

Abstract
Molecular chaperones represent the first line of defense of intracellular protein quality control. As a major constituent of molecular chaperones, heat shock proteins (HSP) are known to confer cardiomyocyte short-term protection against various insults and injuries. Previously, we reported that the small HSP αB-crystallin (CryAB) attenuates cardiac hypertrophic response in mice subjected to 2 weeks of severe pressure overload. However, the long-term role of small HSPs in cardiac hypertrophy and failure has rarely been studied. The present study investigates the cardiac responses to chronic severe pressure overload in CryAB/HSPB2 germ line ablated (KO) and cardiac-specific CryAB overexpressingtransgenic (TG) mice. Pressure overload was induced by transverse aortic constriction in KO, TG, and non-transgenic wild type (NTG) control mice and 10 weeks later molecular, cellular, and whole organ level hypertrophic responses were analyzed. As we previously described, CryAB/HSPB2 KO mice showed abnormal baseline cardiac physiology that worsened into a restrictive cardiomyopathic phenotype with aging. Severe pressure overload in these mice led to rapid deterioration of heart function and development of congestive cardiac failure. Contrary to their short term protective phenotype, CryAB TG mice showed no significant effects on cardiac hypertrophic responses and very modest improvement of hemodynamics during chronic systolic overload. These findings indicate that small HSPs CryAB and/or HSPB2 are essential to maintain cardiac structure and function but overex-pression of CryAB is not sufficient to confer a sustained protection against chronic systolic overload.
Keywords: Small heat shock protein, pressure overload, hypertrophy, fetal genes, cardiac remodeling
Introduction
Intracellular protein quality control (PQC) is carried out by multi-level collaborations between molecular chaperones and targeted proteolysis [1]. The latter is primarily performed by the ubiq-uitin-proteasome system [2]. Most heat shock proteins (HSP) possess molecular chaperone functions and are critical to PQC, especially when the cell is under a stress condition. HSPs play important roles in cardiac pathophysiology [3]. However, the role of HSPs, small HSPs in particular, in cardiac responses to chronic mechanical stress remains to be established. αB-Crystallin (CryAB) belongs to the small HSP subfamily and it is the most abundant chaperone protein in the heart, accounting for 2-3% of the cytosolic proteins in cardiomyocytes [4, 5]. This high abundance is second only to the major sarcomeric proteins that comprise the basic contractile apparatus of cardiomyocytes, and thus underscores its biological importance in the heart.
Clinically, CryAB shows differential expression patterns in various types and stages of human heart disease. CryAB is upregulated in hypertrophic cardiomyopathies and desmin-related myopathies (DRM), but down-regulated in failing human hearts [6-8]. Mutations of the CryAB gene (e.g., CryABR120G) have been linked to human DRM, a disease characterized by intracellular protein aggregation, mitochondrial deficiencies, proteasomal dysfunction, activation of apoptosis and heart failure [9-11]. It has been recently demonstrated that sarcomeric disarray and accumulation of physical aggregates were the cause of significant changes in mechanical properties in CryABR120G cardiomyocytes [12]. Although the consequences of the lack of CryAB in the heart was not well documented, a recent study described the necessity for a mitochondrial chaperone Hsp40 (Dnaja3) in preventing dilated cardiomyopathy (DCM) with a mouse model of cardiac-specific Dnaja3 KO [13].
Both transgenesis and gene targeting techniques have been employed to dissect CryAB function in the heart. CryAB overexpression in the heart was shown to be beneficial in acute ischemia/reperfusion (I/R) recovery while CryAB/HSPB2 null mouse hearts displayed poorer functional recovery, a higher cell death rate, increased stiffness and hence poor relaxation of myocardium following I/R, compared to wild type controls [14-16]. Mitochondrial permeability transition and mitochondrial calcium uptake were increased in cardiomyocytes from CryAB/HSPB2 null mice [17]. Recently, we were able to show that CryAB also interacted with NFAT signaling in the heart in such a way that overexpression of CryAB attenuated and absence of CryAB enhanced the nuclear translocation of NFAT that is an activator of numerous downstream hypertrophy gene targets [18].
The potential role(s) of HSPs in cardiac response to mechanical stress has rarely been investigated. As a bona fide small HSP in the heart, CryAB associates with cytoskeleton and the contractile apparatus in cardiomyocytes [3, 19]. Both cytoskeleton and the contractile apparatus play important roles and undergo dramatic remodeling in cardiac response to mechanical overload. We reported that CryAB suppressed the hypertrophy induced by short term (2 weeks) pressure overload in the heart [18]. However, the effects of CryAB on chronic and severe pressure overload have not been reported. Using CryAB/HSPB2 knockout and car-diomyocyte-restricted CryAB overexpression mouse models, the present study investigates the modulation of CryAB on cardiac responses to chronic systolic overload induced by transverse aortic constriction (TAC). We discovered that mice lacking CryAB/HSPB2 developed a concentric cardiac hypertrophy with features of restrictive cardiomyopathy at a relatively young age and showed rapid progression of pathological cardiac hypertrophy and failure to chronic systolic overload. CryAB overexpression on the other hand was not sufficient to suppress or retard the heart failure progression in mice undergoing chronic systolic overload. Our results suggest that although overexpression of CryAB is insufficient to confer protection against long term cardiac mechanical stress, endogenous CryAB and/or HSPB2 are essential for the heart and the deficiency of these small HSPs produces detrimental outcomes during stressful conditions such as systolic overload. This study not only yields new insights into the physiological significance of small HSPs in the heart but also provide critical information for overexpression of CryAB as a potential therapeutic strategy to protect chronically systolic overloaded hearts.
Materials and methods
Experimental animals
Only male mice were used. Generation of CryAB/HSPB2 KO mice in a 129/Svj genetic background was previously described [20]. KO mice (line 460) used in this study were derived from 8 generations of back-crossing of the 129/Svj CryAB/HSPB2 KO mice into the FVB/N background. Line 11 CryAB transgenic mice (TG) were created and have been maintained in the FVB/N inbred background and the initial characterization of this TG line was reported [11]. Age-matched FVB/N non-transgenic (NTG) mice were used as the controls for both the KO and TG groups. This study conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Transverse aortic constriction (TAC)
TAC was performed on mice at 10 weeks of age under aseptic conditions as previously described with modifications [18]. After inducing anesthesia using 2% Isoflurane and oro-tracheal intubation, the animal was mechanically ventilated with a rodent ventilator (MiniVent Type 845; Harvard Apparatus, MD). Anesthesia was maintained with 1.2-1.5% isoflurane in 100% oxygen with a tidal volume of 200μl at 120 strokes/min. Through a midline partial sternotomy approach, the aortic arch was isolated between the innominate and the left common carotid arteries and ligated with a 7-0 nylon suture (Ethicon, NJ) against a 29G (outer diameter 0.33mm) needle. The needle was immediately withdrawn after the ligation. Sham control mice underwent the same procedure except for aortic constriction. After surgery, the chest wall was closed with 4-0 silk, and the skin was sealed. Pain control was achieved by subcutaneous administration of Buprenorphine. The animals were housed in separate cages in an animal care facility where a 12 hour light/ dark cycle was maintained and ordinary chow and water given ad libitum. Terminal experiments were performed at 10 weeks post-operation.
Transthoracic echocardiography (Echo)
Echo was performed using a high resolution Vevo 660™ Echo system with a 30MHz transducer and 12.7mm focal length (Visual Sonics, Toronto, Canada) both pre-and post-operatively. Mice were kept under light anesthesia with Isoflurane in room air supplemented with 100% oxygen. A 2D guided M-mode was acquired for primary morphometry through the anterior and posterior walls at the papillary muscle level at a frame rate of 140Hz.
Left Ventricular (LV) catheterization and pressure measurement
At the terminal experiments, the mouse right common carotid artery was isolated and cannu-lated with a high-fidelity 1.4F Millar Mikro-Tip® catheter transducer (model SPR-835, Millar Instruments, TX) and advanced to the left ventricular (LV) chamber under anesthesia. After stabilizing for 10 minutes, LV pressure (LVP) and its first derivatives (dP/dt) were recorded using a Powerlab data acquisition system (ADInstruments, Colorado Springs, CO). Heart rate (HR), systolic and diastolic LVP, and LVend-diastolic pressure (LVEDP) were all measured directly from the LVP waveforms. The LV function was evaluated by the maximal positive and negative dP/dt (+dP/dtmax and −dP/dttmax) and the dP/dt at a LVP of 40mmHg (+dP/dt40 and −dP/dt40).
Morphometry of isolated ventricular myocytes
The technique used conforms to the detailed protocols documented [21, 22]. Immediately after hemodynamic measurements, mice were heparinized (5000U/kg) intraperitoneally. After removing the heart, the aorta was retrogradely cannulated and sequentially perfused with modified calcium-free solutions. Myocytes from the LV and ventricular septum were dissociated and filtered through a 250μm mesh and fixed with 2.5% glutaraldehyde. For further measurements, only isolations with at least 75% rod-shaped myocytes were used. AZ2 Coulter Chan-nelyzer (Beckman Coulter Inc., Miami, FL) was employed to measure the cell volume of isolated cardiomyocytes from over 5000 cells/sample. The volumes obtained were corrected for the elliptical geometry of the myocytes by multiplying with a factor of 1.43. A NIH image analysis software was used to measure the length and profile area from at least 80 cells/sample.
RNA dot blot analysis
Total RNA was isolated from the LV of 3 individual mice of each group using Tri-Reagent (Molecular Research Center Inc, OH). For RNA dot blot, 4μg of total RNA was loaded on to a nitrocellulose membrane. After cross-linking, the membrane was hybridized with P32-labeled transcript-specific oligonucleotide probes of the hypertrophic gene panel and detected with the Personal Molecular Imager FX (BioRad) and quantified with the associated Quantity-One software as described [23].
Statistical analysis
All quantitative data are presented as mean+SD, unless indicated otherwise. Significance levels were analyzed by student t-test or one factor analysis of variance (ANOVA) followed by Holm-Sidak post-hoc comparison (SigmaStat 3.0, Systat, Point Richmond, CA), where applicable.
Results
CryAB KO but not overexpression produces cardiomyopathy
We previously examined and reported the phenotype of the CryAB/HSPB2 KO mice after back-crossing them from the 129/Svj to the FVB/N background. At a young age of 10 weeks, KO mice displayed a significant basal level cardiac hypertrophy but relatively normal cardiac function [18]. Here, we continued to monitor these mice for an additional 10 weeks for a maximum of 20 weeks of age to characterize their pheno-type that would allow us to compare and contrast them with the NTG and TG mice.
Serial echocardiography in TG and NTG animals started at 10 weeks of age and continued to 20 weeks at regular intervals, showed almost identical cardiac geometry and function (Table 1). Heart weight to body weight and other organ weight to body weight ratio comparisons showed that NTG and TG mice had similar growth patterns and that there was no indication of cardiac hypertrophy or the consequences thereof (Table 2). At the molecular level, TG and NTG mice displayed comparable gene expression profiles and the TG group showed no evidence of induction of fetal genes characteristic of cardiac hypertrophy (Figure 1). The cell volume, cell length and profile area of isolated cardiomyocytes were not statistically different between the 2 groups (Table 3).
Table 1.
Echocardiographic characterization of NTG, TG and KO mice from 10 to 20 weeks of age
| 10 week |
16 week | 20 week |
|||||
|---|---|---|---|---|---|---|---|
| NTG | TG | KO | KO | KO | NTG | TG | |
| n | 36 | 30 | 23 | 12 | 12 | 12 | 12 |
| LVIDd (mm) | 3.79±0.21 | 3.74±0.18 | 3.58±0.28* | 3.84±0.15 | 3.87±0.19 | 3.77±0.18 | 3.72±0.32 |
| LVIDs (mm) | 2.34±0.21 | 2.28±0.20 | 2.26±0.24 | 2.61±0.14* | 2.64±0.17 | 2.40±0.17 | 2.33±0.29 |
| LVPWd (mm) | 0.77±0.10 | 0.76±0.12 | 0.85±0.12* | 0.88±0.09 | 0.90±0.06* | 0.82±0.08 | 0.76±0.07 |
| HR (bpm) | 438±45 | 415±39 | 443±45 | 403±26 | 444±44 | 432±49 | 445±36 |
| FS (%) | 38.4±3.8 | 39.5±4.6 | 37.2±3.6 | 32.2±1.8* | 31.3±1.9* | 37.4±3.5 | 37.3±4.2 |
| EF (%) | 69.2±4.7 | 70.6±5.5 | 68.0±4.7 | 61.1±2.5* | 59.8±2.7* | 68.0±4.2 | 67.9±5.2 |
| EDV (μl) | 61.8±7.9 | 61.2±8.5 | 54.6±9.5* | 65.2±5.5 | 65.5±7.2 | 63.2±7.5 | 61.0±9.7 |
| ESV (μl) | 19.1±4.1 | 17.9±3.8 | 17.6±4.8 | 25.4±3.0* | 26.3±3.3* | 20.2±3.5 | 19.3±5.5 |
LVIDd: Left ventricular internal diameter atthe end of diastole; LVIDs: Left ventricular internal diameter atthe end of systole; LVPWd: Left ventricular posterior wall thickness at the end of diastole; HR: Heart rate in beats per minute; FS: Percentage fractional shortening; EF: Ejection fraction; EDV: LV volume at end-diastole; ESV: LV volume at end-systole; Mean±SD.
P<0.05; KO compared to NTG and TG at corresponding age. No statistical significant difference was detected for any of the parameters between NTG and TG at anytime point.
Table 2.
Gravimetric changes in NTG, TG and KO mice in chronic pressure overload
| NTG Sham | NTG TAC | TG Sham | TG TAC | KO Sham | KO TAC | |
|---|---|---|---|---|---|---|
| n | 7 | 10 | 6 | 9 | 9 | 7 |
| BW (g) | 29.1±4.5 | 28.3±3.65 | 27.56±2.3 | 29.2±3.2 | 28.6±3.4 | 19.8±2.4¶ |
| HW (mg) | 133.4±19 | 256.8±35* | 124.2±8.7 | 270.8±30* | 155.2±15# | 294.7±37* |
| HW/BW (mg/g) | 4.6±0.1 | 9.15±1.3* | 4.52±0.3 | 9.37±1.5* | 5.44±0.3# | 14.9±1.4*¶ |
| VW (mg) | 120.6±14 | 203.9±21* | 112.7±6.5 | 218.9±20* | 132.4±11 | 214.1±27* |
| VW/BW (mg/g) | 4.17±0.2 | 7.29±1.1* | 4.10±0.3 | 7.51±1.2* | 4.65±0.3# | 10.8±0.7*¶ |
| Lungs (mg) | 163.7±31 | 300.4±105* | 157±11 | 311±124* | 160.7±11 | 349.6±48* |
| Lungs/BW (mg/g) | 5.79±1.3 | 10.2±3.7* | 5.73±0.6 | 10.6±5.2* | 5.5±0.3 | 16.5±4.0*¶ |
| Liver (mg) | 1286±202 | 1326±181 | 1256±144 | 1363±197 | 1368±174 | 947±192*¶ |
| Liver/BW (mg/g) | 44.87±3.3 | 42.89±3.4 | 45.68±5.1 | 45.53±4.5 | 46.04±4.0 | 42.29±3.9 |
| Kidney (mg) | 437±38 | 431±21 | 385±31 | 421±57 | 437±45 | 347±57*¶ |
| Kidney/BW mg/g | 13.8±0.9 | 13.0±0.7 | 14.3±0.7 | 13.6±0.7 | 14.7±1.3 | 15.62±1.3¶ |
| HW/TL (mg/mm) | 7.21±1.1 | 14.17±2.1* | 6.68±0.5 | 14.81±1.5* | 8.77±0.5# | 16.4±1.7¶ |
BW: body weight; HW/BW: heart weight to body weight ratio; VW/BW; ventricular weight to body weight ratio; HW/TL: HW to tibial length ratio. Mean±SD.
P<0.05 or 0.01 compared to the Sham of the same genotype;
P<0.05 compared to NTG TAC and TG TAC groups;
P<0.05 compared to NTG sham.
Figure 1.

Reactivation of the fetal gene program. Quantitative RNA dot blot analysis was utilized to assess the transcript levels of atrial natriuretic factor (ANF), β-myosin heavy chain (β-MyHC), skeletal α-actin (s-Actin), α-MyHC, sar-coplasmic endoplasmic reticulum calcium ATPase 2A (SERCA), phospholamban (PLN), and GAPDH using 32P-labeled transcript-specific oligonucleotide probes. The bound radioactive signal was captured by a phosphor-imaging screen, detected and quantified. (A) Representative RNA dot blot images. Each dot represents an individual animal. After normalizing to the corresponding GAPDH signal, the mean intensity value of the NTG sham group was set at 100 arbitrary units (AU). The intensity signal of each individual dot is then normalized to the mean of the NTG sham. The resultant intensity of each of the 3 animals from the same group was used to derive the mean and standard deviation (error bar) and presented in subsequent panels. (B) The effect of KO on fetal gene reactivation in mouse hearts at 10 weeks after sham or TAC surgery. (C) The effect of TG on fetal gene reactivation in mouse hearts at 10 weeks after sham or TAC surgery. Compared with NTG sham, *: p<0.05, #: p<0.01. NS: not significant.
Table 3.
Morphometry of cardiomyocytes isolated from NTG, TG and KO mice at 10 weeks after TAC or sham surgery
| NTG Sham | NTG TAC | TG Sham | TG TAC | KO Sham | KO TAC | |
|---|---|---|---|---|---|---|
| n | 8 | 8 | 6 | 9 | 7 | 6 |
| Cell Volume (×103 μm3) | 35.5±1.8 | 57.8±5.2* | 33.0±1.6 | 58.5±4.2* | 47.9±2.2# | 65.2±3.0* |
| CSA (μm2) | 259±11 | 351±30* | 242±14 | 359±17* | 309±13# | 388±25* |
| Cell Length (μm) | 137.2±4.2 | 164.7±2.8* | 136.4±2.7 | 163.2±7.0* | 155.0±5.4# | 168.2±4.3* |
| Cell Profile Area (μm2) | 3963±242 | 6157±311* | 3707±172 | 6027±463* | 5087±363# | 6778±496* |
LV cardiomyocytes isolated by collagenase perfusion were fixed in isotonic glutaraldehyde. A Coulter Channelyzer was used to determine the cell volume and microscopic image analyses were used to measure cell dimensions. Mean±SD.
P<0.05 compared to the sham of the same genotype;
P<0.05 compared to NTG sham.
Unlike the TG mice, KO mice showed a distinct abnormal phenotype at all the levels examined.
In comparison to 20-week-old NTG sham mice, the KO sham mice showed a 16%, 18%, and 21% increases in the heart weight, the HW/BW ratio, and HW to tibial length ratio, respectively (p<0.05). The average body weights of the KO and NTG mice did not differ (Table 2). These gravimetric data clearly demonstrated a hyper-trophic growth of the KO hearts.
Assessment of the KO hearts by Echo showed normal function as reflected by unchanged fractional shortening (FS) and ejection fraction (EF), no increase in diastolic LV chamber diameters, but thickened walls (compared to NTG mice) until 16 weeks of age (Table 1). Starting from 16 weeks, the chambers displayed increased end systolic diameters (LVIDs) together with an average 40% expansion of LV end-systolic volume (ESV). However, there were no significant alterations in the LVIDd or LV end-diastolic volume (EDV), compared to NTG hearts, indicating that there was no chamber dilatation taking place in the KO hearts at this point. The FS and EF in the 16 week old KO hearts was reduced by approximately 19% and 14%, respectively, compared to NTG. These cardiac functions were not further impaired in the 20 week old KO mice.
The basal level transcription of the hypertrophic panel genes in KO hearts at 20-weeks of age showed dramatic changes. Atrial natriuretic factor (ANF), skeletal α-actin (s-Actin) and β-myosin heavy chain (MyHC) showed significant upregu-lation while α-MyHC, sarcoplasmic reticulum calcium ATPase 2A (SERCA) and phospholam-ban (PLN) were significantly down-regulated compared to age matched NTG and TG hearts (Figure 1A and 1B).
Cardiac hypertrophy in KO mice is further confirmed at the cellular level. Ventricular myocytes isolated from KO mice showed distinctly significant increases in myocyte volume, length, cross-sectional area (CSA) and profile area, compared with those from the NTG and TG mice (Table 3).
Hemodynamic measurements showed poor diastolic relaxation in the KO hearts (Table 4 and Figure 3). The increase in end diastolic LV pressures (LVEDP), reduced negative dP/dtmax, accompanied by an elevated +dP/dtmax and +dP/dt40 suggest that these mice had increased myocardial stiffness and defects in relaxation.
Table 4.
Hemodynamic characterization of NTG, TG and KO mice 10weeks after TAC or sham surgery
| NTG Sham | NTG TAC | TG Sham | TG TAC | KO Sham | KO TAC | |
|---|---|---|---|---|---|---|
| n | 11 | 13 | 10 | 14 | 11 | 8 |
| LVSP (mmHg) | 89.3±6.1 | 153.9±15.1 | 87.7±4.9 | 168.8±15.1¶ | 87.5±6.6 | 136.3±19.7* |
| LVEDP (mmHg) | 3.92±0.9 | 14.8±9.3 | 4.2±0.8 | 21.9±11.4 | 20.6±5.9 | 32.9±4.8* |
| HR (bpm) | 429±39 | 455±61 | 435±51 | 469±70 | 458±28 | 377±54* |
| + dP/dt40 (mmHg/s) | 5785±623 | 5714±1120 | 6411±979# | 6332±1092 | 7151±685# | 3644±1130 |
| −dP/dt40 (mmHg/s) | −4605±537 | −4387±756 | −5132±456# | −5237±633¶ | −4608±305 | −2973±273 |
| +dP/dtmax (mmHg/s) | 6448±586 | 7997±771 | 7327±666 | 7537±1621 | 8379±705# | 5526±1175 |
| −dP/dtmax (mmHg/s) | −6891±810 | −7491±759 | −7792±779 | −8080±1190 | −6015±668# | −4978±891 |
LVSP: LV systolic pressure; LVEDP: LV end-diastolic pressure; +dP/dtmax and −dP/dtmax: the maximum and minimum first derivative of LV pressure, respectively; +dP/dt40:and −dP/dt40: the positive and negative first derivative of LV pressure when the pressure is at 40mmHg; Mean±SD. P<0.05:
compared to NTG sham;
KO TAC compared to NTG TAC;
TG TAC compared to NTG TAC.
Figure 3.

Hemodynamic analyses of left ventricular systolic pressure (LVSP), end-diastolic pressure (LVEDP, panel A) and maximum and minimum dP/dt at 40mmHg (+dP/dt40 and −dP/dt40, Panel B) in NTG and KO mice after 10 weeks of TAC. *: P<0.05 compared to NTG sham; #: P<0.01compared to KO sham.
Chronic pressure overload induces rapid cardiac failure in CryAB/HSPB2 KO mice
Two weeks of severe pressure overload led to significant cardiac hypertrophy and early features of congestive cardiac failure in the KO mice [18]. In this study, the KO TAC mice were further observed for a maximum of 10 weeks following severe systolic overload. As expected, the 6-, 8- and 10-weeks post-TAC Echo showed significant progressive impairment of function in the KO hearts compared to NTG hearts (Figure 2). The EF and FS were reduced in KO TAC hearts by approximately 20%, LVIDd and LVIDs increased by 6% and 13%, respectively, with corresponding increases in EDV and ESV compared to NTG TAC hearts. They also showed significant bradycardia at 10 weeks of chronic pressure overload.
Figure 2.

Serial echocardiography analyses of left ventricular ejection fraction (EF, panel A) and end diastolic volume (LVEDV, panel B) before and after TAC in NTG and KO mice. Serial Echo analysis was performed on NTG and age-matched KO mice at the day before (marked as 0 week after TAC in the graphs) and 2, 6, 8, and 10 weeks after the TAC or sham surgery. Mean ±SE. *: P<0.05 compared with NTG sham; #: P<0.05 compared with KO sham; $: P<0.05 compared with NTG TAC.
KO hearts had high basal level induction of the fetal gene expression and chronic pressure overload did not significantly alter the already activated fetal gene program in these mice. After 10 weeks of TAC, both the NTG and KO hearts showed similar degrees of transcript level changes characteristic of cardiac hypertrophy with a more pronounced down-regulation of SERCA and PLN in the KO TAC group than NTG TAC (Figure 1A). Interestingly, at 10 weeks after TAC, the steady state transcript level of skeletal α-actin which was significantly up-regulated in KO sham mice, returned to the level comparable to that of NTG sham mice. This suggests that CryAB and HSPB2 may be required for the activation of skeletal α-actin in the heart under mechanical stress.
Unlike NTG mice, KO mice demonstrated significant weight loss after 10 weeks of TAC surgery. Although the reduced body weight of the KO TAC mice may have contributed to the significantly high HW/BW ratio (Table 2 and Figure 4), their total heart weights appeared significantly larger and their HW to tibial length ratios were also significantly greater than those of NTG TAC mice (Table 2), indicating that KO mice showed more severe cardiac hypertrophy than the NTG mice at 10 weeks after TAC. The lung weights and the lung/body weight ratios were all significantly increased in the TAC groups compared with their corresponding Sham groups. The increases of the KO TAC group but not the TG TAC group were significantly greater than those of the NTG TAC (Table 2), suggesting that 10 weeks of TAC induced more severe pulmonary congestion in KO mice than NTG and TG mice.
Figure 4.
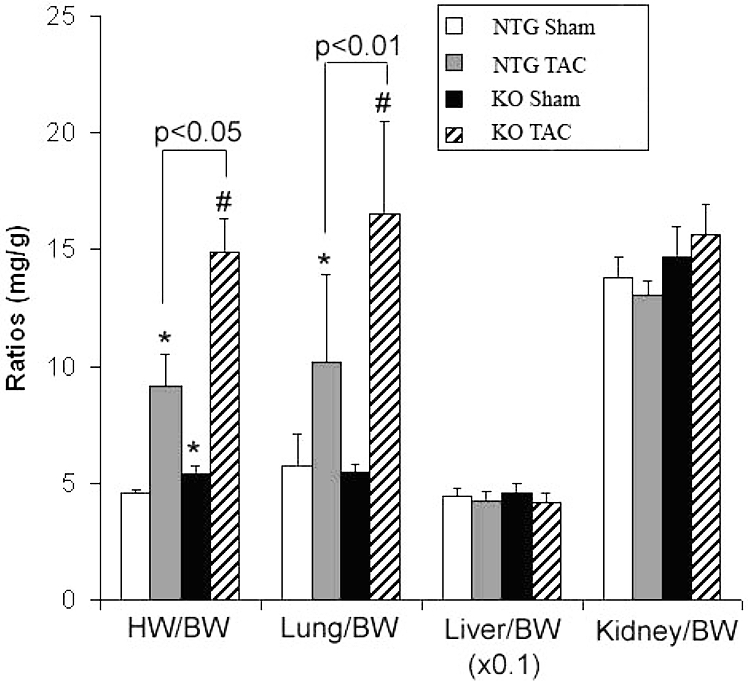
Gravimetric analyses of NTG and KO mice at 10 weeks after TAC. BW: body weight; HW/BW: heart weight to BW ratio; Lung/BW: lung weight to BW ratio; Liver/BW: liver weight to BW ratio; Kidney/BW: kidney weight to BW ratio. *: P<0.05 compared with NTG sham; #: P<0.01 compared with KO sham.
Interestingly, however, KO TAC mice showed shrinkage of liver and kidneys (approximately 30% and 20% respectively, compared to NTG TAC organs). The increased kidney/body weight ratio seen in these mice, despite the reduced kidney weight was obviously due to the significant weight loss.
After prolonged TAC, cardiomyocytes from the KO mice had only 36% increase in cell volume when NTG cells had a 63% increase over their sham controls. This difference was mainly due to the basal level hypertrophy of the cardiomyocytes in the KO shams. Nevertheless, KO cardiomyocytes showed 10% larger profile areas, and 12% larger myocytes volume and CSA compared to NTG TAC myocytes (Table 3). Complementing the Echo findings, after 10 weeks of TAC, the KO mice showed significant bradycardia and deterioration in their heart function by hemody-namic measurements. They demonstrated significantly elevated LVEDP, lower LVSP, and decreased dP/dt at 40mmHg compared to NTG TAC hearts (Table 4 and Figure 3).
Overexpression of CryAB fails to reduce cardiac hypertrophy but modestly improves LV function in chronic systolic overload
We previously reported that CryAB overexpres-sion in the heart significantly attenuated cardiac hypertrophy following a 2 week insult of pressure overload [18]. This was observed at all levels studied, including less growth of the hearts to less re-activation of the fetal genes, and the maintenance of normal cardiac function in TG TAC mice than NTG TAC mice. We anticipated slower progression to cardiac failure of the same hearts when subjected to chronic systolic overload for additional 8 weeks. However, our Echo findings revealed that starting from 6 weeks after TAC, both TG and NTG hearts showed almost parallel changes and equal pace of progression to cardiac failure. The chamber dilatations, reductions in EF and FS and increases in EDV and ESV appeared to be quite comparable in the two TAC groups (Figure 5).
Figure 5.

Serial echocardiography analyses of left ventricle fractional shortening (FS, panel A) and end diastolic volume (panel B) before and after TAC in NTG and TG mice. Mean±SE. NS: not significant between TG sham and NTG sham or between TG TAC and NTG TAC.
Correspondingly, fetal gene expression patterns also became similar in the 2 groups. ANF and β -MyHC transcript levels were elevated to the same degree as the NTG TAC hearts and α-MyHC, SERCA and PLN were down-regulated (Figure 1A, 1C).
Gravimetry showed about 100% increases in HW/BW ratios in the 2 TAC groups and all other parameters studied appeared parallel including the degree of lung congestion (Table 2). Mor-phometry of isolated cardiomyocytes did not reveal any significant difference between the NTG TAC and TG TAC hearts (Table 3). These data consistently indicate that overexpression of CryAB in the heart is not sufficient to confer a long-term protection against cardiac hypertrophic responses and progression to congestive heart failure induced by chronic systolic overload.
Nevertheless, LV hemodynamic measurements showed a slightly mixed picture. Compared with NTG TAC, TG TAC mice had statistically significant higher LVSP and better −dP/dt40 while differences of other parameters were insignificant at 10 weeks after TAC (Table 4).
Discussion
The indispensable role of molecular chaperones has been identified in various cellular stresses. Cardiac hypertrophy and subsequent failure are important pathological phenomena arising from multiple etiologies. The role of small HSP/molecular chaperones in the modulation of the cardiac hypertrophic response has not been well studied. Our recent experiments convincingly showed that CryAB, the most abundant small HSP in the heart that is mainly known for its cytoskeletal chaperoning functions can indeed negatively modulate cardiac responses to a short-term pressure overload condition [18]. We were able to provide evidence that the potential mechanism by which CryAB attenuates cardiac hypertrophic response is by interfering with the pathologic NFAT signaling pathway. The effects of CryAB on long standing cardiac hypertrophy and subsequent failure are not known. In the present study, we tried to address this question by using the cardiac-specific CryAB overex-pressing TG mice and CryAB/HSPB2 KO mice. Cardiac responses were analyzed at the molecular, cellular and whole organ level.
We report here that CryAB and/or HSPB2 are required to maintain normal structure and function in non-stressed or mildly stressed hearts. Lack of these small HSPs led to an abnormal cardiac phenotype at basal state and development of cardiac failure at an accelerated pace to systolic overload. The gravimetric, echocar-diographic and hemodynamic findings of the KO mice featured a restrictive cardiomyopathic phenotype. The moderate increase in cardiac weight, increased wall thickness without chamber dilatation, diastolic dysfunction and normal or reduced global systolic function were compatible with an infiltrative (such as amyloidosis and endomyocardial fibrosis) or idiopathic restrictive cardiomyopathy [24]. Abnormal protein aggregates have been observed in CryAB/ HSPB2 KO skeletal muscle as well as CryABR120G expressing cardiomyocytes [11, 20]. Early fibrosis is also a plausible explanation. CryAB is known to associate with titin filaments that impart elastic properties to the contractile machinery [19]. Hence, poor relaxation of the myocardium could also be due to the stiffness caused by poor structural integrity of titin in the absence of the chaperone molecules. As identified in the 10 weeks old KO mice, persistently activated NFAT signaling due to the lack of CryAB could have led to the hypertrophic phenotype observed in the older mice [18]. CryABR120G TG mice are also known to develop cardiac hypertrophy and failure early in their life while deficiency in HSP40, a mitochondria-associated HSP was reported to cause dilated cardio-myopathy in mice [11, 13]. These reports highlight the importance of CryAB in the maintenance of normal cardiac physiology, and the many potential underlying causative mechanisms that lead to abnormal structure and functioning with loss of function of this small HSP.
The loss of function of the HSPB2 may also have direct contributions to the KO phenotype. In the heart, HSPB2 has been observed to localize to the Z lines and intercalated discs and is believed to protect the actin cytoskeleton and other proteins against oxidative and ischemic stresses [25, 26]. Emerging studies suggest that HSPB2 is also associated with the mitochondria outer layer and plays a role in the stress response [17, 27]. Therefore, it is likely that HSPB2 resembles and shares the roles of CryAB, making its absence as critical as the lack of CryAB in the development of the abnormal hypertrophic response in the KO hearts. Pinz et a l recently reported that HSPB2 has specific roles in the heart to maintain normal systolic performance and normal cardiac energetics whence CryAB protects diastolic performance [28]. The CryAB/HSPB2 KO mice used in this study are a systemic gene deletion model and hence it is likely that other neurohumoral mechanisms also contribute to the observed cardiac phenotype. CryAB is expressed in the central nervous system (CNS) and the kidneys, two systems that are closely associated in the regulation of cardiac function. Unfortunately, the roles of CryAB in these locations are not very well defined. Hypertonicity and electrolyte imbalances have been shown to induce CryAB in the renal tubules and the CNS [29, 30]. The absence of this molecular chaperone from these important systems may reactively induce the renin-angiotensin-aldosterone (RAAS) pathways which could subsequently have a direct impact on cardiac remodeling. Circulating and local angiotensin II in turn, could directly stimulate ANF expression in the heart [31].
This study unveiled that CryAB/HSPB2 KO mice represented a basal level cardiac hypertrophic phenotype with significant abnormalities in cellular morphology, gene transcription and cardiac function within 5 months of age. When these mice were subjected to TAC, they developed cardiac malfunction more rapidly than wild type control mice. It was also observed that the pressure overload stress could not further increase the already strongly induced fetal genes in KO hearts. This suggests that fetal gene reactivation in pressure overload cardiac hypertrophy may be caused by a relative insufficiency in molecular chaperoning activities. This is certainly consistent with the inhibitive action of CryAB on the calcineurin-NFAT pathway because the activation of the latter triggers the expression of some of the fetal genes such as ANF in cardiac pathology [18].
Based on the early phase cardiac hypertrophic response, it was anticipated that TG mice that overexpressed CryAB in the heart would show protection during prolonged pressure overload. However, this was not clearly supported by the evidence collected in the present study. Overall, TG TAC and NTG TAC mice showed almost identical phenotypes until the end of the 10 week observation period. The progression to cardiac insufficiency in the two groups as revealed by echocardiography were at a comparable pace with similar changes at the molecular and cellular levels although the TG TAC hearts displayed few isolated features of better cardiac function. They maintained significantly elevated systolic LV pressures and better isometric relaxation properties compared to the NTG TAC (Table 4). It is possible that the persistent systolic overload might have upregulated the expression of small HSPs including CryAB to a critical level sufficient to fulfill the increased chaperoning demand so that additional overexpression of CryAB would not provide further benefit. Indeed, in our earlier study, we observed a progressive up-regulation of CryAB in the heart during the initial period after TAC [18]. It is also likely that the study was not continued long enough to observe any potential delayed protection of CryAB overexpression in cardiac failure. Protein level examination of the 10 week NTG TAC mice showed that the CryAB proteins were still elevated in the myocardium (data not shown). In human end-stage congestive heart failure (CHF), a significant down-regulation of CryAB levels have been observed [7]. This suggests that the study duration may not be sufficiently long to determine the potential benefits of CryAB over-expression in end-stage heart failure.
Taken together our findings provide evidence that CryAB is an important modulator of the cardiac hypertrophic response. These novel discoveries not only demonstrate the indispensable roles of CryAB in normal and stressed cardio-myocytes, but also suggest the potential malfunctions expected in the cells in the event CryAB proteins are down-regulated.
We have also recently demonstrated that pro-teasome functional insufficiency, the other major player of PQC, is also sufficient to cause cardiac hypertrophy likely via activating the cal-cineurin-NFAT pathway [32]. Taken together, it seems that PQC inadequacy due to the inadequacy of either chaperoning activities or protea-some function can activate the calcineurin-NFAT pathway and trigger or enhance cardiac hyper-trophic responses.
Acknowledgments
This work was supported in part by NIH grants RO1HL072166 and R01HL085629 and an American Heart Association (AHA) Established Investigator Award and Scientist Development Grant (235099N; to X.W.), and an AHA Predoc-toral Fellowship (0510069Z; to A.R.K.K). We thank Rebecca Redetzke for her excellent technical assistance in hemodynamic data acquisition.
References
- 1.Su H, Wang X. The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc Res. 2010;85:253–262. doi: 10.1093/cvr/cvp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng Q, Li J, WangX Interplay between the ubiquitin-proteasome system and autophagy in proteinopathies. Int J Physiol Pathophysiol Pharmacol. 2009;1:127–142. [PMC free article] [PubMed] [Google Scholar]
- 3.Kumarapeli AR, Wang X. Genetic modification of the heart: chaperones and the cytoskeleton. J Mol Cell Cardiol. 2004;37:1097–1109. doi: 10.1016/j.yjmcc.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Kappe G, Franck E, Verschuure P, Boelens WC, Leunissen JA, de Jong WW. The human genome encodes 10 alpha-crystallin-related small heat shock proteins: HspB1-10. Cell Stress Chaperones. 2003;8:53–61. doi: 10.1379/1466-1268(2003)8<53:thgecs>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Longoni S, James P, Chiesi M. Cardiac alpha-crystallin. I. Isolation and identification. Mol Cell Biochem. 1990;99:113–120. [PubMed] [Google Scholar]
- 6.Hwang DM, Dempsey AA, Wang RX, Rezvani M, Barrans JD, Dai KS, Wang HY, Ma H, Cukerman E, Liu YQ, Gu JR, Zhang JH, Tsui SK, Waye MM, Fung KP, Lee CY, Liew CC. A genome-based resource for molecular cardiovascular medicine: toward a compendium of cardiovascular genes. Circulation. 1997;96:4146–4203. doi: 10.1161/01.cir.96.12.4146. [DOI] [PubMed] [Google Scholar]
- 7.Hwang JJ, Allen PD, Tseng GC, Lam CW, Fananapazir L, Dzau VJ, Liew CC. Microarray gene expression profiles in dilated and hypertrophic cardiomyopathic end-stage heart failure. Physiol Genomics. 2002;10:31–44. doi: 10.1152/physiolgenomics.00122.2001. [DOI] [PubMed] [Google Scholar]
- 8.YangJ, Moravec CS, Sussman MA, DiPaola NR, Fu D, Hawthorn L, Mitchell CA, Young JB, Francis GS, McCarthy PM, Bond M. Decreased SLIM1 expression and increased gelsolin expression in failing human hearts measured by high-density oligonucleotide arrays. Circulation. 2000;102:3046–3052. doi: 10.1161/01.cir.102.25.3046. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q, Liu JB, Horak KM, Zheng H, Kumarapeli AR, Li J, Li F, Gerdes AM, Wawrousek EF, Wang X. Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardio-myocytes by compromising substrate uptake. Circ Res. 2005;97:1018–1026. doi: 10.1161/01.RES.0000189262.92896.0b. [DOI] [PubMed] [Google Scholar]
- 10.Maloyan A, Sanbe A, Osinska H, Westfall M, Robinson D, Imahashi K, Murphy E, Robbins J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha-B-crystallin desmin-related cardiomyopathy. Circulation. 2005;112:3451–3461. doi: 10.1161/CIRCULATIONAHA.105.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.WangX, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 12.Maloyan A, Osinska H, Lammerding J, Lee RT, Cingolani OH, Kass DA, Lorenz JN, Robbins J. Biochemical and Mechanical Dysfunction in a Mouse Model of Desmin-Related Myopathy. Circ Res. 2009 doi: 10.1161/CIRCRESAHA.108.193516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi M, Imanaka-Yoshida K, Yoshida T, Wood M, Fearns C, Tatake RJ, Lee JD. A crucial role of mitochondrial Hsp40 in preventing dilated cardiomyopathy. Nat Med. 2006;12:128–132. doi: 10.1038/nm1327. [DOI] [PubMed] [Google Scholar]
- 14.Ray PS, Martin JL, Swanson EA, Otani H, Dillmann WH, Das DK. Transgene overexpression of alphaB crystallin confers simultaneous protection against cardiomyocyte apoptosis and necrosis during myocardial ischemia and reperfusion. Faseb J. 2001;15:393–402. doi: 10.1096/fj.00-0199com. [DOI] [PubMed] [Google Scholar]
- 15.Golenhofen N, Redel A, Wawrousek EF, Drenckhahn D. Ischemia-induced increase of stiffness of alphaB-crystallin/HSPB2-deficient myocardium. Pflugers Arch. 2006;451:518–525. doi: 10.1007/s00424-005-1488-1. [DOI] [PubMed] [Google Scholar]
- 16.Morrison LE, Whittaker RJ, Klepper RE, Wawrousek EF, Glembotski CC. Roles for alphaB-crystallin and HSPB2 in protecting the myocardium from ischemia-reperfusion-induced damage in a KO mouse model. Am J Physiol Heart Circ Physiol. 2004;286:H847–855. doi: 10.1152/ajpheart.00715.2003. [DOI] [PubMed] [Google Scholar]
- 17.Kadono T, Zhang XQ, Srinivasan S, Ishida H, Barry WH, Benjamin IJ. CRYAB and HSPB2 deficiency increases myocyte mitochondrial permeability transition and mitochondrial calcium uptake. J Mol Cell Cardiol. 2006;40:783–789. doi: 10.1016/j.yjmcc.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Kumarapeli AR, Su H, Huang W, Tang M, Zheng H, Horak KM, Li M, WangX Alpha B-crystallin suppresses pressure overload cardiac hypertrophy. Circ Res. 2008;103:1473–1482. doi: 10.1161/CIRCRESAHA.108.180117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golenhofen N, Arbeiter A, Koob R, Drenckhahn D. Ischemia-induced association of the stress protein alpha B-crystallin with I-band portion of cardiac titin. J Mol Cell Cardiol. 2002;34:309–319. doi: 10.1006/jmcc.2001.1513. [DOI] [PubMed] [Google Scholar]
- 20.Brady JP, Garland DL, Green DE, Tamm ER, Giblin FJ, Wawrousek EF. AlphaB-crystallin in lens development and muscle integrity: a gene knockout approach. Invest Ophthalmol Vis Sci. 2001;42:2924–2934. [PubMed] [Google Scholar]
- 21.Gerdes AM. A reliable, efficient, and comprehensive approach to assess myocyte remodeling in cardiac hypertrophy and failure. J Card Fail. 1997;3:63–68. doi: 10.1016/s1071-9164(97)90009-9. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Li F, Gerdes AM. Chronic pressure overload cardiac hypertrophy and failure in guinea pigs: I. Regional hemodynamics and myocyte remodeling. J Mol Cell Cardiol. 1999;31:307–317. doi: 10.1006/jmcc.1998.0884. [DOI] [PubMed] [Google Scholar]
- 23.WangX, Klevitsky R, Huang W, Glasford J, Li F, Robbins J. AlphaB-crystallin modulates protein aggregation of abnormal desmin. Circ Res. 2003;93:998–1005. doi: 10.1161/01.RES.0000102401.77712.ED. [DOI] [PubMed] [Google Scholar]
- 24.Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997;336:267–276. doi: 10.1056/NEJM199701233360407. [DOI] [PubMed] [Google Scholar]
- 25.Shama KM, Suzuki A, Harada K, Fujitani N, Kimura H, Ohno S, Yoshida K. Transient upregulation of myotonic dystrophy protein kinase-binding protein, MKBP, and HSP27 in the neonatal myocardium. Cell Struct Funct. 1999;24:1–4. doi: 10.1247/csf.24.1. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida K, Aki T, Harada K, Shama KM, Kamoda Y, Suzuki A, Ohno S. Translocation of HSP27 and MKBP in ischemic heart. Cell Struct Funct. 1999;24:181–185. doi: 10.1247/csf.24.181. [DOI] [PubMed] [Google Scholar]
- 27.Nakagawa M, Tsujimoto N, Nakagawa H, Iwaki T, Fukumaki Y, Iwaki A. Association of HSPB2, a member of the small heat shock protein family, with mitochondria. Exp Cell Res. 2001;271:161–168. doi: 10.1006/excr.2001.5362. [DOI] [PubMed] [Google Scholar]
- 28.Pinz I, Robbins J, Rajasekaran NS, Benjamin IJ, Ingwall JS. Unmasking different mechanical and energetic roles for the small heat shock proteins CryAB and HSPB2 using genetically modified mouse hearts. Faseb J. 2008;22:84–92. doi: 10.1096/fj.07-8130com. [DOI] [PubMed] [Google Scholar]
- 29.Dihazi H, Asif AR, Agarwal NK, Doncheva Y, Muller GA. Proteomic analysis of cellular response to osmotic stress in thick ascending limb of Henle's loop (TALH) cells. Mol Cell Proteomics. 2005;4:1445–1458. doi: 10.1074/mcp.M400184-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Sadamitsu C, Nagano T, Fukumaki Y, Iwaki A. Heat shock factor 2 is involved in the upregulation of alphaB-crystallin by high extracellular potassium. J Biochem (Tokyo) 2001;129:813–820. doi: 10.1093/oxfordjournals.jbchem.a002924. [DOI] [PubMed] [Google Scholar]
- 31.Focaccio A, Volpe M, Ambrosio G, Lembo G, Pannain S, Rubattu S, Enea I, Pignalosa S, Chiariello M. Angiotensin II directly stimulates release of atrial natriuretic factor in isolated rabbit hearts. Circulation. 1993;87:192–198. doi: 10.1161/01.cir.87.1.192. [DOI] [PubMed] [Google Scholar]
- 32.Tang M, Li J, Huang W, Su H, Liang Q, Tian Z, Horak KM, Molkentin JD, Wang X. Proteasome Functional Insufficiency Activates the Calcineurin-NFAT Pathway in Cardiomyocytes and Promotes Maladaptive Remodeling of Stressed Mouse Hearts. Cardiovasc Res. 2010 doi: 10.1093/cvr/cvq217. Jul 2 (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]