

Abstract
The lack of treatment for worried-well patients with high-grade prostatic intraepithelial neoplasia combined with issues of recurrence and hormone resistance in prostate cancer survivors remains a major public health obstacle. The long latency of prostate cancer development provides an opportunity to intervene with agents of known mechanisms at various stages of disease progression. A number of signaling cascades have been shown to play important roles in prostate cancer development and progression, including the androgen receptor (AR) and PI3K/Akt signaling pathways. Crosstalk between these two pathways is also thought to contribute to progression and hormone refractory prostate disease. Our initial investigations show that the naturally occurring organoselenium compound selenomethionine (SM) and synthetic 1,4-phenylenebis(methylene)selenocyanate (p-XSC) can inhibit human prostate cancer cell viability; however, in contrast to SM, p-XSC is active at physiologically relevant doses. In the current investigation we show p-XSC, but not an equivalent dose of SM, alters molecular targets and induces apoptosis in androgen responsive LNCaP and androgen independent LNCaP C4-2 human prostate cancer cells. p-XSC effectively inhibits AR expression and transcriptional activity in both cell lines. p-XSC also decreases Akt phosphorylation as well as Akt-specific phosphorylation of the AR. Inhibition of Akt, however, does not fully attenuate p-XSC-mediated down-regulation of AR activity suggesting inhibition of AR signaling by p-XSC does not occur solely through alterations in the PI3K/Akt survival pathway. Our data suggest that p-XSC inhibits multiple signaling pathways in prostate cancer, likely accounting for down-stream effects on proliferation and apoptosis.
Keywords: selenium, prostate cancer, androgen receptor, Akt
Introduction
Prostate cancer is the most commonly diagnosed malignancy in men in the United States and the second leading cause of cancer-related death (1). Issues of recurrence and hormone resistance combined with the lack of treatment for men with high-grade prostatic intraepithelial neoplasia (HGPIN), a premalignant condition, present a major public health problem. Thus, mechanism-based alternative and/or adjuvant therapies and strategies for prevention and treatment are critically needed.
Diets rich in selenium, organoselenium compounds or selenized yeast (SeY) have been shown in epidemiologic and preclinical studies, as well as in some clinical intervention trials to have a protective role against prostate cancer (reviewed in 2). Perhaps the most notable and exciting evidence for the protective role of organoselenium in the form of SeY emerged from a clinical study by Clark et al (3). In contrast, preliminary data accrued from the prematurely halted Selenium and Vitamin E Cancer Prevention Trial (SELECT) that investigated the effects of selenomethionine (SM), a major component of SeY, individually and in combination with vitamin E showed no effect of SM on prostate cancer rates (4). Several hypotheses have been offered that may explain the lack of effect of SM in the SELECT study (5). Considering this lack of effect, there is an even more pressing need to develop and test mechanism-based organoselenium compounds. The results of preclinical studies as well as small scale clinical trials using various analogs of organoselenium can assist in making an informed evaluation as to whether selenium supplementation would benefit (or harm) specific populations (5, 6).
Studies in prostate cancer cell lines show that the dose and form of organoselenium can determine its diverse cellular responses. For example, organoselenium can manifest its chemopreventive activity either by conversion to a variety of selenometabolites such as methylselenol or seleno α-keto acids and/or by incorporation into a number of antioxidant selenoproteins, namely, glutathione peroxidase and thioredoxin reductase (7). In this study we compared the effects of two structurally distinct organoselenium compounds, naturally occurring SM and synthetic 1,4-phenylenebis(methylene)selenocyanate (p-XSC) (Figure 1 A), on critical prostate cancer signaling pathways in androgen responsive and androgen independent human prostate cancer cells. Studies in prostate cancer cell lines have shown that SM at non-physiological levels can inhibit growth, induce cell cycle arrest and alter the expression of a number of genes and proteins important for prostate cancer survival (8-11). However, limited studies in animal models of prostate cancer show SM to be largely ineffective at inhibiting tumor incidence and growth (12-14). p-XSC, which was developed in our laboratory, has been shown to be more effective than SM at inhibiting tumorigenesis in a number of preclinical animal models (15-18). We have previously shown that p-XSC is effective at inhibiting both LNCaP and LNCaP C4-2 (here onwards referred to as C4-2) human prostate cancer cell growth (10,11).
Fig. 1.
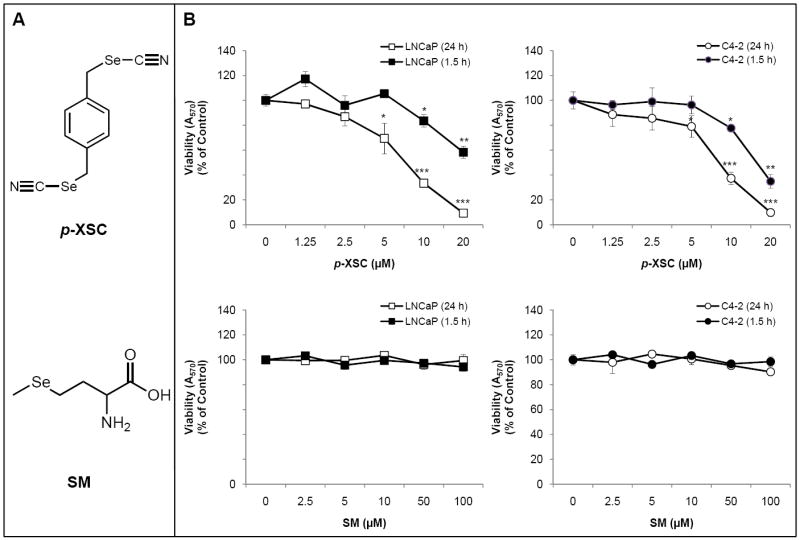
A. Structures of 1,4-phenylenebis(methylene)selenocyanate (p-XSC) and selenomethionine (SM). B. The effects of SM and p-XSC on cell viability. Cell viability was measured by MTT assay in LNCaP and C4-2 human prostate cancer cells treated with a range of doses of SM and p-XSC after 1.5 h and 24 h of exposure. Results are expressed as percent of control. (*p<0.05, **p<0.01, ***p<0.001)
Various selenium compounds have been shown to interfere with androgen receptor (AR) signaling and PI3K/Akt signaling in prostate cancer cells (19-25). Altered activity and crosstalk between these pathways appear to be a prominent feature of prostate cancer progression and the transition to androgen independence (26-29). However studies aimed at determining whether selenium-mediated down-regulation of androgen signaling is a result of inhibiting its crosstalk with Akt are limited. In this study, we investigated the effects of SM and p-XSC on AR and Akt signaling and explored whether crosstalk between these two pathways plays a role in the cellular responses to different forms of organoselenium.
Materials and Methods
Reagents and cell lines
SM was purchased from PharmaSe™ Inc. (Lubbock, TX) and p-XSC was synthesized as reported previously (30). Akt inhibitor VIII was purchased from EMD Chemicals (Gibbstown, NJ). Androgen responsive (AR) LNCaP cells were obtained from the American Type Culture Collection (Manassas, VA) and androgen non-responsive and therefore androgen independent (AI) LNCaP C4-2 cells were obtained from Dr. Warren D.W. Heston, The Lerner Research Institute, The Cleveland Clinic Foundation, OH.
Cell culture and organoselenium treatments
LNCaP cells were maintained in RPMI-1640 medium with 10% heat-inactivated Fetal Bovine Serum (FBS). C4-2 cells were maintained under the same conditions but with 10% FBS. Cells that were to be stimulated by dihydrotestosterone (DHT) were grown and treated in phenol red-free RPMI-1640 media supplemented with 10% charcoal stripped FBS. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and were routinely passaged when they were 70-80% confluent. Following incubation, cells were harvested from plates by either trypsinization or gentle scraping and washed with PBS.
Cells were plated in 10 cm dishes (1 million cells/plate) or 96-well plates (5,000 or 10,000 cells/well) depending on the assay, grown for 48 h and then treated with either SM or p-XSC. Both LNCaP and C4-2 cells were incubated in media containing SM at doses ranging from 0 to 100 μM or p-XSC at doses not exceeding 20 μM. The vehicles for SM and p-XSC were saline and dimethylsulfoxide (DMSO), respectively. Treatments continued for 24 h to examine longer term effects of these compounds on cellular processes or for a shorter exposure time of 1.5 h to evaluate early changes in molecular targets since literature data has shown that organoselenium-mediated alterations of the AR and Akt signaling pathways can been seen as early as 1 hour post-treatment (24,31). After incubation, cells were processed for further analysis.
Cell Viability Assay
Briefly, LNCaP and C4-2 cells were plated in triplicate in 96-well plates. Following treatment for 1.5 or 24 h with a range of doses of SM (2.5 to 100 μM) or p-XSC (1.25 to 20 μM), MTT assay was performed as previously described to determine cell viability (11). A solution of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H tetrazolium bromide (MTT, Sigma, St. Louis, MO) in phenol red-free RPMI-1640 medium at a final concentration of 0.5 mg/ml (50 μg total MTT/well) was added to each well and cells were incubated in the dark at 37°C for 4 hr. The MTT solution was then removed, 100 μl of DMSO was added to each well, and absorbance read at 570 nm using a SPECTRAmax® PLUS384 plate reader (Molecular Devices Corporation, Sunnyvale, CA). The assay was performed in triplicate and results are expressed as percent of untreated or vehicle-only control.
Cell Death ELISA
LNCaP and C4-2 cells were plated in duplicate in 96-well plates. Following treatment for 24 h with SM (10, 50, 100 μM) or p-XSC (2.5, 5, 10 μM), cells were assayed for the presence of cytoplasmic histone-associated DNA fragments characteristic of apoptosis using the Roche Cell Death ELISA kit (Basel, Switzerland), according to the manufacturer’s instructions. Enrichment factor values were calculated as follows: [A405-A490]sample/[A405-A490]control. The assay was performed in triplicate and results are expressed as fold induction of apoptosis compared with untreated or vehicle-only controls.
Immunoblotting
Immunoblotting was performed as previously described to determine changes in molecular markers (10). Briefly, LNCaP and C4-2 cells were treated with SM (5, 10, 50 and 100 μM) or p-XSC (5, and 10 μM) for 24 h, harvested by scraping and washed with phosphate buffered saline. Protein extraction was carried out using cell lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin) with freshly added 1 mM phenylmethylsulfonyl fluoride. Equal amounts of protein (35 μg) were separated on 10% SDS-PAGE gels and transferred to nitrocellulose membranes. Primary antibodies used at a 1:1000 dilution for immunoblotting were Akt, phospho-Akt (Ser473), cleaved PARP (Asp214) and androgen receptor from Cell Signaling Technology (Beverly, MA), phospho-androgen receptor (Ser210) from Abcam, Inc. (Cambridge, MA) and GAPDH from Santa Cruz (Santa Cruz, CA). Anti-mouse and anti-rabbit secondary antibodies (Cell Signaling, Beverly, MA) were used at a dilution of 1:3000. Band expressions were developed using ECL reagents from Amersham (Piscataway, NJ) and density analyzed using VisionWorks™ software (UVP, Inc. Upland, CA). All immunoblotting experiments were repeated three times. The results are presented as representative blots from single experiments and/or in graph form as the average of the measured band densities from three experiments.
QRT-PCR
Total RNA was isolated using the TRIZOL reagent (Gibco BRL, Rockville, MD) from LNCaP and C4-2 cells treated with 10 μM SM or p-XSC and that had been stimulated with AR ligand dihydrotestosterone (DHT) at a final concentration of 10 nM to activate AR signaling. The RNA was pelleted by centrifugation, washed using 75% ethanol and dissolved in RNase-free water. cDNA synthesis was carried out with the Superscript™ First Stand Synthesis System (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions using oligo(dT) as the primer. PCR was performed using the RT2 SYBR Green Master Mix (Superarray Bioscience Corporation, Frederick, MD). Primers were used at a final concentration of 100 nM in 25 μl PCR reactions. cDNA negative controls were run for each target gene. GAPDH expression was determined for each sample and used to normalize expression of the target gene. Relative expressions are depicted as percent of the normalized untreated control. The sequences of the primers were as follows: GAPDH (forward, 5’-AAGGTCGGAGTCAACGGATTTGGT-3’; reverse, 5’-ACAAAGTGGTCGTTGAGGGCAATG-3’) and PSA (forward, 5’-GCCTCTCGTGGCAGGGCAGT-3’; reverse, 5’-CTGAGGGTGAACTTGGGCAC-3’). For PSA, thermocycling conditions were initiated with a 10 min 95°C activation step followed by cycles of 94°C for 15 sec and 56°C for 30 sec and 72°C for 30 sec. For GAPDH, thermocycling conditions were initiated with a 10 min 95°C activation step followed by cycles of 95°C for 15 sec, 62°C for 30 sec, and 72°C for 45 sec. Reactions were run in duplicate and experiments were repeated three times. Relative expressions were calculated using the ΔΔCt method. The results are presented as representative raw data from single experiments and/or in graph form as the average of the relative expressions from three experiments.
Statistics
Results are expressed as mean ± standard error. Statistical significance was analyzed using either the Student’s t test or two-factor analysis of variance (ANOVA). Differences were considered significant at p < 0.05.
Results
The effects of SM and p-XSC on cell viability
We investigated the effects of SM and p-XSC on cell viability by MTT assay after 1.5 and 24 h of treatment. p-XSC (10 μM) began to inhibit LNCaP and C4-2 cell viability after a short duration (1.5 h) of treatment (Figure 1B). SM, at doses up to 100 μM, showed no inhibition of LNCaP or C4-2 cell viability after 1.5 h (Figure 1B). At 24 h, p-XSC dose-dependently inhibited LNCaP and C4-2 cell viability with IC50 of 7.0 and 7.6 μM, respectively (Figure 1B). However, SM still had no effect on both cell types.
Dose-response effects of SM and p-XSC on apoptosis
We investigated the effect of SM and p-XSC on apoptosis in LNCaP and C4-2 cells using Cell Death ELISA. p-XSC treatment resulted in 2.5-, 3.7-, and 5.8-fold increases in apoptosis in LNCaP cells at concentrations of 2.5, 5, and 10 μM, respectively (Figure 2A). Similarly in C4-2 cells, p-XSC induced 2.9-, 3.5-, and 4.4-fold increases in apoptosis at concentrations of 2.5, 5, and 10 μM, respectively (Figure 2B). SM showed no induction of apoptosis in LNCaP cells at concentrations up to 100 μM. SM caused a significant decrease (32%, p < 0.05) in apoptosis in C4-2 cells at the lowest dose tested (10 μM) but had no effect at higher doses (50 or 100 μM). We also analyzed the effects of SM and p-XSC on apoptosis by examining Poly (ADP-ribose) polymerase (PARP) cleavage. PARP is a major target of caspases in vivo (32,33). Immunoblot analysis of cell lysates from LNCaP and C4-2 cells showed increased levels of cleaved PARP (Asp 214) in cells treated with 5 and 10 μM p-XSC (Figure 2A,B). LNCaP and C4-2 cells treated with doses of SM ranging from 5 to 100 μM showed no detectable PARP cleavage, supporting the above finding that SM is not inducing apoptosis in these cells. No induction of PARP cleavage was seen after 1.5 h of treatment with either p-XSC or SM in LNCaP and C4-2 cells (data not shown). Taken together these results show that p-XSC significantly and dose-dependently induces apoptosis similarly in LNCaP and C4-2 cells and that inhibition of cell viability by p-XSC is due, at least in part, to programmed cell death.
Fig. 2.

The effects of SM and p-XSC on induction of apoptosis. Apoptosis was detected by cleaved PARP (cl-PARP) and measured by Cell Death ELISA in A. LNCaP and B. C4-2 cells treated for 24 h with a range of doses of SM and p-XSC. GAPDH levels were assessed as a loading control. ELISA results are represented by bar graph as fold induction of apoptosis compared with vehicle-only controls. (*p<0.05, **p<0.01)
The effects of SM and p-XSC on androgen receptor and Akt pathway proteins
To determine the effect of SM and p-XSC on AR signaling, we first examined the effects of these compounds on AR protein levels in LNCaP and C4-2 cells. SM and p-XSC significantly reduced AR protein levels in LNCaP cells after 24 h though p-XSC was superior to SM (Figure 3 A,B). p-XSC also significantly reduced AR protein levels in C4-2 prostate cancer cells, while SM showed a non-significant increase in AR protein expression (Figure 3 A,B).
Fig. 3.

The effects of SM and p-XSC on protein expression and phosphorylation. Androgen receptor (AR) protein levels in whole cell lysates from LNCaP and C4-2 cells treated for 24 h with 5 or 10 μM A. SM and B. p-XSC were measured by immunoblot analysis. Results are presented as representative blots from single experiments and in graph form as the average band density (normalized to GAPDH protein levels) from three experiments relative to control samples. (*p<0.05, **p<0.01) C. Levels of phosphorylated Akt and phosphorylated androgen receptor in whole cell lysates of LNCaP and C4-2 cells treated for 1.5 h with 5 or 10 μM SM and p-XSC were measured by immunoblotting. Fold change in band densities of phosphorylated proteins were normalized to the band densities of their respective total protein and to GAPDH levels.
We also investigated, by western blot analysis, the effects of SM and p-XSC on Akt phosphorylation and phosphorylation of AR at the major Akt-specific phosphorylation site, Ser 210, in LNCaP and C4-2 cells (34). After 24 h of treatment both p-XSC and SM at doses of 10 μM and higher reduced the levels of AR phosphorylated at Ser 210 in both cell types (data not shown). No inhibitory effects were seen in C4-2 cells treated with SM. However, this down-regulation of AR phosphorylation correlated with the decreased levels of total AR protein. Therefore, we next examined AR phosphorylation after 1.5 h of exposure. SM decreased total AR proteins levels after 1.5 h of treatment, but caused non-significant changes in AR and Akt phosphorylation (Figure 3C). SM did, however, dose-dependently increase AR phosphorylation in C4-2 cells at this time point (Figure 3C). After 1.5 h, p-XSC did not alter total AR levels in LNCaP or C4-2 cells but did decrease Akt-mediated phosphorylation of the AR at Ser 210 as well as Akt phosphorylation at Ser 473 in both cell lines (Figure 3C). These effects on Akt phosphorylation were undetectable after 24 h of treatment with these compounds (data not shown), suggesting alteration of the PI3K/Akt pathway may be an early event in selenium-mediated modulation of prostate cancer cell growth.
The effects of SM and p-XSC on androgen receptor activity
We examined the effects of SM and p-XSC on AR transcriptional activity by measuring the RNA expression of PSA, an androgen-regulated gene. p-XSC (10 μM) significantly decreased PSA mRNA levels in both LNCaP and C4-2 cells (Figure 4A,B). SM (10 μM) showed no significant change in PSA expression in either LNCaP or C4-2 cells (Figure 4A,B).
Fig. 4.

The effects of SM and p-XSC on androgen target gene PSA. PSA mRNA levels were measured by quantitative real time PCR in A. LNCaP and B. C4-2 cells treated with 10 μM SM or p-XSC (1.5 h) and stimulated with 10 nM dihydrotestosterone (DHT). The results are presented as representative raw data (fluorescence vs. cycle number) from single experiments and/or in graph form as the average of the relative expressions (normalized to GAPDH mRNA levels) from three experiments. C. PSA mRNA levels were measured in LNCaP and C4-2 cells treated with 10 μM p-XSC and stimulated with DHT and the percent inhibition (compared with untreated controls) was compared to treated cells not stimulated with DHT. (*p<0.01, **p<0.001)
To determine whether p-XSC specifically inhibits androgen-induced PSA expression we further compared the effects on PSA mRNA levels in unstimulated cells with cells stimulated with the AR ligand DHT. Inhibition of PSA expression was significantly enhanced in LNCaP cells stimulated with DHT compared with unstimulated cells (Figure 4C), suggesting the decrease in PSA mRNA levels is due, at least in part, to inhibition of AR transcriptional activity.
Akt inhibition and p-XSC-mediated inhibition of LNCaP and C4-2 cells
In order to determine whether the inhibition of Akt by p-XSC was contributing to the downstream effects on cell viability we first treated cells with an Akt specific inhibitor and then exposed them to p-XSC for 1.5 h. The inhibitor alone at a final concentration of 2 μM (at which there is a dramatic inhibition of Akt phosphorylation in LNCaP and C4-2 cells) decreased cell viability in LNCaP cells by less than 10% and in C4-2 cells by close to 20% (Figure 5A). p-XSC decreased cell viability similarly in both untreated LNCaP cells and cells pre-treated with the Akt inhibitor suggesting that inhibition of Akt by p-XSC does not solely account for the decrease in cell viability. Inhibition of cell viability by p-XSC in the C4-2 cells was slightly, albeit significantly, attenuated by pre-treatment with the Akt inhibitor suggesting a more important role for p-XSC-mediated inhibition of Akt signaling in these cells. However, it is clear that in both LNCaP and C4-2 cells p-XSC may be inhibiting additional targets/pathways contributing to prostate cancer cell death.
Fig. 5.

The effects of Akt inhibitor on selenium-mediated inhibition of cell viability and PSA expression. A. Cell viability in LNCaP and C4-2 cells pre-treated with an Akt-specific inhibitor (Akti, 2 μM) for 1 h and/or treated with selenium (10 μM SM or p-XSC) for 1.5 h was measured by MTT assay. Results are expressed as percent of vehicle-only treated control. (*p<0.05, **p<0.001) B. PSA mRNA levels in LNCaP and C4-2 cells pre-treated with an Akt-specific inhibitor (Akti, 2 μM) for 1 h and/or treated with p-XSC (10 μM) for 1.5 h and stimulated with 10 nM DHT were measured by QRT-PCR. Results are normalized to GAPDH mRNA levels and are expressed as percent of vehicle-only treated control. (*p<0.05, **p<0.01, ***p<0.001)
To investigate whether inhibition of Akt is a factor in the down regulation of AR activity by p-XSC, we measured the effects of SM and p-XSC on PSA mRNA levels in the presence of an Akt inhibitor. LNCaP and C4-2 cells were exposed to the inhibitor then treated with either SM or p-XSC (10 μM) and subsequently with DHT to stimulate AR activity. Treatment of both LNCaP and C4-2 cells with the Akt inhibitor alone significantly decreased PSA mRNA levels showing that Akt does affect AR transcriptional activity in these cells (Figure 5B). Results showed that inhibiting Akt signaling prior to exposure to p-XSC had no attenuating effect on the AR inhibiting activity of the compound. In fact, the combination of the Akt inhibitor and p-XSC seems to enhance inhibition of PSA expression suggesting that p-XSC may target AR signaling via mechanisms in addition to or other than Akt down-regulation.
Discussion
In this study we found marked differences in the responses of both LNCaP and C4-2 prostate cancer cells to the structurally distinct organoselenium compounds SM and p-XSC. Comparison of growth effects of SM and p-XSC on LNCaP and C4-2 cells highlighted the significant role structure and dose play in mediating cellular response to organoselenium compounds. p-XSC is superior to SM at inhibiting prostate cancer cell viability. At the doses examined, only p-XSC was able to induce apoptosis, a critical cellular event in cancer prevention by selenium compounds (35). Though SM has been the supplemental form of selenium used in a handful of clinical prostate cancer trials including the most recent and largest ever conducted SELECT study, it was not able to achieve significant inhibition of LNCaP or C4-2 cells at physiologically relevant doses after 24 h of treatment and even appeared protective in C4-2 cells. By contrast, p-XSC can achieve significant growth inhibition of both LNCaP and C4-2 prostate cancer cells at concentrations as low as 5 μM. SM was able to down-regulate AR protein levels in LNCaP cells after 24 h of treatment, however had no effect on AR activity and therefore did not alter cell growth. It is possible that inhibition of AR by SM may be occurring at a later time point and thus longer exposures to SM may elicit inhibitory effects on cell growth that were missed after only 24 h of treatment. These findings underscore the importance of determining efficacy and understanding the mechanisms of organoselenium compounds as they may possess often quite diversified function in their ability to prevent or control prostate cancer progression.
It is increasingly evident that crosstalk between AR and other signaling pathways (e.g., PI3K/Akt) may play an important role in advanced prostate cancer. Cell viability analyses in our study showed an increased sensitivity of the C4-2 cells to an Akt-specific inhibitor, which may be due to an increased reliability of androgen refractory cells on the PI3K/Akt pathway. To our knowledge the potential for selenium compounds to affect the crosstalk between Akt and AR signaling has not been previously explored. Figure 6 shows our proposed scheme for p-XSC-mediated inhibition of LNCaP and C4-2 human prostate cancer cells.
Figure 6.
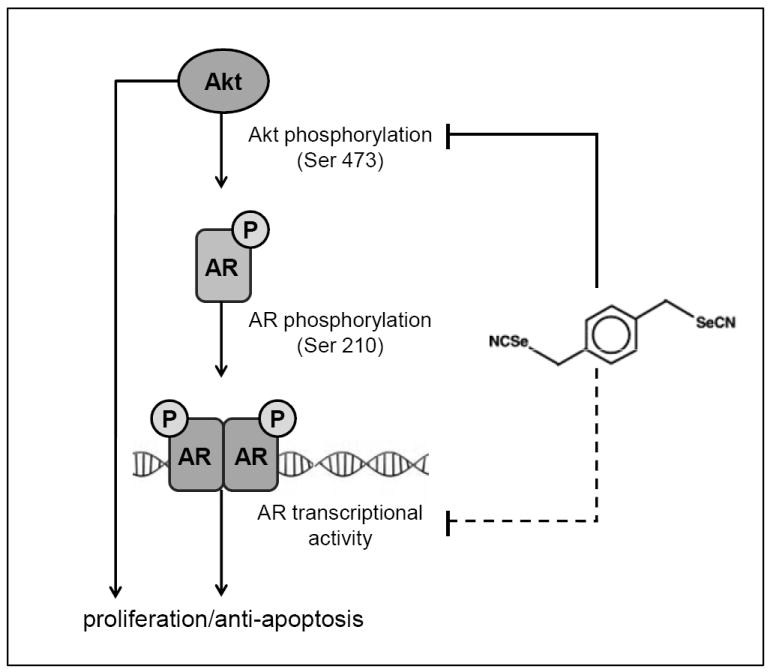
Proposed scheme for p-XSC-mediated inhibition of LNCaP and C4-2 human prostate cancer cells. p-XSC inhibits both Akt signaling and AR signaling, contributing to its downstream effects of decreased cell viability and increased apoptosis. Akt-mediated regulation of AR (via phosphorylation at Ser 210) may contribute to, but does not solely account for p-XSC-mediated inhibition of AR activity and cell viability as inhibition of Akt does not fully attenuate the effects of p-XSC. The alternate mechanisms by which p-XSC down-regulates AR transcriptional activity have yet to be determined (represented by broken line).
AR phosphorylation by several kinases including Akt is thought to play a role in the regulation of its function (27,34,36). We have shown, for the first time that an organoselenium compound can down-regulate Akt-specific phosphorylation of the AR, a potentially pivotal regulatory mechanism and player in androgen independence. Though p-XSC inhibited PSA expression in a manner similar to that of an Akt-specific inhibitor, inhibition of Akt prior to treatment with p-XSC did not attenuate the effect of p-XSC on PSA mRNA levels. This suggests that p-XSC inhibits AR activity via additional or distinct mechanisms and the inhibition of AR and Akt signaling by this agent may occur independently. We have considered the possibility that p-XSC directly inhibits AR activity. In our previous study we showed that the covalent binding of p-XSC to cysteinyl moieties within the p50 subunit of NFκB may potentially account for its inhibition of the transcription factor (37). Organoselenium compounds can exhibit higher nucleophilicity than organosulfur (cysteinyl) moieties and thus can facilitate disruption of the charge relay system that involves zinc finger motifs (38). Selenium compounds have been shown to inhibit DNA binding and induce zinc release from DNA repair proteins (38). The DNA binding domain of the AR, which contains two zinc finger motifs each with a four-cysteine coordination site, may be a target for p-XSC.
This study compared the effects of SM and p-XSC on molecular markers at equal doses less than or equal to 10 μM (which include physiological selenium levels) even though SM showed no clear inhibition of LNCaP or C4-2 cells at these concentrations. The concentrations of SM required to achieve significant inhibition are exceedingly higher than those used in the clinic. Preliminary data from our laboratory indicates differences in mechanisms of action between SM and p-XSC. For example, p-XSC causes cell cycle arrest in G1 whereas SM treatment causes cells to accumulate in G2/M, which has also been previously shown by others (39).
Our findings that p-XSC inhibits both LNCaP and C4-2 prostate cancer cell growth and modulates clinically relevant signaling pathways lend support for the evaluation of this agent in well-defined animal models of prostate cancer and ultimately for its potential use in the management of prostate cancer. Future studies may benefit from exploring the effects of organoselenium at stages beyond localized prostate cancer as evidence supports a potential role for p-XSC and various other selenium compounds in mediating metastasis and androgen independence, events inherent to increased mortality. With the goal of increasing survivorship and improving quality of life issues, investigators should consider the efficacy of organoselenium compounds in future exploration of primary or supplemental treatment options for advanced prostate cancer. However, caution should be exercised since it has been shown that high levels of serum selenium were associated with a slightly elevated risk of aggressive prostate cancer in individuals carrying a certain variant form of the superoxide dismutase (SOD2) gene (6). Clearly, not all individuals appear to benefit from selenium supplementation and the future design of clinical trials should carefully consider the form and dose of selenium as well as the population’s baseline selenium levels and their selenium-dependent genetic polymorphism.
Acknowledgments
Studies conducted in the authors’ laboratory were supported in part by NCI CA111842, CA127729, DOD W81XWH-08-1-0297, and Penn State Hershey Cancer Institute seed funds
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Facompre N, El-Bayoumy K. Potential stages for prostate cancer prevention with selenium: Implications for cancer survivors. Cancer Res. 2009;69:2699–703. doi: 10.1158/0008-5472.CAN-08-4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark LC, Combs GF, Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin: a randomized controlled trial. JAMA. 1996;276:1957–63. [PubMed] [Google Scholar]
- 4.Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers. JAMA. 2009;309:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Bayoumy K. The negative results of the SELECT study do not necessarily discredit the selenium-cancer hypothesis. Nutr Cancer. 2009;61:285–6. doi: 10.1080/01635580902892829. [DOI] [PubMed] [Google Scholar]
- 6.Chan JM, Oh WK, Xie W, et al. Plasma selenium, manganese superoxide dismutase, and intermediate- or high-risk prostate cancer. J Clin Oncol. 2009;27:3577–83. doi: 10.1200/JCO.2008.18.8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee JI, Nian H, Cooper AJL, et al. α-Keto acid metabolites of naturally-occurring organoselenium compounds as inhibitors of histone deacetylase in human prostate cancer cells. Cancer Prev Res. 2009;2:683–93. doi: 10.1158/1940-6207.CAPR-09-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao H, Domann FE, Zhong W. Apoptosis induced by selenomethionine and methioninase is superoxide mediated and p53 dependent in human prostate cancer cells. Mol Cancer Ther. 2006;5:3275–84. doi: 10.1158/1535-7163.MCT-06-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao H, Brooks HD. Selenomethionine induced transcription programs in human prostate cancer cells. J Urol. 2007;177:743–50. doi: 10.1016/j.juro.2006.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pinto JT, Sinha R, Papp K, Facompre ND, Desai D, El-Bayoumy K. Differential effects of naturally occurring and synthetic organoselenium compounds on biomarkers in androgen responsive and androgen independent human prostate carcinoma cells. Int J Cancer. 2007;120:1410–17. doi: 10.1002/ijc.22500. [DOI] [PubMed] [Google Scholar]
- 11.Sinha R, Pinto JT, Facompre N, Kilheffer J, Baatz JE, El-Bayoumy K. Effects of naturally occurring and synthetic organoselenium compounds on protein profiling in androgen responsive and androgen independent human prostate cancer cells. Nutr Cancer. 2008;60:267–75. doi: 10.1080/01635580701630479. [DOI] [PubMed] [Google Scholar]
- 12.Corcoran NM, Najdovska M, Costello AJ. Inorganic selenium retards progression of experimental hormone refractory prostate cancer. J Urol. 2004;171:907–10. doi: 10.1097/01.ju.0000092859.16817.8e. [DOI] [PubMed] [Google Scholar]
- 13.Li GX, Lee HJ, Wang Z, et al. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis. 2008;29:1005–12. doi: 10.1093/carcin/bgn007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCormick DL, Rao KVN, Johnson WD, Bosland MC, Lubet RA, Steele VE. Null activity of selenium and vitamin E as cancer chemopreventive agents in the rat prostate. Cancer Prev Res. 2010;3:381–92. doi: 10.1158/1940-6207.CAPR-09-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reddy BS, Wynn TT, El-Bayoumy K, Upadhyaya P, Fiala E, Rao CV. Evaluation of organoselenium compounds for potential chemopreventive properties in colon cancer. Anticancer Res. 1996;16:1123–7. [PubMed] [Google Scholar]
- 16.Prokopczyk B, Amin S, Desai DH, Kurtzke C, Upadhyaya P, El-Bayoumy K. Effects of 1,4-phenylenebis(methylene)selenocyanate and selenomethionine on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced tumorigenesis in A/J mouse lung. Carcinogenesis. 1997;18:1855–67. doi: 10.1093/carcin/18.9.1855. [DOI] [PubMed] [Google Scholar]
- 17.Das A, Desai D, Pittman B, Amin S, El-Bayoumy K. Comparison of the chemopreventive efficacies of 1,4-phenylenebis(methylene)selenocyanate and selenium-enriched yeast on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induced lung tumorigenesis in A/J mouse. Nutr Cancer. 2003;46:179–85. doi: 10.1207/S15327914NC4602_11. [DOI] [PubMed] [Google Scholar]
- 18.El-Bayoumy K, Sinha R. Mechanisms of mammary cancer chemoprevention by organoselenium compounds. Mutat Res. 2004;551:181–97. doi: 10.1016/j.mrfmmm.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 19.Cho SD, Jiang C, Malewicz B, et al. Methyl selenium metabolites decrease prostate-specific antigen expression by inducing protein degradation and suppressing androgen-stimulated transcription. Mol Cancer Ther. 2004;3:605–11. [PubMed] [Google Scholar]
- 20.Dong Y, Lee SO, Zhang H, Marshall J, Gao AC, Ip C. Prostate specific antigen expression is down-regulated by selenium through disruption of androgen receptor signaling. Cancer Res. 2004;64:19–22. doi: 10.1158/0008-5472.can-03-2789. [DOI] [PubMed] [Google Scholar]
- 21.Chun JY, Nadiminty N, Lee SO, Onate SA, Lou W, Gao AC. Mechanisms of selenium down-regulation of androgen receptor signaling in prostate cancer. Mol Cancer Ther. 2006;5:913–8. doi: 10.1158/1535-7163.MCT-05-0389. [DOI] [PubMed] [Google Scholar]
- 22.Husbeck B, Bhattacharyya RS, Feldman D, Knox SJ. Inhibition of androgen receptor signaling by selenite and methylseleninic acid in prostate cancer cells: two distinct mechanisms of action. Mol Cancer Ther. 2006;5:2078–85. doi: 10.1158/1535-7163.MCT-06-0056. [DOI] [PubMed] [Google Scholar]
- 23.Hu H, Jiang C, Li G, Lu J. PKB/AKT and ERK regulation of caspase-mediated apoptosis by methylseleninic acid in LNCaP prostate cancer cells. Carcinogenesis. 2005;26:1374–81. doi: 10.1093/carcin/bgi094. [DOI] [PubMed] [Google Scholar]
- 24.Wu Y, Zu K, Warren MA, Wallace PK, Ip C. Delineating the mechanism by which selenium deactivates Akt in prostate cancer cells. Mol Cancer Ther. 2006;5:246–52. doi: 10.1158/1535-7163.MCT-05-0376. [DOI] [PubMed] [Google Scholar]
- 25.Lee SO, Chun JY, Nadiminty N, et al. Monomethylated selenium inhibits growth of LNCaP human prostate cancer xenograft accompanied by a decrease in the expression of androgen receptor and prostate-specific antigen (PSA) Prostate. 2006;66:1070–5. doi: 10.1002/pros.20329. [DOI] [PubMed] [Google Scholar]
- 26.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 27.Edwards J, Bartlett JM. The androgen receptor and signal-transduction pathways in hormone-refractory prostate cancer. Part 1: Modifications to the androgen receptor. BJU Int. 2005;95:1320–6. doi: 10.1111/j.1464-410X.2005.05526.x. [DOI] [PubMed] [Google Scholar]
- 28.Malik SN, Brattain M, Ghosh PM, et al. Immunohistochemical demonstration of phospho-Akt in high Gleason grade prostate cancer. Clin Cancer Res. 2002;8:1168–71. [PubMed] [Google Scholar]
- 29.Kreisberg JI, Malik SN, Prihoda TJ, et al. Phosphorylation of Akt (Ser473) is an excellent predictor of poor clinical outcome in prostate cancer. Cancer Res. 2004;64:5232–6. doi: 10.1158/0008-5472.CAN-04-0272. [DOI] [PubMed] [Google Scholar]
- 30.El-Bayoumy K, Chae YH, Upadhyaya P, Meschter C, Cohen LA, Reddy BS. Inhibition of 7,12-dimethylbenz(a)anthracene-induced tumors and DNA adduct formation in the mammary glands of female Sprague-Dawley rats by the synthetic organoselenium compound 1,4-phenylenebis(methylene)selenocyanate. Cancer Res. 1992;52:2402–7. [PubMed] [Google Scholar]
- 31.Dong Y, Zhang H, Gao AC, Marshall JR, Ip C. Androgen receptor signaling intensity is a key factor in determining the sensitivity of prostate cancer cells to selenium inhibition of growth and cancer-specific biomarkers. Mol Cancer Ther. 2005;4:1047–55. doi: 10.1158/1535-7163.MCT-05-0124. [DOI] [PubMed] [Google Scholar]
- 32.Nicholson DW, Ali A, Thornberry NA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 33.Tewari M, Quan LT, O’Rourke K, et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–9. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 34.Lin HK, Yeh S, Kang HY, Chang C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. PNAS. 2001;98:7200–5. doi: 10.1073/pnas.121173298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinha R, El-Bayoumy K. Apoptosis is a critical cellular event in cancer chemoprevention and chemotherapy by selenium compounds. Curr Cancer Drug Targets. 2004;4:13–28. doi: 10.2174/1568009043481614. [DOI] [PubMed] [Google Scholar]
- 36.Lin HK, Hu YC, Yang L, et al. Suppression versus induction of androgen receptor functions by the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer LNCaP cells with different passage numbers. J Biol Chem. 2003;278:50902–7. doi: 10.1074/jbc.M300676200. [DOI] [PubMed] [Google Scholar]
- 37.Chen KM, Spratt TE, Stanley BA. Inhibition of nuclear factor-kappaB DNA binding by organoselenocyanates through covalent modification of the p50 subunit. Cancer Res. 2007;67:10475–83. doi: 10.1158/0008-5472.CAN-07-2510. [DOI] [PubMed] [Google Scholar]
- 38.Blessing H, Kraus S, Heindl P, Bal W, Hartwig A. Interaction of selenium compounds with zinc finger proteins involved in DNA repair. Eur J Biochem. 2004;271:3190–9. doi: 10.1111/j.1432-1033.2004.04251.x. [DOI] [PubMed] [Google Scholar]
- 39.Menter DG, Sabichi AL, Lippman SM. Selenium effects on prostate cell growth. Cancer Epidemiol Biomarkers Prev. 2000;9:1171–82. [PubMed] [Google Scholar]