

Abstract
Crohn’s Disease (CD) is caused by a loss of the regulatory capacity of the immune apparatus. Nod2 is an intracellular bacterial sensor and its mutations are associated with the development of CD. Here we summarize recent and controversial findings about the role of the Nod2 mutants in the disease process.
Keywords: Crohn’s disease, Inflammation, Homeostasis, Interleukin-10, Monocytes, Macrophages, Nod2, 1007fs, R702W, G908R, Interleukin-12, Interleukin-23, Mucosal immunity, TLR, Commensals
1. Crohn’s disease
Crohn’s disease (CD) is a chronic inflammatory bowel disease (IBD) that can affect any portion of the gastrointestinal tract (GI), but most commonly in terminal ileum, cecum, perianal area and colon. CD is manifested histologically in a transmural, dense infiltration of lymphocytes and macrophages, the presence of granulomas (in up to 60% of patients), fissuring ulceration and submucosal fibrosis. CD can cause significant morbidity such as diarrhea, pain, narrowing of the gut lumen leading to strictures and bowel obstruction, abscess formation, and fistulization to skin and internal organs. CD has a prevalence rate of 10–200 per 100,000 people in North America and Europe, with the highest disease incidence in develop, urbanized countries and increased rate of disease occurrence in the last four decades.
IBD is thought to result from inappropriate and continuing activation of the mucosal immune system driven by the presence of normal luminal flora. This aberrant response is likely facilitated by defects in both the barrier function of the intestinal epithelium and the mucosal immune system. These defects are in turn caused by genetic and environmental factors [1]. Enteric bacteria are important for the structural and functional integrity of the gut, and are essential for the development of a competent immune defense system [2]. Mice raised under germ-free conditions display architectural abnormalities with crypt hyperplasia and lack of lymphoid follicle development. Introduction of segmental filamentous bacteria into the intestine of germ-free mice raises the numbers of lymphoid cells in the lamina propria and increases the numbers of IgA-producing cells with elevated IgA secretion [3]. Two important immunological functions of the gut are the secretion of polymeric immunoglobulin A to protect the intestinal surface against harmful stimuli and inhibition of the systemic response to commensal bacteria and food proteins (e.g., oral tolerance) to prevent chronic inflammation. A representative study by Rakoff-Nahoum et al. strongly demonstrates that recognition of commensal microflora by Toll-like receptors (TLRs) is required for intestinal epithelial homeostasis and protection from dextran sulfate sodium-induced injury [4].
In CD, there is a sustained activation of mucosal immune responses of the Th1 type caused by either intrinsic defects such as constitutive activation or the failure of regulatory mechanisms, or by continued stimulation resulting from changes in the epithelial mucosal barrier [5].
2. Nucleotide-binding oligomerization domain 2 and host defense
Innate immune responses are initiated with the detection of microbial invaders by two distinct host systems. One system comprises a family of membrane-bound receptors, termed TLRs, while the other family consists of molecules that are found in the cytoplasmic compartment, termed the nucleotide-binding site/leucine-rich repeat (NBS/LRR) proteins. These two detection systems recognize conserved molecular components of microbes including such structural motifs as lipopolysaccharide (LPS) from the Gram-negative bacterial cell wall and peptidoglycan (PGN) found in the cell wall of both Gram-negative and Gram-positive bacteria.
Two members of the NBS/LRR family of proteins, nucleotide-binding oligomerization domain (Nod) 1 and 2, are believed to be the cytoplasmic “sensors” of microbial products [6]. The microbial motifs sensed by these two molecules have been tentatively characterized. Both Nod1 and Nod2 recognize PGN, however, each requires distinct molecular motifs to attain sensing. Nod1 recognizes a naturally occurring muropeptide of PGN that presents a unique amino acid at its terminus called diaminopilemic acid (DAP). This amino acid is found mainly in the PGN of Gram-negative bacteria designating Nod1 as a sensor of Gram-negative bacteria. In contrast, Nod2 can detect the minimal bioactive fragment of PGN, called muramyl dipeptide (MDP). Thus Nod2 is a general sensor of bacterial PGN [7].
Nod2 expression is generally restricted to monocytes [8], and is recognized as an important mediator of inflammatory induction. Current models hypothesize that upon binding to MDP, Nod2 oligomerizes and binds to the caspase recruitment domain (CARD)-containing serine/threonine kinase RICK (also known as RIP2, CARDIAK, CCK and Ripk2). RICK then oligomerizes to transmit Nod2’s signal directly to the IKK complex, leading to activation of the NF-κB signaling pathway, by inducing ubiquitination of a site on NEMO [9]. An additional Nod2-interacting protein, Erbin, was identified in a yeast two-hybrid screen, which negatively regulates Nod2-dependent activation of NF-κB and cytokine secretion [10].
3. Nod2 mutants and CD
The gene encoding Nod2 is the first susceptibility gene that has been identified for CD [11,12]. In a study by mutational analyses of Nod2 in 453 patients with CD, 159 patients with ulcerative colitis (UC), and 103 healthy control subjects, three main variants of the six polymorphisms that was identified: the arginine to tryptophan conversion at amino acid residue 702 (R702W), the glycine to arginine change at 908 (G908R), and the frame shift-stop codon mutation at 1007 (1007fs), represented 32%, 18%, and 31%, respectively, of the total CD mutations, and were found to be independently associated with susceptibility to CD [13]. The allele and genotype frequencies of these mutations in 3575 Caucasian healthy controls recruited by 15 groups distributed on three continents were 4.3% (3.6–4.9), 1.2% (0.8–1.6), and 2.3% (1.8–2.8), respectively, with large geographic fluctuations of the G908R, 1007fs, and wild-type alleles (P < 0.0001) [14]. All three mutations are located within the LRR domain of Nod2 (Fig. 1). It is postulated that the LRR domain of CD-associated variants is likely to be impaired, possibly to various degrees, in its recognition of microbial components and/or in the physiological inhibition of Nod2 dimerization, thus resulting in the inappropriate activation of NF-κB in monocytes [12].
Fig. 1.
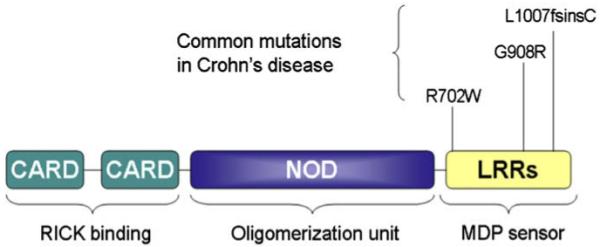
Structure of human Nod2 and the three common CD-associated mutations. CARD, caspase activation recruitment domain; NOD, nucleotide oligomerization domain; LRR, leucine rich repeats. The number coordinates refer to amino acid residues. Note that the L1007fsinsC mutation is the same as the 1007fs mutation.
Between 30% and 50% of CD patients in the Western hemisphere carry Nod2 mutations of at least one allele of the disease-causing mutations (DCMs). Individuals who carry two of these mutated Nod2 alleles have a 20–40–fold increased risk in developing CD [11,12]. The patients with double-dose mutations are characterized by a younger age at onset, a more frequent stricturing phenotype, and a less frequent colonic involvement than are seen in those patients who had no mutation [13]. However, it should be cautioned that normal individuals might occasionally have Nod2 mutations on both chromosomes in the absence of disease [15] and none of the three mutations in the Nod2 gene was found in 483 Japanese patients with CD [16]. The latter provides strong evidence for the presence of genetic heterogeneity among patients of different ethnic groups. It is of great interest and relevance to note that the three LRR domain mutations are also associated with graft-vs-host disease [17], which is like CD in the sense that both are thought to be mediated by Th1 cellular immunity.
The 1007fs mutation in the Nod2 gene associated with CD results from an insertion mutation at nucleotide position 3020 that leads to a partial deletion of the terminal LRR of the protein. The genotype relative risk (GRR) for developing CD in heterozygotes and homozygotes of this mutation alone is 1.5–2.6 and 17.6–42.1, respectively [11]. A recent retrospective study reviewing the records of 202 patients with confirmed CD found that G908R heterozygosity is associated with ileal involvement and smoking, whereas 1007fs homozygosity is strongly associated with gastroduodenal CD and younger age at diagnosis [18]. Overall, 1007fs homozygous patients demonstrate a much more severe disease phenotype than other patients with Crohn’s disease and have an increased risk for ileal stenoses and surgical interventions [19,20].
4. The controversies regarding the role of 1007fs in CD
1007fs is unable to detect MDP to initiate NF-κB activation [7]. By definition, 1007fs would represent a “loss-of-function” mutation. However, three important mouse studies have revealed significant information about the role of Nod2 in host defense against microbial pathogenesis that render the issue of how 1007fs contributes to the development to CD more complex than it appears.
In the first study in Nod2−/− mice, Watanabe et al. showed that intact Nod2 signaling inhibited TLR2-driven activation of NF-κB, particularly of the NF-κB subunit c-Rel [21]. Moreover, Nod2 deficiency or the presence of a CD-like mutation in Nod2 increased TLR2-mediated activation of NF-κB-c-Rel, and Th1 responses were enhanced. These observations suggest that Nod2 may be a negative regulator of NF-κB c-Rel activation and IL-12 gene expression/production, and that mutations in Nod2 may render the protein non-functional, thus unable to control the production of immunostimulatory cytokines such as IL-12 and IL-12-mediated inflammation in response to intracellular microbial stimulation. These in vivo data, coupled with the previous biochemical evidence, suggest that Nod2 may function to both positively and negatively regulate NF-κB signaling, depending on the circumstance surrounding its activation [22]. The biochemical basis for this dichotomous property of Nod2 function remains unknown and controversial. It is worth noting that mice genetically deficient in Nod2 do not develop CD spontaneously [23], indicating that other perturbations must also occur collectively to bring about CD.
It should also be pointed out that Nod2’s role in the NF-κB pathway has been controversial. Although Nod2 strongly activates NF-κB in transient transfection assays [8], the clinical pathophysiology of increased Th1 responses characteristic of CD suggests that the opposite is true in patients carrying these so called “loss-of-function” mutations in Nod2 [24]. CD patients show increased NF-κB activity, and NF-κB inhibitors are used clinically to ameliorate CD symptoms [1].
In the second study, Kobayashi et al. showed that Nod2-deficicient mice were susceptible to bacterial infection via the oral route but not through intravenous or peritoneal delivery. Nod2 was required for the expression of a subgroup of intestinal anti-microbial peptides, known as cryptidins produced by paneth cells in crypts [25]. This study supports the notion that the Nod2 deficiency represents a “loss-of-function” mutation and the epithelial barrier becomes more permissive to the entrance of solutes, including microbial products, or becomes less resistant to microbial invasion. This suggests that Nod2 mutations can directly influence intestinal epithelial barrier function.
In the third study, Maeda et al. demonstrated that the “knock-in” mice in which the wild-type Nod2 alleles were replaced by the Nod22939iC mutant gene (the corresponding mutant to 1007fs in humans, herein referred to as 2939iC) exhibited elevated NF-κB activation in response to MDP and more efficient processing and secretion of IL-1β, associated with increased susceptibility to bacterium-induced intestinal inflammation [26]. This study suggests that 2939iC is a “gain-of-function” allele, whose product induces elevated IKK and caspase-1 activation in response to MDP. However, the mouse study was contradicted by two human studies in which a strongly decreased production of IL-1β by peripheral mononuclear cells was found upon exposure to either PGN or PGN-derived MDP in homozygous patients bearing the 1007fs mutation [27,28].
5. Role of IL-10 in homeostatic regulation of inflammation and immune response
IL-10 is a pleiotropic cytokine produced by both T/B cells and macrophages and possesses both anti-inflammatory and immunosuppressive properties [29]. Extensive research shows that IL-10 is an inhibitor of a broad spectrum of monocyte/macrophage functions, including cytokine synthesis, nitric oxide production, and expression of MHC class II and costimulatory molecules such as CD80/CD86 [30]. Investigations in numerous inflammatory disease models including chronic enterocholitis, cutaneous inflammatory condition, endotoxic shock and Shwartzman reaction, and autoimmune encephalomyelitis in IL-10-deficient mice have yielded strong evidence that IL-10 plays a central role in vivo in restricting inflammatory responses [31]. However, endogenous IL-10 production and systemic administration can also exacerbate macrophage- and T-cell dysfunction, decrease T-cell apoptosis, blunt antimicrobial activity, and increase mortality in other less acute bacterial models of sepsis or after thermal injury [32]. In addition, IL-10 also processes immunostimulatory effects that have not attracted sufficient attention. IL-10 is a potent growth factor for B lymphocytes. It promotes B cell proliferation, antibody production, and class II expression [33]. IL-10 enhances, paradoxically, the development of cytotoxic T lymphocytes (CTL) [34]. It induces NK cytotoxicity against NK-resistant tumor cells in vitro and increases IL-2-induced NK cell proliferation [35]. It acts as a co-factor for colony formation by mast cell progenitors [36] and thymocytes [34].
6. IL-10 and CD
IL-10-deficient (Il10−/−) mice remain healthy under germ-free conditions, but some IL-10-deficient strains develop colitis when given a normal specific pathogen-free (SPF) bowel flora. This is due to a poorly controlled immune response to the bowel flora. Interestingly, colonization of germ-free Il10−/− mice with Lactobacillus plantarum does not induce colitis and can protect from subsequent introduction of SPF flora. Moreover the lactobacillus could also alleviate the established colitis in SPF Il10−/− mice [37]. Similarly, antigen preparations from anaerobic bowel commensals given by gavage to BALB/c mice can alleviate colitis induced by dextran sulfate, which like that seen in Il10−/− mice, is also attributable to an immune response to gut contents [38].
The first studies examining the ability of IL-10 administration to alleviate colitis in animal models were encouraging. Early clinical studies in untreated, as well as steroid-refractory CD patients, suggested that IL-10 might have strong potential in the treatment of human disease [39]. However, the biology of IL-10 is complex because it possesses both immunosuppressive and immunostimulatory effects. The understanding that is emerging regarding the in vivo effects of IL-10 is one of both anti- and pro-inflammatory effects, depending on the local concentrations of IL-10 achieved, the types of antigens present in the microenvironment, and the activation state of the immune cells in the vicinity. It is possible that high systemic doses of IL-10 alters the balance between its immunoregulatory and immunostimulatory effects, which would explain the bell-shaped curve in therapeutic efficacy reported in the clinical trials. In addition, it is entirely possible that the pharmacodynamics of daily systemic IL-10 administration does not allow for efficient delivery of the cytokine to the local sites of inflammation. The current challenges for researchers using gene-transfer techniques will be to determine the localization and duration of the expression of the transduced gene, and to ensure that delivery of immunoregulatory proteins is indeed local, tissue-specific, and therapeutic.
7. Nod2 and IL-10 production
The relationship between Nod2 and IL-10 has rarely been looked at. However, Netea and colleagues repeatedly showed that the 1007fs mutation in the Nod2 gene associated with CD caused defective release of IL-10 from blood mononuclear cells (PBMC) after stimulation with the TLR2 ligands, PGN and Pam3Cys-KKKK, but not with LPS, a TLR4 ligand [40]. Consistent with these findings, CD patients homozygous for 1007fs displayed decreased anti-inflammatory cytokine release when cells from these patients were stimulated with different species of Bacteroides, an enteric microorganism implicated in the pathogenesis of CD. Furthermore, activation of Nod2 has strong synergistic effects on TLR2-mediated production of both pro- and anti-inflammatory cytokines [41]. Two other relevant studies are worth mentioning. Li et al., analyzing PBMC from CD patients by microarray-based mRNA expression profiling, found that MDP treatment neither significantly induced TGFβ nor IL-10 [28]. Thus, it is not possible from this study to determine the effect of the 1007fs mutation on IL-10 production. A second study using a proteomic approach to characterize genes regulated by wild-type Nod2 vs 1007fs was done in HEK293 cells, which are epithelial in origin expressing neither Nod2 nor IL-10 [42].
The findings from these human studies are at odds with the study by Maeda et al. in the “knock-in” mice in which the wild-type Nod2 alleles were replaced by the 2939iC mutant gene [26]. In these animals, IL-10 levels in macrophages stimulated by LPS/PGN/MDP were comparable to those of wild-type mice but IL-1β production was greatly boosted, which was contrasted by patient based studies that immediately followed [27,28]. One possible explanation is that human CD patients carrying the 1007fs allele and mice expressing 2939iC are different due to species-specific physiology (see below).
8. Challenge to dogmas
To explore the differences underlying the various models our group undertook a study, which resulted in the uncovering of a novel regulatory role of 1007fs in determining how IL-10 levels are maintained to sustain the immunological homeostasis [43]. This study revealed an “acquired” activity of the three major Nod2 LRR-domain mutants in inhibiting steady-state and activated IL-10 gene transcription. They do so by blocking the phosphorylation of heterogeneous nucleor-ibonuclear protein A1 (hnRNP A1) via p38 MAPK [43]. hnRNP A1 serves as a novel transcription factor for IL-10 in both resting and activated monocytes and macrophages. In this study, the impairment of hnRNP A1’s phosphorylation and binding in 1007fs-CD patients deficient in IL-10 synthesis was confirmed [43].
Interestingly, this effect of 1007fs was seen in both LPS as well as TLR2 ligand-stimulated monocytes, which is inconsistent with a previous study showing that 1007fs in human cells only affected TLR2 signaling pathways [40]. A possible explanation for this discrepancy is that in contrast to our study in which mostly one type of cells were used: monocytes or macrophages, all previous patient studies published to date used peripheral blood mononuclear cells without further purification. Since Nod2 is primarily expressed in myeloid cells, which account for 5 to 10% of PMBCs, it is possible that the 1007fs effect is primarily manifested in monocytes. When T and B cells are present in large portions, they may be able to respond to TLR 2 and TLR 4 ligands, directly or indirectly, through Nod2-independent pathways. For example, if B cells respond more to TLR4 signaling than to TLR2, with respect to IL-10 synthesis, to such an extent that it may obscure the deficit seen in 1007fs CD patients in PBMCs stimulated with LPS. In support of this view, there was a report that TLR4-mediated triggering promotes B cell maturation and activation while TLR2 engagement arrests/retards B cell maturation [44]. This possibility can be formally tested.
Our findings strongly challenge the current paradigms represented by some of the studies described above. It calls into question of the validity of the Nod2-null mouse model and the model targeting the artificial mouse version of the human Nod2 mutant for mimicking the human disease. In other words, we believe that mouse 2939iC ≠ 1007fs, and mouse Nod2 deficiency ≠ human 1007fs.
9. A new working model
We hypothesize that 1007fs is also a gain-of-function mutant that can block the transcriptional activity of hnRNP A1 in IL-10 gene expression in the absence or presence of microbial stimuli by interfering with p38 MAPK’s ability to phosphorylate hnRNP A1. Additionally, 1007fs, when expressed, is susceptible to proteolytic destruction, rendering it functionally absent (loss of function) (our unpublished results). This gain-of-function property of 1007fs may result in chronically impaired IL-10 levels, and could accumulatively cause loss of homeostatic regulation and persistently elevated inflammation in the intestinal mucosa leading to the development and/or pathogenesis of CD, particularly in collaboration with other contributory genetic and environmental factors (Fig. 2).
Fig. 2.
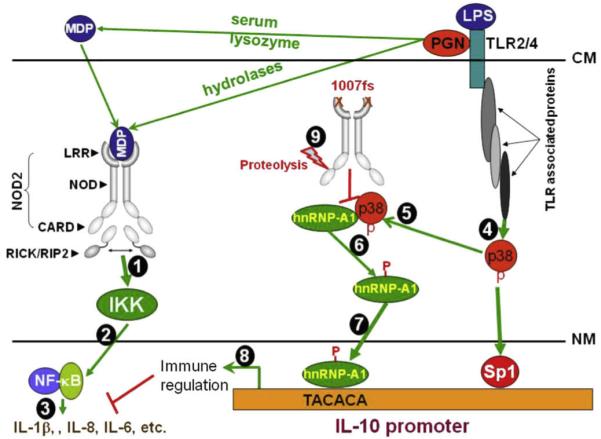
Model of 1007fs-mediated inhibition of IL-10 expression and its own regulation. MDP can be generated from PGN by serum lysozyme digestion extracellularly, or by hydrolases intracellularly. MDP then binds to the leucine-rich repeats (LRR) of NOD2 homodimers. MDP binding activates NOD2 and results in recruitment of the serine-threonine kinase RICK (also known as RIP2 or CARDIAK) through CARD-CARD domain interactions (Step 1). This triggers a cascade of downstream events that lead to the activation of IKK-NF-κB (Step 2) and expression of proinflammatory cytokine and induction of anti-microbial responses (Step 3). P38 MAPK is activated via TLR stimulation (Step 4). Activated p38 MAPK interacts with hnRNP-A1 in the cytosol, forming a molecular complex (Step 5). This interaction is critical for the phosphorylation of hnRNP-A1 (Step 6), which may be important for the cleavage of hnRNP-A1 into the transcriptionally active 26 kDa cleavage product. The cleaved hnRNP-A1 then moves into the nucleus, binds the IL10 TACACA element (NRE), and together with other transcription factors such as Sp1, drives IL10 transcription (Step 7), which impacts on inflammatory reactions (Step 8). The 1007fs mutant, however, has acquired a property that wt NOD2 does not have, i.e. interfering with the interaction between p38 and hnRNP-A1 (Step 9). By doing so, it reduces p38-mediated phosphorylation of hnRNP-A1, as well as hnRNP-A1 cleavage, nuclear translocation and binding to the IL10 NRE. 1007fs itself is susceptible to proteolytic degradation (our unpublished data). P, phosphorylated serine residues; CM, cell membrane; NM, nuclear membrane.
We further hypothesize that it should be possible to block this acquired activity of 1007fs using small molecule inhibitors (SMIs). Because Nod2 does not have this activity, these specific SMIs should act discriminately. The potential benefit of this type of research in the long run is to the therapy of GI inflammation. If the signaling mechanism(s) of IL-10 gene expression regulated by 1007fs could be elucidated more specific and sensitive points of the regulatory pathway(s) might be identified as potential therapeutic targets in the treatment of GI disorders by lessening the exacerbated inflammation driven by the breaching of the homeostatic balance in the intestines. The benefit may also be extended to other immune disorders such as graft-vs-host disease, transplant rejection, non-Hodgkin lymphoma, and spondyloarthropathy in which 1007fs has been strongly implicated.
10. R702W and G908R mutants show enhanced inflammatory cytokine response
In contrast to the 1007fs mutation, little is known about the functional characteristics of the R702W and G908R mutations. A recent Italian study examining the functional properties of monocyte-derived dendritic cells (MoDCs) from CD patients revealed that MoDCs carrying the R702W mutation displayed an increased basal level of IL-8 release, which, after a bacterial encounter, equilibrated to the levels similar to healthy controls. The compound R702W/G908R MoDCs, however, produced significantly higher levels of IL-12 upon stimulation with whole bacteria (Salmonella enteric serovar typhimurium SL strain), compared to controls of other genotypes including heterozygotes of the three mutations in non-compound forms [45].
The role of IL-12/IL-23 in CD has been strongly implicated in several studies. Kobayashi et al. showed that Stat3 deficiency in myeloid cells led to the induction of chronic enterocolitis in mice. IL-12/IL-23p40/Stat3 double-mutant mice, however, showed normal Th1 responses and no inflammatory change in the colon [46], indicating that overproduction of IL-12p40, which promotes potent Th1 and Th17 responses through the production of IL-12 and IL-23, is essential for the development of chronic enterocolitis in Stat3 mutant mice. In humans, a genome-wide association study identified IL23R as a strong inflammatory bowel disease gene [47]. The IL-23/IL-17-mediated immunity in lamina propria has been described as a hallmark of CD and is believed to be fundamentally connected to the etiology of CD [48]. Kamada et al described a unique subset of CD14+ intestinal macrophages that expressed both macrophage (CD14, CD33, CD68) and DC markers (CD205, CD209) and produced larger amounts of proinflammatory cytokines, such as IL-23, TNF-α, and IL-6, than typical intestinal resident macrophages (CD14−CD33+), and which contribute to the pathogenesis of CD via the IL-23/IFN-γ axis [49]. Last but not the least, a randomized trial showed that ustekinumab, a monoclonal antibody against the p40 subunit of IL-12 and IL-23, induced a clinical response in patients with moderate-to-severe CD, especially in patients previously given infliximab [50].
11. Conclusion
Dynamic interactions between the gastrointestinal epithelium, the commensal microflora of the gut and the mucosal immune system normally contribute to ensuring intestinal homeostasis and optimal immunosurveillance. Subversion of these interactions can lead to the development of chronic inflammatory diseases. The mechanisms by which the gut microbiota influences innate and adaptive mucosal immune responses leading to chronic inflammatory disease and gastrointestinal carcinogenesis are still poorly understood. Similarly, the mechanisms by which the Nod2 mutants contribute to the loss of control of homeostatic equilibrium in the intestinal mucosa have only been partially revealed. IL-10 clearly plays a pivotal role in maintaining the immunological balance in the GI tract; however, it is very likely that the impact of the Nod2 mutants could go well beyond IL-10. It is of great interest to explore those novel targets and pathways, especially in the context of multi-allelic and multi-genic interactions that collectively lead to the manifestation of the disease.
Acknowledgement
This work was in part supported by a grant from the Broad Medical Research Program of the Broad Foundation (IBD-0210R2).
References
- [1].Podolsky DK. Inflammatory bowel disease. N. Engl. J. Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- [2].Umesaki Y, Setoyama H. Structure of the intestinal flora responsible for development of the gut immune system in a rodent model. Microbes Infect. 2000;2:1343–1351. doi: 10.1016/s1286-4579(00)01288-0. [DOI] [PubMed] [Google Scholar]
- [3].Klaasen HL, Van der Heijden PJ, Stok W, Poelma FG, Koopman JP, Van den Brink ME, Bakker MH, Eling WM, Beynen AC. Apathogenic, intestinal, segmented, filamentous bacteria stimulate the mucosal immune system of mice. Infect. Immun. 1993;61:303–306. doi: 10.1128/iai.61.1.303-306.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- [5].Mashimo H, Wu DC, Podolsky DK, Fishman MC. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science. 1996;274:262–265. doi: 10.1126/science.274.5285.262. [DOI] [PubMed] [Google Scholar]
- [6].Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 2003;3:371–382. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- [7].Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- [8].Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J. Biol. Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- [9].Abbott DW, Wilkins A, Asara JM, Cantley LC. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr. Biol. 2004;14:2217–2227. doi: 10.1016/j.cub.2004.12.032. [DOI] [PubMed] [Google Scholar]
- [10].McDonald C, Chen FF, Ollendorff V, Ogura Y, Marchetto S, Lecine P, Borg JP, Nunez G. A role for Erbin in the regulation of Nod2-dependent NF-{kappa}B signaling. J. Biol. Chem. 2005;280:40301–40309. doi: 10.1074/jbc.M508538200. [DOI] [PubMed] [Google Scholar]
- [11].Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- [12].Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- [13].Lesage S, Zouali H, Cezard JP, Colombel JF, Belaiche J, Almer S, Tysk C, O’Morain C, Gassull M, Binder V, Finkel Y, Modigliani R, Gower-Rousseau C, Macry J, Merlin F, Chamaillard M, Jannot AS, Thomas G, Hugot JP. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am. J. Hum. Genet. 2002;70:845–857. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hugot JP, Zaccaria I, Cavanaugh J, Yang H, Vermeire S, Lappalainen M, Schreiber S, Annese V, Jewell DP, Fowler EV, Brant SR, Silverberg MS, Cho J, Rioux JD, Satsangi J, Parkes M. Prevalence of CARD15/NOD2 mutations in Caucasian healthy people. Am. J. Gastroenterol. 2007;102:1259–1267. doi: 10.1111/j.1572-0241.2007.01149.x. [DOI] [PubMed] [Google Scholar]
- [15].Linde K, Boor PP, Houwing-Duistermaat JJ, Kuipers EJ, Wilson JH, de Rooij FW. Card15 and Crohn’s disease: healthy homozygous carriers of the 3020insC frameshift mutation. Am. J. Gastroenterol. 2003;98:613–617. doi: 10.1111/j.1572-0241.2003.07287.x. [DOI] [PubMed] [Google Scholar]
- [16].Yamazaki K, Takazoe M, Tanaka T, Kazumori T, Nakamura Y. Absence of mutation in the NOD2/CARD15 gene among 483 Japanese patients with Crohn’s disease. J. Hum. Genet. 2002;47:469–472. doi: 10.1007/s100380200067. [DOI] [PubMed] [Google Scholar]
- [17].Holler E, Rogler G, Herfarth H, Brenmoehl J, Wild PJ, Hahn J, Eissner G, Scholmerich J, Andreesen R. Both donor and recipient NOD2/CARD15 mutations associate with transplant-related mortality and GvHD following allogeneic stem cell transplantation. Blood. 2004;104:889–894. doi: 10.1182/blood-2003-10-3543. [DOI] [PubMed] [Google Scholar]
- [18].Mardini HE, Gregory KJ, Nasser M, Selby L, Arsenescu R, Winter TA, de Villiers WJ. Gastroduodenal Crohn’s disease is associated with NOD2/CARD15 gene polymorphisms, particularly L1007P homozygosity. Dig. Dis. Sci. 2005;50:2316–2322. doi: 10.1007/s10620-005-3054-2. [DOI] [PubMed] [Google Scholar]
- [19].Seiderer J, Brand S, Herrmann KA, Schnitzler F, Hatz R, Crispin A, Pfennig S, Schoenberg SO, Goke B, Lohse P, Ochsenkuhn T. Predictive value of the CARD15 variant 1007fs for the diagnosis of intestinal stenoses and the need for surgery in Crohn’s disease in clinical practice: results of a prospective study. Inflamm. Bowel Dis. 2006;12:1114–1121. doi: 10.1097/01.mib.0000235836.32176.5e. [DOI] [PubMed] [Google Scholar]
- [20].Seiderer J, Schnitzler F, Brand S, Staudinger T, Pfennig S, Herrmann K, Hofbauer K, Dambacher J, Tillack C, Sackmann M, Goke B, Lohse P, Ochsenkuhn T. Homozygosity for the CARD15 frameshift mutation 1007fs is predictive of early onset of Crohn’s disease with ileal stenosis, entero-enteral fistulas, and frequent need for surgical intervention with high risk of re-stenosis. Scand. J. Gastroenterol. 2006;41:1421–1432. doi: 10.1080/00365520600703900. [DOI] [PubMed] [Google Scholar]
- [21].Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat. Immunol. 2004;5:800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- [22].O’Neill LA. How NOD-ing off leads to Crohn disease. Nat. Immunol. 2004;5:776–778. doi: 10.1038/ni0804-776. [DOI] [PubMed] [Google Scholar]
- [23].Pauleau AL, Murray PJ. Role of nod2 in the response of macrophages to toll-like receptor agonists. Mol. Cell. Biol. 2003;23:7531–7539. doi: 10.1128/MCB.23.21.7531-7539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat. Rev. Immunol. 2003;3:521–533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- [25].Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- [26].Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, Eckmann L, Karin M. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- [27].Netea MG, Ferwerda G, de Jong DJ, Girardin SE, Kullberg BJ, van der Meer JW. NOD2 3020insC mutation and the pathogenesis of Crohn’s disease: impaired IL-1beta production points to a loss-of-function phenotype. Neth. J. Med. 2005;63:305–308. [PubMed] [Google Scholar]
- [28].Li J, Moran T, Swanson E, Julian C, Harris J, Bonen DK, Hedl M, Nicolae DL, Abraham C, Cho JH. Regulation of IL-8 and IL-1beta expression in Crohn’s disease associated NOD2/CARD15 mutations. Hum. Mol. Genet. 2004;13:1715–1725. doi: 10.1093/hmg/ddh182. [DOI] [PubMed] [Google Scholar]
- [29].Moore KW, Vieira P, Fiorentino DF, Trounstine ML, Khan TA, Mosmann TR. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein-Barr virus gene BCRFI. Science. 1990;248:1230–1234. doi: 10.1126/science.2161559. [DOI] [PubMed] [Google Scholar]
- [30].Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- [31].Fuss IJ, Boirivant M, Lacy B, Strober W. The interrelated roles of TGF-beta and IL-10 in the regulation of experimental colitis. J. Immunol. 2002;168:900–908. doi: 10.4049/jimmunol.168.2.900. [DOI] [PubMed] [Google Scholar]
- [32].Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: A complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit. Care Med. 2002;30:S58–S63. [PubMed] [Google Scholar]
- [33].Go NF, Castle BE, Barrett R, Kastelein R, Dang W, Mosmann TR, Moore KW, Howard M. Interleukin 10, a novel B cell stimulatory factor: unresponsiveness of X chromosome-linked immunodeficiency B cells. J. Exp. Med. 1990;172:1625–1631. doi: 10.1084/jem.172.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].MacNeil IA, Suda T, Moore KW, Mosmann TR, Zlotnik A. IL-10, a novel growth cofactor for mature and immature T cells. J. Immunol. 1990;145:4167–4173. [PubMed] [Google Scholar]
- [35].Carson WE, Lindemann MJ, Baiocchi R, Linett M, Tan JC, Chou CC, Narula S, Caligiuri MA. The functional characterization of interleukin-10 receptor expression on human natural killer cells. Blood. 1995;85:3577–3585. [PubMed] [Google Scholar]
- [36].Thompson LM, Rubin RT, McCracken JT. Neuroendocrine aspects of primary endogenous depression: XII. Receiver operating characteristic and kappa analyses of serum and urine cortisol measures in patients and matched controls. Psychoneuroendocrinology. 1992;17:507–515. doi: 10.1016/0306-4530(92)90009-v. [DOI] [PubMed] [Google Scholar]
- [37].Schultz M, Veltkamp C, Dieleman LA, Grenther WB, Wyrick PB, Tonkonogy SL, Sartor RB. Lactobacillus plantarum 299V in the treatment and prevention of spontaneous colitis in interleukin-10-deficient mice. Inflamm. Bowel Dis. 2002;8:71–80. doi: 10.1097/00054725-200203000-00001. [DOI] [PubMed] [Google Scholar]
- [38].Verdu EF, Bercik P, Cukrowska B, Farre-Castany MA, Bouzourene H, Saraga E, Blum AL, Corthesy-Theulaz I, Tlaskalova-Hogenova H, Michetti P. Oral administration of antigens from intestinal flora anaerobic bacteria reduces the severity of experimental acute colitis in BALB/c mice. Clin. Exp. Immunol. 2000;120:46–50. doi: 10.1046/j.1365-2249.2000.01170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].van Deventer SJ, Elson CO, Fedorak RN, Crohn’s Disease Study Group Multiple doses of intravenous interleukin 10 in steroid-refractory Crohn’s disease. Gastroenterology. 1997;113:383–389. doi: 10.1053/gast.1997.v113.pm9247454. [DOI] [PubMed] [Google Scholar]
- [40].Netea MG, Kullberg BJ, de Jong DJ, Franke B, Sprong T, Naber TH, Drenth JP, Van der Meer JW. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn’s disease. Eur. J. Immunol. 2004;34:2052–2059. doi: 10.1002/eji.200425229. [DOI] [PubMed] [Google Scholar]
- [41].Netea MG, Ferwerda G, de Jong DJ, Jansen T, Jacobs L, Kramer M, Naber TH, Drenth JP, Girardin SE, Kullberg BJ, Adema GJ, Van der Meer JW. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J. Immunol. 2005;174:6518–6523. doi: 10.4049/jimmunol.174.10.6518. [DOI] [PubMed] [Google Scholar]
- [42].Weichart D, Gobom J, Klopfleisch S, Hasler R, Gustavsson N, Billmann S, Lehrach H, Seegert D, Schreiber S, Rosenstiel P. Analysis of NOD2-mediated proteome response to muramyl dipeptide in HEK293 cells. J. Biol. Chem. 2006;281:2380–2389. doi: 10.1074/jbc.M505986200. [DOI] [PubMed] [Google Scholar]
- [43].Noguchi E, Homma Y, Kang X, Netea MG, Ma X. A Crohn’s disease-associated NOD2 mutation suppresses transcription of human IL10 by inhibiting activity of the nuclear ribonucleoprotein hnRNP-A1. Nat. Immunol. 2009;10:471–479. doi: 10.1038/ni.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hayashi EA, Akira S, Nobrega A. Role of TLR in B cell development: signaling through TLR4 promotes B cell maturation and is inhibited by TLR2. J. Immunol. 2005;174:6639–6647. doi: 10.4049/jimmunol.174.11.6639. [DOI] [PubMed] [Google Scholar]
- [45].Salucci V, Rimoldi M, Penati C, Sampietro GM, van Duist MM, Matteoli G, Saibeni S, Vecchi M, Ardizzone S, Porro GB, Rescigno M. Monocyte-derived dendritic cells from Crohn patients show differential NOD2/CARD15-dependent immune responses to bacteria. Inflamm. Bowel Dis. 2008;14:812–818. doi: 10.1002/ibd.20390. [DOI] [PubMed] [Google Scholar]
- [46].Kobayashi M, Kweon MN, Kuwata H, Schreiber RD, Kiyono H, Takeda K, Akira S. Toll-like receptor-dependent production of IL-12p40 causes chronic enterocolitis in myeloid cell-specific Stat3-deficient mice. J Clin Invest. 2003;111:1297–1308. doi: 10.1172/JCI17085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Holtta V, Klemetti P, Sipponen T, Westerholm-Ormio M, Kociubinski G, Salo H, Rasanen L, Kolho KL, Farkkila M, Savilahti E, Vaarala O. IL-23/IL-17 immunity as a hallmark of Crohn’s disease. Inflamm. Bowel Dis. 2008;14:1175–1184. doi: 10.1002/ibd.20475. [DOI] [PubMed] [Google Scholar]
- [49].Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, Sakuraba A, Kitazume MT, Sugita A, Koganei K, Akagawa KS, Hibi T. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J. Clin. Invest. 2008;118:2269–2280. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sandborn WJ, Feagan BG, Fedorak RN, Scherl E, Fleisher MR, Katz S, Johanns J, Blank M, Rutgeerts P. A Randomized Trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate to severe Crohn’s disease. Gastroenterology. 2008 doi: 10.1053/j.gastro.2008.07.014. [DOI] [PubMed] [Google Scholar]