc Natl Acad Sci U S A. 2002;99:955–960. doi: 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7:1313–1319. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- 20.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 21.Bache KG, Slagsvold T, Cabezas A, Rosendal KR, Raiborg C, Stenmark H. The growth-regulatory protein HCRP1/hVps37A is a subunit of mammalian ESCRT-I and mediates receptor down-regulation. Mol Biol Cell. 2004;15:4337–4346. doi: 10.1091/mbc.E04-03-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goff A, Ehrlich LS, Cohen SN, Carter CA. Tsg101 control of human immunodeficiency virus type 1 Gag trafficking and release. J Virol. 2003;77:9173–9182. doi: 10.1128/JVI.77.17.9173-9182.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pornillos O, Alam SL, Davis DR, Sundquist WI. Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat Struct Biol. 2002;9:812–817. doi: 10.1038/nsb856. [DOI] [PubMed] [Google Scholar]
- 24.VerPlank L, Bouamr F, LaGrassa TJ, Agresta B, Kikonyogo A, Leis J, Carter CA. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55 (Gag) Proc Natl Acad Sci U S A. 2001;98:7724–7729. doi: 10.1073/pnas.131059198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991;222:581–597. doi: 10.1016/0022-2836(91)90498-u. [DOI] [PubMed] [Google Scholar]
- 26.Razai A, Garcia-Rodriguez C, Lou J, Geren IN, Forsyth CM, Robles Y, Tsai R, Smith TJ, Smith LA, Siegel RW, Feldhaus M, Marks JD. Molecular evolution of antibody affinity for sensitive detection of botulinum neurotoxin type A. J Mol Biol. 2005;351:158–169. doi: 10.1016/j.jmb.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Zhou Y, Drummond DC, Zou H, Hayes ME, Adams GP, Kirpotin DB, Marks JD. Impact of single-chain Fv antibody fragment affinity on nanoparticle targeting of epidermal growth factor receptor-expressing tumor cells. J Mol Biol. 2007;371:934–947. doi: 10.1016/j.jmb.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. PRODUCTION OF ACQUIRED IMMUNODEFICIENCY SYNDROME-ASSOCIATED RETROVIRUS IN HUMAN AND NON-HUMAN CELLS TRANSFECTED WITH AN INFECTIOUS MOLECULAR CLONE. Journal of Virology. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen MS, Singh MK, Balachandran R, Gupta P. Isolation and characterization of two divergent infectious molecular clones of HIV type 1 longitudinally obtained from a seropositive patient by a progressive amplification procedure. Aids Research and Human Retroviruses. 1997;13:743–750. doi: 10.1089/aid.1997.13.743. [DOI] [PubMed] [Google Scholar]
- 30.Condra JH, Schleif WA, Blahy OM, Gabryelski LJ, Graham DJ, Quintero JC, Rhodes A, Robbins HL, Roth E, Shivaprakash M, Titus D, Yang T, Teppler H, Squires KE, Deutsch PJ, Emini EA. IN-VIVO EMERGENCE OF HIV-1 VARIANTS RESISTANT TO MULTIPLE PROTEASE INHIBITORS. Nature. 1995;374:569–571. doi: 10.1038/374569a0. [DOI] [PubMed] [Google Scholar]
- 31.Panchal RG, Ruthel G, Kenny TA, Kallstrom GH, Lane D, Badie SS, Li L, Bavari S, Aman MJ. In vivo oligomerization and raft localization of Ebola virus protein VP40 during vesicular budding. Proc Natl Acad Sci U S A. 2003;100:15936–15941. doi: 10.1073/pnas.2533915100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheets MD, Amersdorfer P, Finnern R, Sargent P, Lindquist E, Schier R, Hemingsen G, Wong C, Gerhart JC, Marks JD. Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proc Natl Acad Sci U S A. 1998;95:6157–6162. doi: 10.1073/pnas.95.11.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huie MA, Cheung MC, Muench MO, Becerril B, Kan YW, Marks JD. Antibodies to human fetal erythroid cells from a nonimmune phage antibody library. Proc Natl Acad Sci U S A. 2001;98:2682–2687. doi: 10.1073/pnas.051631798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Connell D, Becerril B, Roy-Burman A, Daws M, Marks JD. Phage versus phagemid libraries for generation of human monoclonal antibodies. J Mol Biol. 2002;321:49–56. doi: 10.1016/s0022-2836(02)00561-2. [DOI] [PubMed] [Google Scholar]
- 35.Welsch S, Habermann A, Jager S, Muller B, Krijnse-Locker J, Krausslich HG. Ultrastructural analysis of ESCRT proteins suggests a role for endosome-associated tubular-vesicular membranes in ESCRT function. Traffic. 2006;7:1551–1566. doi: 10.1111/j.1600-0854.2006.00489.x. [DOI] [PubMed] [Google Scholar]
- 36.Balasubramanian K, Mirnikjoo B, Schroit AJ. Regulated externalization of phosphatidylserine at the cell surface: implications for apoptosis. J Biol Chem. 2007;282:18357–18364. doi: 10.1074/jbc.M700202200. [DOI] [PubMed] [Google Scholar]
- 37.Soares MM, King SW, Thorpe PE. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nature Medicine. 2008;14:1357–1362. doi: 10.1038/nm.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Japour AJ, Mayers DL, Johnson VA, Kuritzkes DR, Beckett LA, Arduino JM, Lane J, Black RJ, Reichelderfer PS, Daquila RT, Crumpacker CS, Balfour H, Erice A, Coombs R, Katzenstein D, Lathey J, Richman D, McIntosh K, Rangan S, Reichman R, Scott W, Ussery M, Abrams L, McCutchan F, Burke D, Gardner L, Roberts C, Chung R, Hicks C, Shellie E, Fowler A, Merritt L, Fujimurajustice M, Ruiz N, Wagner K, Gail M. STANDARDIZED PERIPHERAL-BLOOD MONONUCLEAR CELL-CULTURE ASSAY FOR DETERMINATION OF DRUG SUSCEPTIBILITIES OF CLINICAL HUMAN-IMMUNODEFICIENCY-VIRUS TYPE-1 ISOLATES. Antimicrobial Agents and Chemotherapy. 1993;37:1095–1101. doi: 10.1128/aac.37.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krowicka H, Robinson JE, Clark R, Hager S, Broyles S, Pincus SH. Use of tissue culture cell lines to evaluate HIV antiviral resistance. Aids Research and Human Retroviruses. 2008;24:956–966. doi: 10.1089/aid.2007.0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bashirova AA, Bleiber G, Qi Y, Hutcheson H, Yamashita T, Johnson RC, Cheng J, Alter G, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, May M, Maldarelli F, Jacobson L, O'Brien SJ, Telenti A, Carrington M. Consistent effects of TSG101 genetic variability on multiple outcomes of exposure to human immunodeficiency virus type 1. Journal of Virology. 2006;80:6757–6763. doi: 10.1128/JVI.00094-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu F, Zhou ZH, Liao YH. The renin-angiotensin system and therapeutic vaccines for hypertension. Current Opinion in Investigational Drugs. 2008;9:286–294. [PubMed] [Google Scholar]
- 42.Tissot AC, Maurer P, Nussberger J, Sabat R, Pfister T, Ignatenko S, Volk HD, Stocker H, Muller P, Jennings GT, Wagner F, Bachmann MF. Effect of immunisation against angiotensin II with CYT006-AngQb on ambulatory blood pressure: a double-blind, randomised, placebo-controlled phase IIa study. Lancet. 2008;371:821–827. doi: 10.1016/S0140-6736(08)60381-5. [DOI] [PubMed] [Google Scholar]
- 43.Miller SA, Accardi JR, Onge ELS. Angiotensin II vaccine: a novel approach in the treatment of hypertension. Expert Opinion on Biological Therapy. 2008;8:1669–1673. doi: 10.1517/14712598.8.11.1669. [DOI] [PubMed] [Google Scholar]