

Abstract
Calmodulin (CaM) is an essential eukaryotic calcium receptor that regulates many kinases, including CaMKII. Calcium-depleted CaM does not bind to CaMKII under physiological conditions. However, binding of (Ca2+)4-CaM to a basic amphipathic helix in CaMKII releases auto-inhibition of the kinase. The crystal structure of CaM bound to CaMKIIp, a peptide representing the CaM-binding domain (CaMBD) of CaMKII, shows an anti-parallel interface: the C-domain of CaM primarily contacts the N-terminal half of the CaMBD. The two domains of calcium-saturated CaM are believed to play distinct roles in releasing auto-inhibition. To investigate the underlying mechanism of activation, calcium-dependent titrations of isolated domains of CaM binding to CaMKIIp were monitored using fluorescence anisotropy. The binding affinity of CaMKIIp for the domains of CaM increased upon saturation with calcium, with a 35-fold greater increase observed for the C-domain than the N-domain. Because the interdomain linker of CaM regulates calcium-binding affinity and contribute to conformational change, the role of each CaM domain was explored further by investigating effects of CaMKIIp on site-knockout mutants affecting the calcium-binding sites of a single domain. Investigation of the thermodynamic linkage between saturation of individual calcium-binding sites and CaM-domain binding to CaMKIIp showed that calcium binding to sites III and IV was sufficient to recapitulate the behavior of (Ca2+)4-CaM. The magnitude of favorable interdomain cooperativity varied depending on which of the four calcium-binding sites were mutated, emphasizing differential regulatory roles for the domains of CaM, despite the high degree of homology among the four EF-hands of CaM.
Keywords: Thermodynamic, anisotropy, fluorescence, cooperativity, mutation, calcium binding
Calmodulin (CaM) is a small (148 a.a.), ubiquitous calcium signaling protein that regulates the activities of many cellular proteins. It has four calcium-binding sites (I and II in the N-domain, and III and IV in the C-domain; Fig. 1A). Favorable calcium binding depends on a glutamate at position 12 in each site, which provides bidentate coordination of the calcium ion 1–3. Studies of a site IV mutant of CaM lowered the calcium-binding affinity in the C-domain, but raised the affinity of the N-domain 4. Mutagenesis and structural studies by Grabarek and coworkers emphasized the key role of the glutamate at position 12 in determining secondary and tertiary structure, as well as coordinating calcium 5.
Figure 1. Ribbon diagrams of CaM and regulatory and kinase domains of CaMKII.

(A) Ribbon diagram of rat calmodulin refined at 2.2 Å resolution (3CLN.pdb) 68. The N-domain of CaM (residues 1–75) is highlighted in blue, the linker region (residues 76–80) in black, and the C-domain (residues 81–148) in red. Calcium-binding sites are shown in yellow. Residues 31, 67, 104 and 140 are shown in black ball-and-stick representation.
(B) Ribbon diagram of CaMKII regulatory and kinase domains refined at 1.8 Å resolution (2BDW.pdb) 32. Regulatory domain is in orange, kinase domain in green, and CaM-binding region on the regulatory domain is in gray. Tyr 286, 305 and 306 are shown as black spheres.
(C) Schematic representation of CaMKII regulatory and kinase domains showing a stacked set of 2 six-membered rings; hexagons match those shown in part (B).
(D) Ribbon diagram of CaM in complex with CaMBD of CaMKII (1CDM.pdb) 17. The CaMBD of CaMKII is shown in gray. The N-domain of CaM (residues 1–75) is highlighted in blue, the linker region (residues 76–80) in black, and the C-domain (residues 81–148) in red. Calcium-binding sites are shown in yellow. All figures were made with MacPymol (DeLano Scientific).
Upon calcium binding to CaM, methionine-rich hydrophobic patches are exposed to solvent 6,7, promoting association with the CaM-binding domain (CaMBD) of its target proteins 8–10. CaM bound to a target protein varies from calcium-free (apo) 11–13, to having only one domain calcium-saturated 14,15, to having both domains saturated 13,16–22. CaMBDs in ion channels, kinases, synthases, cyclases and motor proteins typically are basic amphipathic α-helical (BAA) motifs 8. They are classified according to the positions of anchoring hydrophobic residues that make a high number of contacts with CaM 10. Generally, an aromatic residue of the target protein fills the C-domain pocket of CaM, while occupancy of the N-domain pocket is variable 8,23–25
CaM-dependent kinase II (CaMKII) is a multifunctional serine/threonine kinase found in many tissue types with highly homologous α, β, and δ isoforms (for review, see 26). Activation of CaMKII contributes to synaptic plasticity and long-term potentiation (LTP) in the brain 26–28, and normal and pathological conditions in the heart 29,30. CaMKII contains 12 subunits 31, each consisting of regulatory, catalytic and self-association domains. The regulatory domain of CaMKII contains an autophosphorylation site (Thr286 in CaMKIIα and Thr287 in CaMKIIβ), which is essential for its calcium-dependent activation.
In the absence of calcium-saturated CaM, autophosphorylation of Thr286 is inhibited via interactions of the regulatory domain with the catalytic domain, thereby preventing auto-activation of CaMKII. A high resolution structure of the regulatory and catalytic domains shows how the interface between two adjacent regulatory subunits separates the catalytic domains from one another, preventing non-specific Thr autophosphorylation (Fig. 1B and 1C) 32. Binding of calcium-saturated CaM disrupts the interactions between the regulatory and catalytic domains of CaMKII, allowing the catalytic domain of one subunit to phosphorylate the adjacent subunit at Thr286, leading to activation. Once Thr286 becomes phosphorylated, the enzyme remains active, even in the absence of calcium-saturated CaM 33.
The CaMBD of CaMKII (residues 290–309) is in the regulatory region of each subunit 34. The crystal structure of (Ca2+)4-CaM bound to the CaMBD of CaMKII represents a classical “wrap-around” CaM-target conformation, in which both calcium-saturated domains of CaM engulf the CaMBD in an anti-parallel orientation 20 (Fig. 1D).
CaM is initially bound to CaMKII via one domain in an extended conformation depending on whether ATP is bound or CaMKII is phosphorylated, as shown by FRET analysis 35 and ITC 36. Mayo and coworkers computationally designed mutants of CaM that maintain an apo-like conformation in either the N- or C-domain, and showed that the C-domain of CaM was sufficient to regulate the enzymatic activity of CaMKII 37.
Binding of CaMKII to CaM alters the calcium-binding affinity of individual CaM domains 38, which is necessary for a quick response to changes in the intracellular calcium levels. Previous studies showed that binding to peptides representing the CaMBDs of MLCK and CaMKII produced dramatic decreases in the rate of calcium dissociation from the N-domain of CaM relative to its C-domain 39, thereby decreasing the differences in affinities for the domains. By analogy with the effect of melittin on CaM 40, interdomain cooperativity, as well as effects on individual calcium-binding sites of CaM may play a role in the regulation of CaMKII.
In the present work, fluorescence anisotropy was used to determine the dissociation constant (Kd) of a 20-residue synthetic peptide (CaMKIIp) representing the CaMBD of CaMKII (residues 290–309) for CaM1–80, CaM76–148 and CaM1–148. Mutants of CaM1–148 with dramatically reduced affinity for calcium in the N-domain (E31Q/E67Q) or C-domain (E104Q/E140Q) were used to probe the domain-specific effects of calcium-binding on the CaM–CaMKIIp interaction. In order to probe changes in interdomain cooperativity, we performed equilibrium calcium titrations of WT and mutant CaM in the absence and presence of CaMKIIp.
Our results indicate that the C-domain of CaM mediates binding to CaMKII, in agreement with a previous report 37. In addition, we have determined that CaM binds to CaMKII at sub-micromolar calcium concentration (~0.3 μM calcium) but the binding affinity at the basal calcium concentration was very weak. CaMKIIp-binding caused a greater increase in the calcium-binding affinities of the N- and C-domains of full-length CaM than in the individual domains and the increase in the calcium-binding affinity of the N-domain was much greater than that of the C-domain within the full-length CaM. These findings suggest that domain cooperativity occurs within CaM1–148 when bound to CaMKIIp. Studies of mutants with point mutations in the CaM calcium-binding sites (sites I, II, III and IV) reveal how each of the calcium-binding sites contributes to the CaMKIIp mediated domain cooperativity within CaM.
These results, in combination with the reported calcium-affinities of the domains of CaM and the role of each of the calcium-binding sites within CaM, provide a detailed thermodynamic linkage for the formation of a CaM-CaMKIIp complex. The results from this study can be applied for the analysis of other CaM-target interactions with opposing binding affinities under apo and calcium-saturated conditions.
Materials and Methods
Overexpression and Purification of CaM
DNA encoding the rat calmodulin fragments rCaM1–80, rCaM76–148, rCaM1–148 was cloned into a pT7-7 bacterial vector and overexpressed in Escherichia coli Lys-S cells (U.S. Biochemicals, Cleveland, OH) as previously described 41–43. The QuikChange II site-directed mutagenesis kit (Stratagene) was used to make six point mutants of CaM1–148: E31Q in site I, E67Q in site II, E31Q/E67Q, E104Q in site III, E140Q in site IV, and E140Q/E140Q. Forward and reverse primers were purchased from Integrated DNA Technologies (IDT, Coralville, IA). PCR products were extracted, digested and inserted into a pT7-7 bacterial vector. The vector was then transformed into BL21 (DE3) cells for overexpression. CaM (WT and mutant forms) was purified using Phenyl Sepharose CL-4B (Amersham Pharmacia Biotech, Piscataway, NJ) chromatography as previously described 44. Some protein samples required subsequent purification using a Superdex 75 (Pharmacia size exclusion resin) or ammonium sulfate precipitation of contaminants. Purified proteins were dialyzed into 50 mM HEPES, 100 mM KCl and 50 μM EGTA pH 7.4. The purity of each recombinant protein was 97–99 % as assessed by reversed-phase HPLC or silver staining. Protein concentrations were determined from UV absorbance in 0.1 N NaOH 45 and aliquots were stored at −20°C.
Preparation of Peptides
CaMKIIp (L-K-K-F-N-A-R-R-K-L-K-G-A-I-L-T-T-M-L-A), corresponding to residues 290–309 from the CaMBD of CaMKII, and Fl-CaMKIIp (CaMKIIp labeled with 5,6-carboxyfluorescein at its N-terminus) were synthesized by GenScript Corporation (Piscataway, NJ). Peptides were dissolved in distilled/autoclaved water to make stock solutions up to 1 mM. The purity of each peptide was determined by reversed-phase HPLC and their molecular weights were confirmed by MALDI-TOF. The amino acid content of each peptide was confirmed by amino acid analysis at the Molecular Analysis Facility at the Univ. of Iowa.
Analysis of CaMKIIp Binding to CaM by Fluorescence Anisotropy
Association of CaM with Fl-CaMKIIp was observed as an increase in the fluorescence anisotropy of Fl-CaMKIIp measured with a Fluorolog 3 fluorimeter (Jobin Yvon) with a λex of 496 nm, a λem of 520 nm, slit widths of 3 nm (excitation) and 10 nm (emission), and a temperature of 22°C. Anisotropy (R) was calculated using Eq. 1,
| (1) |
where IVV is the intensity of vertically emitted light when vertically excited, IVH is the intensity of horizontally emitted light when vertically excited. G equals IHV/IHH where IHV is the intensity of vertically emitted light when excited horizontally and IHH is the intensity of horizontally emitted light when horizontally excited. The G value was calculated before each experiment and was consistently found to be 0.85 for Fl-CaMKIIp. Each signal after addition of CaM was monitored for 1 sec and averages of 3 readings were calculated. Aliquots of concentrated CaM (0.5–1.2 mM) were added to titrate 0.1 μM of Fl-CaMKIIp in a calcium-depleted (apo) or Ca2+-saturated buffer. Apo buffer contained 50 mM HEPES, 100 mM KCl, 1 mM MgCl2, 0.05 mM EGTA and 5 mM NTA (pH 7.4). Ca2+-saturated buffer contained all components of the apo buffer and 10 mM CaCl2. The total dilution of the peptide solution was < 3% over the course of the titrations.
To determine the affinity of each CaM for Fl-CaMKIIp, the anisotropy data from the titration curves were fit to a simple binding model, treating the peptide-CaM complex as having a 1:1 stoichiometry. Normalized anisotropy values were plotted against the total concentration of CaM ([CaMtotal]). The association constant was determined by fitting [CaMtotal] against normalized fractional change in anisotropy (Ȳ1) to a one-site Langmuir binding isotherm as described by Eq. 2,
| (2) |
where Ka represents the intrinsic association constant (the reciprocal of the dissociation constant, Kd) for CaM binding to a peptide and [CaMfree] is the concentration of unbound CaM, calculated from the two independent variables, [CaMtotal] and the total concentration of Fl-CaMKIIp, according to the quadratic equation (Eq. 3)
| (3) |
where b=1+Ka[Fl-CaMKIIptotal]-Ka[CaMtotal]. Under apo conditions, the affinity of CaM1–148, CaM1–80 and CaM76–148 for Fl-CaMKIIp was weak and [CaMfree] ≈ [CaMtotal]. However, the affinity of calcium-saturated CaM1–148 for Fl-CaMKIIp was high enough that [Fl-CaMKIIptotal] ≥ 10(Kd). While this condition is appropriate for determining the stoichiometry of binding, it is not appropriate for resolving accurate affinities of binding because [CaMfree] is limiting. To obtain limiting values for the affinity under those conditions, [CaMfree] was estimated iteratively in the nonlinear least-squares analysis 46 as the difference between [CaMtotal] and [CaMbound] (calculated as [Fl-CaMKIIptotal] • Ȳ1). To account for the effect of change in volume over the titration, the value of [Fl-CaMKIIptotal] was corrected for dilution and included as a second independent variable in the nonlinear least squares analysis.
Experimental variations in the observed fluorescence signal at the plateau of individual titrations are accounted for by Eq. 4
| (4) |
where Ȳ1 refers to average fractional saturation of the peptide and Y[X]low corresponds to the intrinsic fluorescence anisotropy of Fl-CaMKIIp in the absence of CaM. The Span describes the magnitude and direction of signal change upon titration and describes the difference between the high (Y[X]high) and low (Y[X]low) endpoints. The Span is defined as positive for the increasing signals and negative for the decreasing signals. The endpoint or upper limit of data was fitted in analysis of titrations that were done under calcium-saturated conditions.
Equilibrium titrations of Fl-CaMKIIp with apo-CaM did not reach a well-defined upper plateau for fitting the experimental data to Eq. 4; therefore, the final apo CaM:CaMKIIp complex was titrated with CaCl2 up to a final concentration of 10 mM. The final anisotropy obtained for the Ca2+-saturated CaM:CaMKIIp complex was used as a fixed endpoint to represent 100% saturation in the nonlinear least squares analysis of the titration of peptide with of apo CaM1–148. Because of the relatively weak affinity of Ca2+-saturated CaM1–80 and CaM76–148 for Fl-CaMKIIp, the addition of CaCl2 to a titration of peptide with an apo domain fragment failed to fully saturate the peptide even though CaM was in excess. The upper endpoints were determined by comparing the anisotropy of titrations conducted with calcium-saturated domains. The average final fractional saturation reached was 76% for CaM1–80 and 97% for CaM76–148.
Fluorescence Monitored Equilibrium Calcium Titrations
Equilibrium calcium titrations were conducted with a PTI fluorimeter (Photon Technology International, Lawrenceville, NJ) with a xenon lamp using 8 nm band passes to measure changes in the calcium affinity of CaM in the absence and presence of CaMKIIp. For the titrations in the absence of CaMKIIp, 2 μM CaM (CaM1–148, CaM1–80 and CaM76–148) in 50 mM HEPES, 100 mM KCl, 1 mM MgCl2, 0.05 mM EGTA, 5 mM NTA, pH 7.4 at 22°C were titrated with a concentrated calciumsolution (~5 mM, 50 mM or 500 mM CaCl2 in the same buffer that was used throughout the titrations) to achieve 1 nM – 0.01 M calcium in the reaction cuvette using a microburet (Micro-Metric Instrument Co., Cleveland, OH) fitted with a 250 μl Hamilton syringe (Hamilton Co., Reno, NV). For the titrations in the presence of CaMKIIp, 2 μM CaM and 10 μM of CaMKIIp (1:5 CaM:CaMKIIp molar ratio) was used to ensure the saturation of CaM with CaMKIIp. Calcium binding to the N-domain (sites I and II) was monitored by intrinsic phenylalanine fluorescence (λex of 250 nm, λem of 280 nm), and calcium binding to the C-domain (sites III and IV) was monitored by intrinsic tyrosine fluorescence (λex of 277 nm, λem of 320 nm) as previously described 47. For each calcium addition, the free calcium concentration was determined using Eq. 5 to relate the degree of saturation of a fluorescent dye (0.1 μM Oregon Green for experiments with no peptide or 0.1 μM XRhod5F (Molecular Probes, Eugene, OR) for experiments in the presence of CaMKIIp) present in the sample to the concentration of free-calcium.
| (5) |
The Kd values of Oregon Green and XRhod5F were determined experimentally to be 34.24 μM and 1.78 μM, respectively, in 50 mM HEPES, 100 mM KCl, 1 mM MgCl2, pH 7.4 at 22 °C using a calcium titrant with concentration determined by atomic absorption. XRhod5F was used as an indicator for CaM calcium titrations in the presence of CaMKIIp because of its ~20-fold lower Kd (higher affinity for calcium), to make it possible to resolve the data points early in the titrations. Each calcium titration of CaM in the presence and absence of CaMKIIp was repeated at least three times. The fluorescence readings for each titration were normalized to the high and low endpoints before the performance of a nonlinear least squares analysis using NONLIN 46, which determined the free energies of calcium-binding (ΔG1 and ΔG2) to N-domain sites I and II and C-domain sites III and IV.
Analysis of the Free Energy of Calcium Binding (ΔG2) to CaM
The Gibbs free energy of calcium binding to two sites in a domain was obtained from fits of the titration data to the two-site Adair function, as shown in Eq. 6.
| (6) |
where the pair of sites within a domain (i.e., sites I and II in the N-domain or sites III and IV in the C-domain) are allowed to be non-identical and cooperative 43. The macroscopic equilibrium constant K1 (ΔG1= −RT ln K1) in Eq. 6 represents the sum of two intrinsic constants (k1 and k2) that may or may not be equal. The macroscopic constant K2 (ΔG2= −RT ln K2) is the equilibrium constant for calcium binding to both sites (the product of k1, k2 and k12) and accounts for any positive or negative cooperativity.
In the absence of CaMKIIp, Phe residues within the C-domain of CaM were quenched by Tyr residues 48. Hence, the overall change in the Phe signal (λex of 250 nm, λem of 280 nm) represented calcium-binding solely to the N-domain (sites I and II) and the signal from the equilibrium calcium-titration was fit to a function [f(X)] as shown in Eq. 7.
| (7) |
where Y[X]low corresponds to the value of the fluorescence intensity at the lowest calcium concentration, and Span accounts for the magnitude and direction of signal change upon increasing calcium concentration (usually increasing for Tyr signal, decreasing for Phe signal).
In the presence of CaMKIIp, Tyr no longer fully quenched the steady-state fluorescence intensity of the C-domain Phe residues, and the intensity of the Phe signal for CaM76–148 increased in a calcium-dependent manner in parallel with the Tyr signal (Fig. 5B-inset). However, the net change was a negative deflection, indicating that the Phe intensity of CaM1–148 in the presence of CaMKIIp (AT) was the sum of an increasing intensity change from the C-domain (AC) and a decreasing intensity change arising from the N-domain (AN) upon calcium titration.
Figure 5.

Equilibrium calcium titrations of WT CaM with and without 1:5 molar ratio of CaMKIIp. Phenylalanine fluorescence of CaM1–80 (A), the N-domain of CaM1–148 (C) and tyrosine fluorescence of CaM76–148 (B), the C-domain of CaM1–148 (D) were monitored with (■, ●) and without (□, ○) CaMKIIp. CaM alone (dashed) and (1:5) CaM:CaMKIIp (solid) were simulated using values in Table III and Eq. 6, Eq. 8b and Eq. 8c.
| (8a) |
The free energy of calcium binding to the N-domain of CaM1–148 was determined using Eq. 8b,
| (8b) |
which takes into account the sum of two Adair equations (ȲN and ȲC) multiplied by the corresponding fractional contribution of each domain to the total amplitude of the intensity change (i.e., AN/AT • ȲN and AC/AT • ȲC). The fractional contribution of the N-domain to the total intensity change is simply (1-AC/AT). Therefore, Eq. 8b can be rewritten as follows:
| (8c) |
The contribution that calcium binding to sites III and IV of CaM1–148 made to the Phe fluorescence intensity was calculated by comparing the Phe signal intensity, as determined from equilibrium calcium titrations (same solution conditions and instrumental settings) of CaM1–148 (AT) and CaM76–148 (AC). The value of AC was assumed to be the same as the amplitude of the intensity change for CaM76–148, and the fractional contribution of the C-domain Phe signal to the total amplitude intensity (AC/AT) was determined to be 0.78. Because of the opposite (increasing) direction of the Phe signal from CaM76–148 relative to the Phe signal from CaM1–148 (decreasing), AC/AT is calculated to be negative (−0.78).
Results
Association of CaM with Fl-CaMKIIp
The affinity of CaM for fluorescently labeled CaMKIIp (Fl-CaMKIIp) was determined using fluorescence anisotropy at a series of calcium concentrations. Under calcium-saturating conditions (10 mM CaCl2), CaM1–148 was found to have a very high affinity for Fl-CaMKIIp (Fig. 2A). The concentration of Fl-CaMKIIp used in the experiment was 0.1 μM and therefore the binding of (Ca2+)4-CaM1–148 to Fl-CaMKIIp was not at equilibrium but was in the stoichiometric range 49. The Kd for (Ca2+)4-CaM1–148 for Fl-CaMKIIp was estimated by comparing the experimentally obtained binding data with the curves created by simulations using several Kd values (Fig. 2A, inset). This allowed us to determine an upper limit for the dissociation constant to be ≤ 10 nM (Table I) for the binding of Fl-CaMKIIp to (Ca2+)4-CaM1–148. The actual value of apparent Kd may be lower, or more favorable.
Figure 2.

Change in the fluorescence anisotropy of Fl-CaMKIIp upon titration with CaM1–148. The normalized anisotropy of 0.1 μM Fl-CaMKIIp (λex of 496 nm, λem of 520 nm) increased upon titrating with CaM1–148 in 10 mM CaCl2 (A) or apo (○), (▲) 23 nM, (△) 311 nM, (□) 346 nM, and (◇) 494 nM calcium (B). The buffer was 50 mM HEPES, 100 mM KCl, 1 mM MgCl2, 0.05 mM EGTA, 5 mM NTA (pH 7.4) at 22°C. Arrow indicates the fixed-point for the titrations that are done under apo conditions and 23 nM of calcium. A comparison of simulated one-site binding curves of Fl-CaMKIIp binding to CaM1–148 under equilibrium conditions (dashed lines-apo,—·— in 311 nM calcium, and ● in 10 mM calcium) is shown in (C).
Table I.
Calcium-Dependent Dissociation Constants for Fl-CaMKIIp Binding to WT CaM1–148
| [Ca2+] | Kd (μM) |
|---|---|
| 0 | 142 ± 32 |
| 23 nM | 89 ± 6 |
| 311 nM | 0.32 ± 0.08 |
| 346 nM | 0.29 ± 0.15 |
| 494 nM | 0.27 ± 0.13 |
| 10 mM | ≤0.010 |
The binding of CaM1–148 to Fl-CaMKIIp under apo conditions was assessed in the presence of 50 μM EGTA and 5 mM NTA as calcium-chelators. Under these conditions, CaM1–148 titrations showed less than 45% of the overall change in anisotropy when compared to the titration of CaM1–148 in 10 mM CaCl2 (Fig. 2B). As described in Methods, the final anisotropy of the peptide titrated with calcium-saturated CaM was used as the upper endpoint. Kd of apo CaM1–148 binding to Fl-CaMKIIp was determined to be 142 ± 32 μM (Table I).
Although it is known that there are large differences in the affinities of CaM for CaMKIIp under calcium-saturated and apo conditions in vivo, CaMKII experiences a wide range of calcium concentrations during calcium-mediated signaling events. This prompted us to investigate binding using a series of calcium buffers with free calcium concentrations of 23, 311, 346 or 494 nM (Fig. 2B; Table I). At 10 μM of CaM1–148, the level of saturation of CaMKIIp by CaM1–148 in the presence of 23 nM of calcium was less than 10%. However, the level of saturation of CaMKIIp at 10 μM of CaM1–148 in the presence of 311 nM of calcium was more than 95% (Fig. 2B). The simulated lines representing the calcium-dependent differences in the affinity of CaM1–148 for CaMKIIp under apo (dashed lines) or calcium-saturating (solid line) conditions, and at 311 nM calcium (—·—) are shown in Fig. 2C under equilibrium conditions.
The affinities of the N-domain (CaM1–80) and C-domain (CaM76–148) of CaM for Fl-CaMKIIp were determined under both calcium-saturating and apo conditions. Under calcium-saturating conditions, the Kd of Fl-CaMKIIp was 32.82 μM for CaM1–80 and 0.95 μM for CaM76–148 (Fig. 3A and 3B). For the apo studies with the domains of CaM, Fl-CaMKIIp was titrated up to ~100 μM of apo CaM; however, due to a weak affinity between Fl-CaMKIIp and the domains of CaM, the increases in raw anisotropy values were only 19% for the apo CaM1–80, and 28% for apo CaM76–148 of the overall anisotropy signal change under calcium-saturated conditions at the highest level of CaM tested (Fig. 3A and 3B). From this analysis, the apparent Kd of Fl-CaMKIIp was 697 μM for apo CaM1–80, and 88 μM for apo CaM76–148 (Table I).
Figure 3.

Change in the fluorescence anisotropy of Fl-CaMKIIp upon titration with CaM1–80 and CaM76–148. The normalized anisotropy of 0.1 μM Fl-CaMKIIp (λex of 496 nm, λem of 520 nm) increased upon titration with apo (open) and calcium-saturated (closed) CaM1–80 (A) and CaM76–148 (B) in 50 mM HEPES, 100 mM KCl, 1 mM MgCl2, 0.05 mM EGTA, 5 mM NTA with or without 10 mM CaCl2 (pH 7.4). Simulated one-site binding curves are shown as dashed (apo) and solid (calcium-saturated) lines. Arrows indicate the fixed-point for the titrations that are done under apo conditions.
Association of CaM Mutants with Fl-CaMKIIp
To determine how disrupted calcium-binding properties within each CaM domain affect the affinity of Fl-CaMKIIp for CaM, we tested double site-knockout mutants of CaM1–148 (E31Q/E67Q at sites I and II and E104Q/E140Q at sites III and IV) in association studies. The 12-residue EF-hand calcium-binding loops of CaM (two present within in each domain) bind a total four calcium ions, whose positions are coordinated by conserved Asp and Glu residues (Fig. 1A, yellow sites). To significantly perturb calcium binding, the terminal Glu residues of the two EF-hand calcium-binding loops in each domain were mutated to Gln. The E31Q/E67Q mutations within the N-domain completely abolished calcium binding to sites I and II, whereas C-domain calcium binding was not affected by the mutations (data not shown). Likewise, E104Q/E140Q mutations within the C-domain dramatically reduced calcium binding to sites III and IV (~100-fold decrease in the calcium-binding affinity), and there was only a modest shift in the calcium-binding affinity of the N-domain relative to that of the N-domain of WT CaM1–148 (data not shown).
The titration of Fl-CaMKIIp with E104Q/E140Q CaM1–148 (WT N-domain) under calcium-saturated conditions is shown in Fig. 4A and the Kd was 6.51 ± 2.29 μM (Table II). The apparent affinity of E31Q/E67Q CaM1–148 (WT C-domain) for Fl-CaMKIIp was very similar to the affinity of CaM76–148 (dashed lines) for Fl-CaMKIIp (Kd of 1.22 ± 0.23 μM) (Fig. 4B).
Figure 4.

Change in the fluorescence anisotropy of Fl-CaMKIIp upon titration with calcium site knockout mutants of CaM1–148. Normalized anisotropy of Fl-CaMKIIp (λex of 496 nm and λem of 520 nm) upon titration with E104Q/E140Q CaM1–148 (●) (A) and E31Q/E67Q CaM1–148 (●) (B) in 50 mM HEPES, 100 mM KCl, 1 mM MgCl2, 0.05 mM EGTA, 5 mM NTA, 10 mM CaCl2 (pH 7.4). Simulated one-site binding curve for WT Ca2+4-(CaM1–148) binding to Fl-CaMKIIp is shown as solid line. Simulated 1-site binding curves of the titration of Fl-CaMKIIp by (Ca2+)2-CaM1–80 (A) and (Ca2+)2-CaM76–148 (B) are shown in dashed lines. The total CaM concentration at 50% saturation of the titrations with the calcium site knockout mutants is shown as vertical lines.
Table II.
Dissociation Constants for Fl-CaMKIIp Binding to CaM
| Protein | Kd, Apoa | Kd, Ca2+b |
|---|---|---|
| WT CaM1–148 | 142 ± 32 | ≤0.010 |
| WT CaM1–80 | 697 ± 44 | 32.82 ± 1.06 |
| WT CaM76–148 | 88 ± 10 | 0.95 ± 0.08 |
| E31Q/E67Q CaM1–148 | N.D. | 1.22 ± 0.23 |
| E104Q/E140Q CaM1–148 | N.D. | 6.51 ± 2.29 |
Kd reported in μM; apo buffer: 50 mM HEPES, 100 mM KCl, 1 mM MgCl2, 5 mM NTA, 50 μM EGTA pH 7.4
apo buffer with 10 mM CaCl2
Equilibrium Calcium Titrations of CaM1–80, CaM76–148 and CaM1–148
Equilibrium calcium titrations were performed as described in Materials and Methods. For the calcium titration of CaM in the absence of a peptide, we have previously shown that a decreasing Phe signal (Ȳ2) is associated exclusively with calcium binding to the N-domain, whereas an increasing Tyr signal is associated exclusively with calcium binding to the C-domain 48. Titrations of WT CaM1–80, CaM76–148 and CaM1–148 in the absence and presence of CaMKIIp are shown in Fig. 5.
As shown in Table III, the free energy (ΔG2) of calcium binding to CaM1–80 was −12.76 ± 0.09 kcal/mol in the absence of CaMKIIp (Fig. 5A). This was within the margin of error for the ΔG2 of calcium binding to the N-domain (sites I and II) of CaM1–148, which is −12.82 ± 0.09 kcal/mol (Fig. 5C). CaM76–148 bound calcium with a higher affinity than CaM1–80, with a ΔG2 of −14.66 ± 0.13 kcal/mol (Fig. 5B). However, the ΔG2 of calcium binding to CaM76–148 was slightly less favorable than that for the C-domain (sites III and IV) of CaM1–148, which is −15.06 ± 0.03 kcal/mol (Fig. 5D).
Table III.
Effect of CaMKIIp on Free Energies of Calcium Binding to WT CaM
| Protein | Peptide Equivalent | ΔG1app | ΔG2app | ΔGc | ΔΔG2a |
|---|---|---|---|---|---|
| Sites I and II | |||||
| CaM1–80 | 0 | −5.92 ± 0.38 | −12.76 ± 0.09 | −1.73 ± 0.68 | -- |
| 5 | −5.27 ± 0.49 | −13.12 ± 0.17 | −3.38 ± 1.18 | −0.36 | |
| CaM1–148 | 0 | −5.98 ± 0.02 | −12.82 ± 0.09 | −1.67 ± 0.08 | -- |
| 5 | −6.00b | −17.98 ± 0.16 | −6.79 ± 0.16 | −5.16 | |
| Sites III and IV | |||||
| CaM76–148 | 0 | −5.45 ± 0.51 | −14.66 ± 0.13 | −4.57 ± 1.15 | -- |
| 5 | −7.08 ± 0.25 | −16.19 ± 0.03 | −2.81 ± 0.47 | −1.53 | |
| CaM1–148 | 0 | −6.40 ± 0.19 | −15.06 ± 0.03 | −3.05 ± 0.35 | -- |
| 5 | −6.40c | −18.26 ± 0.05 | −6.27 ± 0.05 | −3.20 | |
ΔΔG2= ΔG2app (CaM+CaMKIIp) − ΔG2app (CaM)
ΔΔG1 for N was fixed to −6.00 kcal/mol while ΔG1 of C was fixed to −6.40 kcal/mol.
ΔG1 of C was fixed to −6.40 kcal/mol.
Due to the low affinity of apo CaM for CaMKIIp, the presence of 5 eq. of CaMKIIp did not lead to CaM saturation under apo conditions (i.e., the concentration of apo CaM-CaMKIIp was ≤ 10%). Therefore, the ΔG2 of calcium binding to CaM in the presence of CaMKIIp is reported here as the apparent free energy (ΔG2app). The addition of 5 eq. of CaMKIIp did not significantly change the calcium-binding affinity of CaM1–80, resulting in a ΔG2app of −13.12 ± 0.17 kcal/mol (ΔΔG2app of 0.36 kcal/mol) (Fig. 5A). However, the addition of CaMKIIp increased the calcium-binding affinity of CaM76–148 with a ΔG2app of −16.17 ± 0.06 kcal/mol (ΔΔG2app of 1.51 kcal/mol) (Fig. 5B).
In the presence of CaMKIIp, ΔG2app of calcium binding was −17.96 ± 0.11 kcal/mol for the N-domain of CaM1–148 (Fig. 5C) and −18.17 ± 0.05 kcal/mol for the C-domain of CaM1–148 (Fig. 5D). The magnitude of change in free energy (ΔΔG2app) of calcium binding to the N-domain of CaM1–148 (−5.14 kcal/mol) was greater than the C-domain (−3.11 kcal/mol). Therefore, in the presence of CaMKIIp, the two domains of CaM1–148 have more similar affinities (Fig. 6). These results suggested that domains of CaM communicate in the presence of CaMKIIp.
Figure 6.
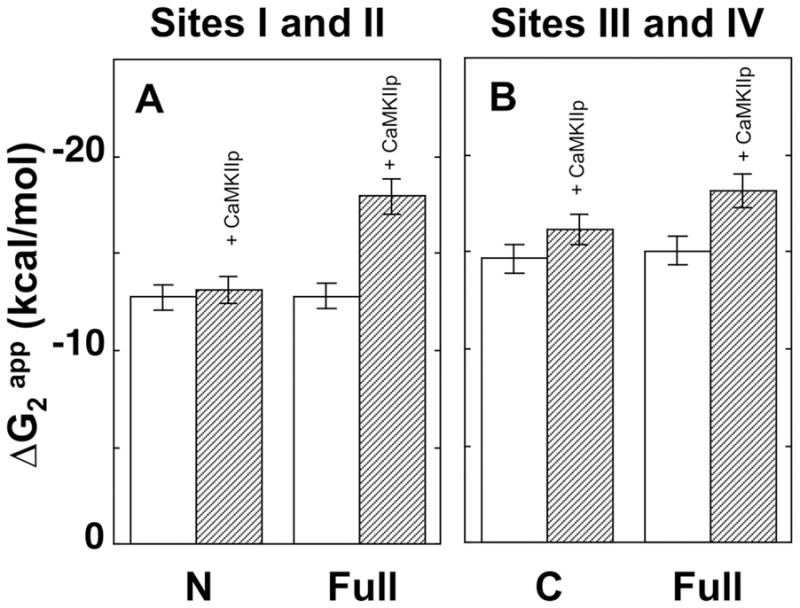
Bar graph for the apparent free energy (ΔG2app) of calcium binding to CaM. ΔG2app of calcium binding to WT CaM1–80 and WT CaM1–148 at sites I and II (A) and ΔG2app of calcium binding to WT CaM76–148 and WT CaM1–148 at sites III and IV (B) with (dashed bars) and without (open bars) CaMKIIp.
Equilibrium Calcium-Titrations of N- and C-Domain Point Mutants of CaM1–148
To further explore the effect of calcium binding to sites I, II, III and IV on the interactions with CaMKIIp and domain cooperativity within CaM, we used point mutants of CaM. We substituted Gln for the terminal Glu residues in position 12 of each CaM EF-hand calcium-binding sites to produce a series of CaM1–148 mutants at site I (E31Q), site II (E67Q), site III (E104Q) and site IV (E140Q).
In the absence of CaMKIIp, the N-domain of E31Q CaM1–148 displayed a ~3-fold lower calcium-binding affinity relative to the N-domain of WT CaM1–148 as calculated by the mid-point (Ȳ0.5) of the calcium-binding curve (Fig. 7A). The Ka of calcium-binding to the N-domain of E31Q CaM1–148 was determined using Eq. 2 and the Kd was 36 μM. Calcium binding to sites III and IV within the C-domain of E31Q CaM1–148 was not significantly affected by the site I (E31Q) mutation and the ΔG2 of calcium binding to the C-domain was −14.88 ± 0.08 kcal/mol (Fig. 7B).
Figure 7.

Equilibrium calcium titrations of site I (E31Q) and II (E67Q) mutants of CaM1–148. Phenylalanine fluorescence of the N-domain of E31Q (A), E67Q (C) CaM1–148 and tyrosine fluorescence of the C-domain of E31Q (B) and E67Q (D) CaM1–148 were monitored with (■, ●) and without (□, ○) CaMKIIp.
In the presence of saturating CaMKIIp (5 eq.), the calcium-binding curve for the N-domain of E31Q CaM1–148 was biphasic (Fig. 7A). The increasing signal represented Phe signal contribution from the C-domain of E31Q CaM1–148, and the decreasing signal represented the calcium-dependent decrease of Phe within the N-domain of E31Q CaM1–148 (as discussed in Materials and Methods). ΔG2app of calcium binding to the N-domain of E31Q CaM1–148 in the presence of CaMKIIp was determined by fitting the biphasic data to a function that accounts for the contributing Phe signals from both the N- and C-domains of CaM (Eq. 8c). ΔG2app of calcium-binding to the N-domain of E31Q CaM1–148 was −15.25 ± 0.13 kcal/mol in the presence of CaMKIIp. CaMKIIp binding to E31Q CaM1–148 also increased the calcium-binding affinity of the C-domain of E31Q CaM1–148 with a ΔG2app of −17.38 ± 0.04 kcal/mol (ΔΔG2app of −2.50 kcal/mol) (Fig. 7B).
The site II (E67Q) mutation within the N-domain of CaM1–148 had a more damaging effect on the spectral properties of the N-domain of CaM than any other mutations. Calcium-binding studies for the N-domain of E67Q CaM1–148 showed a very small change in the Phe signal upon addition of calcium (Fig. 7C). This finding was also observed in previous studies monitoring the calcium binding to the N-domain with a site II mutation 1,2. Although the E67Q mutation resulted in reduced calcium binding to the N-domain of CaM1–148, the ΔG2 of calcium binding to its C-domain was not affected by the mutation (ΔG2 of −15.10 ± 0.01 kcal/mol). CaMKIIp binding did not change the Phe fluorescence of the N-domain of E67Q CaM1–148 (Fig. 7C) (i.e., the signal was still noisy and low) but the ΔG2app of calcium binding to the C-domain of E67Q CaM1–148 increased to −17.00 ± 0.01 kcal/mol (ΔΔG2app of −1.90 kcal/mol) (Fig. 7D).
Mutation of site III (E104Q) within the C-domain of CaM1–148 reduced its calcium-binding affinity by ~25-fold compared to that of the C-domain of WT CaM1–148 (Fig. 8B). The Kd of calcium binding to the C-domain of E104Q CaM1–148 was ~19 μM (Table IV). Calcium binding to the N-domain of CaM1–148 (Fig. 8A) was not significantly affected by the E104Q mutation (ΔG2 of −13.13 ± 0.04 kcal/mol) (Table IV). In the presence of CaMKIIp, the calcium-binding affinity of the C-domain of E104Q CaM1–148 increased ~25-fold (Fig. 8B). The data resulted in a better fit with a 2-site binding isotherm (Eq. 6) rather than with a 1-site (Eq.2) and the ΔG2app was −16.53 ± 0.01 kcal/mol (Table IV). The ΔG2app of calcium binding to the N-domain of E104Q CaM1–148 was more favorable in the presence of CaMKIIp (ΔΔG2app of −1.14 kcal/mol) (Fig. 8A).
Figure 8.

Equilibrium calcium titrations of sites III (E104Q) and IV (E140Q) mutants of CaM1–148. Phenylalanine fluorescence of the N-domain of E104Q (A), E140Q (C) CaM1–148 and tyrosine fluorescence of the C-domain of E104Q (B) and E140Q (D) CaM1–148 were monitored with (■,●) and without (□, ○) CaMKIIp.
Table IV.
Effect of CaMKIIp on Free Energies of Calcium Binding to Mutants of CaM1–148
| Protein | Peptide Equivalent | ΔG1app | ΔG2app | ΔGc | ΔΔG2a |
|---|---|---|---|---|---|
| Sites I and II | |||||
| WT CaM1–148 | 0 | −5.98 ± 0.02 | −12.82 ± 0.09 | −1.67 ± 0.08 | -- |
| 5 | −6.00b | −17.98 ± 0.16 | −6.79 ± 0.16 | −5.16 | |
| E31Q CaM1–148 | 0 | −6.00 ± 0.14 | -- | -- | -- |
| 5 | −7.54 ± 0.11 | −15.25 ± 0.13 | −2.39 ± 0.32 | ||
| E67Q CaM1–148 | 0 | -- | -- | -- | -- |
| 5 | −8.21 ± 0.84 | −17.14 ± 0.33 | −1.52 ± 1.28 | -- | |
| E104Q CaM1–148 | 0 | −6.36 ± 0.24 | −13.13 ± 0.04 | −1.21 ± 0.45 | -- |
| 5 | −7.75 ± 0.06 | −14.17 ± 0.11 | 0.51 ± 0.18 | −1.14 | |
| E140Q CaM1–148 | 0 | −5.93 ± 0.46 | −13.13 ± 0.16 | −2.07 ± 0.92 | -- |
| 5 | −7.19 ± 0.19 | −14.52 ± 0.11 | −0.94 ± 0.28 | −1.39 | |
| Sites III and IV | |||||
| WT CaM1–148 | 0 | −6.40 ± 0.19 | −15.06 ± 0.03 | −3.05 ± 0.35 | -- |
| 5 | −6.40c | −18.26 ± 0.05 | −6.27 ± 0.05 | −3.20 | |
| E31Q CaM1–148 | 0 | −5.98 ± 0.18 | −14.88 ± 0.08 | −3.73 ± 0.45 | -- |
| 5 | −7.90 ± 0.15 | −17.38 ± 0.04 | −2.38 ± 0.31 | −2.50 | |
| E67Q CaM1–148 | 0 | −6.14 ± 0.52 | −15.10 ± 0.01 | −3.62 ± 1.04 | -- |
| 5 | −6.79 ± 0.39 | −17.00 ± 0.01 | −4.22 ± 0.79 | −1.90 | |
| E104Q CaM1–148 | 0 | −6.37 ± 0.05 | -- | -- | -- |
| 5 | −7.97 ± 0.04 | −16.53 ± 0.01 | −1.39 ± 0.07 | ||
| E140Q CaM1–148 | 0 | −5.39 ± 0.11 | -- | -- | -- |
| 5 | −6.38 ± 1.06 | −14.57 ± 0.02 | −2.60 ± 2.11 | ||
ΔΔG2=ΔG2app (CaM+CaMKIIp) − ΔG2app (CaM)
ΔG1 for N was fixed to −6.00 kcal/mol while ΔG1 of C was fixed to −6.40 kcal/mol.
ΔG1 of C was fixed to −6.40 kcal/mol.
The site IV (E140Q) mutation within CaM1–148 had a stronger impact on calcium binding to the C-domain than the site III (E104Q) mutation, causing a ~45-fold decrease in its affinity relative to the C-domain of WT CaM1–148 as calculated from the mid-point (Ȳ0.5) of the titration curve. The Kd for calcium binding to the C-domain of E140Q CaM1–148 was ~101 μM using the 1-site binding equation (Eq.2) (Fig. 8D). As was observed with the site III (E104Q) mutation, the site IV (E140Q) mutation failed to make a significant change in the ΔG2 of calcium binding to the N-domain (ΔG2= −13.13 ± 0.16 kcal/mol). In the presence of CaMKIIp, the calcium-binding affinity of the C-domain of E140Q CaM1–148 increased ~30-fold (Fig. 8D). Data for calcium binding to the C-domain of E140Q CaM1–148 was fit to Eq. 6 (a 2-site binding isotherm) with a ΔG2app of −14.57 ± 0.02 kcal/mol (Table IV). CaMKIIp binding increased the ΔG2app of calcium binding to the N-domain of E140Q CaM1–148 (ΔΔG2app= −1.39 kcal/mol).
Discussion
In the study presented here, thermodynamic linkage of CaM binding to CaMKIIp and calcium binding to CaM was evaluated by titrations of CaM with CaMKIIp or calcium. The roles of calcium binding sites I, II, III and IV of CaM on the interactions with CaMKIIp and the domain cooperativity were investigated.
A peptide representing the CaMBD of CaMKII (CaMKIIp) was chosen due to the size and complexity of CaMKII, a dodecamer of ~600 kDa. Although the use of a peptide fragment rather than a whole protein as a CaM target may result in differences in the calcium-binding affinity of CaM 38, HSQC-NMR studies with 15N-labeled CaM failed to detect significant structural differences in CaM when in complex with either the CaMBD of CaM/Dependent Kinase I (CaMKI) or full-length CaMKI 50, suggesting that our approach would give representative results.
Domain-Specific Interactions of CaM with CaMKIIp
Binding of CaM1–148 to Fl-CaMKIIp indicated that there were 4 orders of magnitude difference in the affinity under calcium-saturated and apo conditions. Other association studies of (Ca2+)4-CaM1–148 and CaMKIIp using orthogonal methods report tight binding of CaM to holo CaMKII or a peptide representing the CaMBD of CaMKII and CaM (from pM to nM) 31,32,34–36,51. Due to the high affinity of (Ca2+)4-CaM1–148 for CaMKIIp (Kd ≤ 10 nM) and signal-to-noise limitations, an accurate equilibrium constant could not be assessed using the fluorescence anisotropy method because binding was stoichiometric. However, this can be calculated on the basis of the coupled equilibrium and the principle of conservation of energy. A thermodynamic linkage diagram depicting these interactions for a single domain is shown below:
The depicted linkage diagram for the free energy of calcium-binding (ΔGa and ΔGc) or free energy of CaMKIIp-binding (ΔGd) was utilized to calculate the ΔG of CaM1–148 binding to CaMKIIp (ΔGb) under calcium-saturated conditions and estimated to be −13.90 kcal/mol, with a corresponding Kd of 50 pM.
The binding affinity of Fl-CaMKIIp for CaM1–148 at 311 nM of calcium was ~30-fold weaker than the binding affinity when CaM was calcium-saturated (Table I). This was significantly more favorable than its binding affinity determined under lower levels of calcium (23 nM or zero calcium) (Fig. 2B). Calcium concentrations at this level have been shown to be sufficient for the autophosphorylation of CaMKII in the presence of CaM 37. At basal concentrations in the cell (~22 nM calcium), CaM1–148 had a very weak affinity for CaMKIIp, which suggests that CaM1–148 does not bind to the CaM binding site of CaMKII under these conditions (Fig. 2, Table I). Simulated equilibrium binding curves for the interaction of CaM1–148 and CaMKIIp under apo and calcium-saturating conditions, as well as at 311 nM calcium, illustrate the effect of calcium levels on the interaction between CaM and CaMKIIp (Fig. 2).
In order to dissect the roles that the domains of CaM play in the interactions with CaMKIIp, we determined the affinity of Fl-CaMKIIp for CaM1–80 and CaM76–148. Similar to CaM1–148, CaM1–80 and CaM76–148 both had a very weak affinity for Fl-CaMKIIp under apo conditions (Fig. 3). However, apo CaM76–148 had ~8-fold higher affinity for Fl-CaMKIIp than apo CaM1–80 (697 μM versus 88 μM). This trend was consistent with studies that were done under calcium-saturating conditions, where CaM76–148 had a ~35-fold higher affinity (0.95 μM) for Fl-CaMKIIp than CaM1–80 (32.82 μM) (Table II). Because of the weaker affinity, interaction of the domains of CaM with Fl-CaMKIIp was under equilibrium conditions. In contrast to the affinity of calcium-saturated CaM1–148 for Fl-CaMKIIp, the weak affinity between domains of CaM and Fl-CaMKIIp exclude the possibility of non-specific interactions and suggest the stoichiometry to be 1:1. The higher affinity of CaM76–148 for Fl-CaMKIIp both under apo and calcium-saturating conditions suggests that the C-domain of CaM may be sufficient for the interactions between CaM and CaMKIIp. However, the high affinity of (Ca2+)4-CaM1–148 for CaMKIIp (50 pM) compared to the affinity of (Ca2+)2-CaM76–148 for CaMKIIp (0.93 μM) suggests that the N-domain of full-length CaM plays an essential role in the high-affinity interactions with CaMKIIp, despite the relatively low affinity of CaM1–80 under both apo and calcium-saturated conditions.
Previous studies have shown that CaM can bind to and activate CaMKII with the C-domain calcium-saturated and the N-domain calcium-depleted 37. Therefore, we hypothesize that the initial binding of the C-domain of CaM1–148 increases the local concentration of the N-domain near the CaMBD of CaMKII, thereby dramatically increasing the overall affinity of CaM1–148 for CaMKIIp (50 pM). This model of kinase binding for CaM has been described for another CaM kinase target, the skeletal myosin light chain kinase (skMLCK) 52. However, a recent report showed that the activation of CaMKII may be mediated by the initial binding of the N-domain of CaM 53. In our study, we determined the affinity of CaM1–80 for Fl-CaMKIIp to be poor. This suggests that, if the N-domain is loosening the coiled-coil structure to initiate the kinetic process of attachment, it is acting by interacting with parts of CaMKII that are not represented by the CaMBD. Overall, both domains of CaM are required to disrupt interactions between regulatory and kinase domains or between the two regulatory domains that are under the calcium levels higher than the basal calcium concentrations.
To determine how calcium binding to the domains of CaM1–148 contributes to CaMKIIp association, we utilized mutants of CaM1–148 with severely disrupted calcium-binding properties, either at the N-domain (E31Q/E67Q) or C-domain (E104Q/E140Q). To determine the effect that each domain had on the overall affinity of CaM1–148 for CaMKIIp, we compared the affinities of these two CaM mutants with those of the individual domains of CaM (Fig. 4). Loss of calcium-binding in the C-domain of CaM1–148 (E104Q/E140Q) decreased its affinity for CaMKIIp relative to that of WT CaM1–148 under calcium-saturating conditions; nevertheless, its affinity for CaMKIIp was ~ 5-fold higher than that of WT CaM1–80 (Fig. 4A). This suggests that cooperativity between the domains of full-length CaM influences the recognition of CaMKIIp; in other words, CaM1–148 with a C-domain defective in calcium binding is not equivalent to CaM1–80. In comparison, a mutant with loss of calcium binding in the N-domain of CaM1–148 (E31Q/E67Q) and WT CaM76–148 had similar affinities for CaMKIIp with overlapping binding curves (Fig. 4B). The similarity in the affinities of E31Q/E67Q CaM1–148 and CaM76–148 suggests that the C-domain of E31Q/E67Q CaM1–148 is sufficient for initial association of CaM to CaMKIIp, even in the absence of a calcium-saturated N-domain. However, the ~ 102-fold reduced affinity of E31Q/E67Q CaM1–148 for CaMKIIp with respect to that of the WT CaM1–148 indicates that calcium binding to the N-domain is required for the high-affinity complex formation (Kd ≤ 50 pM). Overall, our results suggest that domain cooperativity regulates the affinity CaM1–148 for various targets, and calcium binding to each domain plays a role in the tight binding to CaMKIIp.
The Effect of CaMKIIp-Binding on the Calcium Affinity of WT CaM
Thermodynamic linkage of CaM interactions of CaMKIIp requires the determination of the effects of target association on the calcium-binding affinity of CaM. The binding of some target peptides or proteins lead to enhanced calcium-binding affinity 38,54,55, whereas the binding of others can decrease the calcium-binding affinity 56,57. CaMKIIp binding to CaM1–148 resulted in an increase in the calcium-affinity of both CaM domains, which was greater than that observed for either of the individual CaM domains, providing evidence for domain cooperativity within CaM1–148 in the presence of CaMKIIp. CaMKIIp-binding led to a greater increase in the calcium-binding affinity of the N-domain of CaM1–148 relative to that of the C-domain (Fig. 5, Table III), resulting in the affinities of the two CaM domains becoming more similar to each other. The bar graph in Fig. 6 shows the ΔG2app of calcium binding to CaM in the absence and presence of CaMKIIp. The similar calcium-binding affinity of the domains of (Ca2+)4-CaM suggest that these two domains have equivalent roles when bound to CaMKIIp. Our studies of calcium-binding affinity of each CaM domains agree with the macroscopic findings from a dialysis study by Peersen et. al, in which CaM binding to the CaMBD of CaMKII was inferred to result in calcium-binding affinities of ~−20 kcal/mol for the domains of CaM1–148 38.
The Effect of Calcium Binding to the Sites of CaM1–148 on the Interactions with CaMKIIp
The effect of defective calcium binding at four distinct sites in CaM1–148 on calcium binding to non-mutated sites was investigated by equilibrium calcium-titrations in the absence and presence of CaMKIIp (Fig. 7 and 8). In the absence of CaMKIIp, equilibrium calcium-titration studies of the calcium binding site knockout mutants of CaM1–148 showed that the mutations had a small but reproducible impact on the calcium-binding affinity of the non-mutated domain as shown by the ΔG2 obtained from calcium-titration studies, which were very similar to WT CaM1–148 (Table IV). The results that we obtained by ESI-Mass spectroscopy of these CaM site knockout mutants suggested that disruption of calcium binding to sites I (E31Q), III (E104Q) or IV (E140Q) affect calcium binding to individual sites. However, disruption of calcium binding to site II (E67Q) affects calcium binding to both sites I and II by reducing the calcium-binding stoichiometry of CaM from 4 to 2 (data not shown). Previous studies which investigated these CaM mutations using 1-D 1H-NMR, UV difference spectra or flow dialysis techniques determined that mutations of residues 67 and 140 at sites II and IV had more deleterious effects on calcium-binding than did mutations of residues 31 and 104 at sites I and III 1,2. This difference was attributed to the presence of more severe structural changes in the case of mutations at sites II and IV, as determined by electrophoretic mobility studies 1. NMR studies on site III (E104Q) and site IV (E140Q) CaM mutants showed that, under apo conditions, these mutants did not exhibit significant changes in the chemical shift differences of the backbone amide nuclei 58,59. These same studies also suggested that each mutant caused a major conformational change of the calcium saturated state and affected calcium binding to site IV. In addition, some site knockout mutants have also been reported to contribute to domain cooperativity by altering the conformation of the protein 1,60,61.
Previous studies have shown that the reduction of calcium-binding affinity observed for site knockout CaM mutants can be reversed upon target binding 62–64. For example, Findlay et al. reported that target binding compensated for changes in the secondary and tertiary structures of CaM mutants, thereby improving the calcium-binding affinity of these mutated sites or rescuing their calcium-binding properties 62.
In the current study, the effect of CaMKIIp binding on the calcium-binding affinity of both mutated and non-mutated sites was investigated. Titrations were compared with WT CaM1–148 titrations in the presence of CaMKIIp (Fig. 7 and Fig. 8 dashed lines). The presence of a 5-fold molar excess of CaMKIIp led to an increase in the calcium-binding affinity of all the mutated sites—the only exception being the site II mutant (E67Q). In the presence of CaMKIIp, the calcium-binding affinity of sites III and IV of CaM1–148 modified in site I (E31Q) and site II (E67Q) increased by 8-fold and 5-fold, respectively (Table IV). However, the magnitude of the increases was lower than that observed for sites III and IV of WT CaM1–148 in the presence of CaMKIIp (14-fold) (Fig. 7). A similar observation was made for CaM1–148 mutated in sites III (E104Q) and IV (E140Q) where the calcium-binding affinity of sites I and II in the N-domain of increased (by 13- and 3-fold) but to a lower level than that of sites I and II of WT CaM1–148 in the presence of CaMKIIp (60-fold) (Fig. 8). These results suggest that when both domains of CaM are bound to a target sequence, N-domain mutations (E31Q, E67Q) affect the calcium-binding affinity of the WT sites in the C-domain, and the C-domain mutations (E104Q, E140Q) affect the calcium-binding affinity of the WT sites in the N-domain. The effects of these point mutations on the calcium-binding affinity of non-mutated domains provide further support for domain cooperativity within CaM when bound to CaMKIIp. Results related to the domain cooperativity of calcium-binding site mutations are summarized in Fig. 9.
Figure 9.

Bar graphs showing the concentration of calcium required to reach 50% saturation (Ȳ0.5). Sites I and II of E31Q, E67Q (A) and E104Q, E140Q (C) and sites III and IV of E31Q, E67Q (B) and E104Q, E140Q (D) CaM1–148 with (dashed bars) and without (white bars) CaMKIIp.
Domain cooperativity was previously reported for viable Paramecium CaM mutants in which either the calcium-binding affinity or the target association properties were determined to be altered 65. One of the mutations in the C-domain (E104K) changed the calcium-binding affinity of the N-domain, whereas four others (D95G, S101F, E104K and H135R) affected the calcium-binding affinity of only the mutated C-domain 65.
Our study illustrates the difficulty in predicting binding energetics from structural information alone. Residues of the domains of CaM which are within 4.5 Å of the peptide are shown in Fig. 10A and Fig. 10C (contacts with the N- and the C-domain, respectively) based on the crystal structure of CaM in complex with the CaMBD of CaMKII (1CDM.pdb, 20). A computational analysis using Contacts of Structural Units (CSU) 66 determined that a total of 66 CaM residues contact CaMBD of CaMKIIp (28 in the N-domain and 38 in the C-domain) (Fig. 10B). The higher number of interacting residues in the C-domain of CaM correlates well with our association studies, which showed that CaM76–148 has a much higher affinity for CaMKIIp than does CaM1–80, both under apo and calcium-saturating conditions. A recent study comparing the thermodynamic energetics of CaM binding to 3 peptides representing CaMBD of CaMKII suggested that a single domain of CaM is sufficent for binding to CaMKII before ATP binds and autophosphorylation of Thr286 takes place. The highest affinity CaM-CaMKII complex was formed when both domains of CaM engaged with the CaMBD of CaMKII after autophosphorylation 36.
Figure 10.

Ribbon diagram of CaM bound to CaMBD of Ca2+/CaM-dependent kinase II (1CDM.pdb) 20. CaM residues in the N-domain (A) and C-domain (C) within 4.5 Å of CaMKIIp are shown in blue (N-domain) and red (C-domain) ball-and-stick; CaMKIIp T305 and T306 are shown as black spheres (made with PyMol, DeLano Scientific LLC). (B) Sequence map showing CaM residues that are within 4.5 Å of each residue of the CaMKII CaMBD peptide calculated with CSU66.
Conclusion
CaM participates in complex signal transduction pathways in which differential calcium-binding affinities of the domains of CaM regulate targets such as calcium channels 67, which in turn are regulated by CaMKII, a CaM-stimulated kinase. The studies reported here explored the domain-specific energetics of CaM binding to the fully exposed CaMBD of CaMKII. The binding of CaMKIIp to individual domains of CaM showed that the high-affinity of CaM1–148 for the kinase recognition sequence is not a simple sum of the affinities of the N- and C-domains under calcium-saturated conditions but include contributions from CaMKIIp-induced interdomain interactions. Thermodynamic linkage analysis showed that the dissociation constant of calcium-saturated CaM1–148 binding to CaMKIIp was as low as 50 pM. Studies with calcium-binding site knockout mutants of CaM suggest that in vivo, the C-domain of CaM may play a greater role in CaMKII activation; however, covalent linkage of both domains is necessary for the high affinity CaM-CaMKII complex formation.
CaMKIIp binding to CaM increased and equalized the calcium-binding affinity of both domains of CaM1–148. To determine how residues within the calcium-binding sites alter the CaMKIIp-induced changes of the other calcium-binding sites, we used single calcium-binding site knockout mutants of CaM. The calcium-binding site knockout mutation decreased the calcium-binding affinity of the non-mutated domains when CaM was bound to CaMKIIp. These results provide insight into linkage between the association of the domains of CaM with the CaMBD of CaMKII and the calcium-binding affinity of CaM. This lays the foundation to better understand the mechanism of attachment, in which it will be necessary to determine interactions of each CaM domain with subsets of the CaMKIIp sequence representing the partially exposed CaMBD. These will provide greater insight into the allosteric mechanisms of target protein regulation by CaM.
Acknowledgments
This study was supported by a Postdoctoral Training Fellowship from the Iowa Cardiovascular Center (T32 HL 07121-30) and a National Research Service Award from NIH (F32 GM 077927) to T.I.A.E. and a grant from NIH (R01 GM 57001) to M.A.S.
Abbreviations
- CaM1–148
Full-length mammalian calmodulin, residues 1–148
- CaM1–80
N-domain fragment of calmodulin, residues 1–80
- CaM76–148
C-domain fragment of calmodulin, residues 76–148
- CaMBD
Calmodulin binding domain
- CaMKII
Calcium/CaM dependent kinase II
- CaMKIIp
CaMBD of CaMKII, spanning residues 290 to 309
- EGTA
Ethylene glycol bis(aminoethyl ether)-N′,N′,N′,N′-tetraacetic acid
- Fl-CaMKIIp
CaMKIIp fluoresceinated at the N-terminus
- Ka
Association constant
- Kd
Dissociation constant
- NONLIN
Nonlinear least squares analysis
- NTA
Nitrilotriacetic acid
- pdb
Protein data bank
- Phe
Phenylalanine
- skMLCK
Skeletal myosin light chain kinase
- Tyr
Tyrosine
- WT
Wild type
References
- 1.Maune JF, Klee CB, Beckingham K. Ca2+ binding and conformational change in two series of point mutations to the individual Ca2+-binding sites of calmodulin. J Biol Chem. 1992;267:5286–5295. [PubMed] [Google Scholar]
- 2.Starovasnik MA, Su DR, Beckingham K, Klevit RE. A series of point mutations reveal interactions between the calcium-binding sites of calmodulin. Protein Sci. 1992;1(2):245–253. doi: 10.1002/pro.5560010206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black DJ, Tikunova SB, Johnson JD, Davis JP. Acid Pairs Increase the N-Terminal Ca2+ Affinity of CaM by Increasing the Rate of Ca2+ Association. Biochemistry. 2000;39(45):13831–13837. doi: 10.1021/bi001106+. [DOI] [PubMed] [Google Scholar]
- 4.Shea MA, Verhoeven AS, Pedigo S. Calcium-Induced Interactions of Calmodulin Domains Revealed by Quantitative Thrombin Footprinting of Arg37 and Arg106. Biochemistry. 1996;35:2943–2957. doi: 10.1021/bi951934g. [DOI] [PubMed] [Google Scholar]
- 5.Grabarek Z. Structure of a trapped intermediate of calmodulin: calcium regulation of EF-hand proteins from a new perspective. J Mol Biol. 2005;346(5):1351–1366. doi: 10.1016/j.jmb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Yuan T, Ouyang H, Vogel H. Surface exposure of the methionine side chains of calmodulin in solution. A nitroxide spin label and two-dimensional NMR study. J Biol Chem. 1999;274(13):8411–8420. doi: 10.1074/jbc.274.13.8411. [DOI] [PubMed] [Google Scholar]
- 7.LaPorte DC, Wierman BM, Storm DR. Calcium-induced exposure of a hydrophobic surface on calmodulin. Biochemistry. 1980;19:3814–3819. doi: 10.1021/bi00557a025. [DOI] [PubMed] [Google Scholar]
- 8.Yamniuk AP, Vogel HJ. Calmodulin’s flexibility allows for promiscuity in its interactions with target proteins and peptides. Mol Biotechnol. 2004;27:33–57. doi: 10.1385/MB:27:1:33. [DOI] [PubMed] [Google Scholar]
- 9.Vetter SW, Leclerc E. Novel aspects of calmodulin target recognition and activation. Eur J Biochem. 2003;270:404–414. doi: 10.1046/j.1432-1033.2003.03414.x. [DOI] [PubMed] [Google Scholar]
- 10.Rhoads AR, Friedberg F. Sequence motifs for calmodulin recognition. FASEB J. 1997;11(5):331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 11.Schumacher MA, Crum M, Miller MC. Crystal structures of apocalmodulin and an apocalmodulin/SK potassium channel gating domain complex. Structure. 2004;12:849–860. doi: 10.1016/j.str.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 12.Houdusse A, Gaucher JF, Krementsova E, Mui S, Trybus KM, Cohen C. Crystal structure of apo-calmodulin bound to the first two IQ motifs of myosin V reveals essential recognition features. Proc Natl Acad Sci U S A. 2006;103(51):19326–19331. doi: 10.1073/pnas.0609436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menetrey J, Bahloul A, Wells AL, Yengo CM, Morris CA, Sweeney HL, Houdusse A. The structure of the myosin VI motor reveals the mechanism of directionality reversal. Nature. 2005;435:779–785. doi: 10.1038/nature03592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schumacher MA, Rivard AF, Bachinger HP, Adelman JP. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 2001;410(6832):1120–1124. doi: 10.1038/35074145. [DOI] [PubMed] [Google Scholar]
- 15.Drum CL, Yan S, Bard J, Shen Y, LUD, Soelaiman S, Grabarek Z, Bohm A, Tang W. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature. 2002;415:396–402. doi: 10.1038/415396a. [DOI] [PubMed] [Google Scholar]
- 16.Aoyagi M, Arvai AS, Tainer JA, Getzoff ED. Structural basis for endothelial nitric oxide synthase binding to calmodulin. EMBO J. 2003;22(4):766–775. doi: 10.1093/emboj/cdg078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meador WE, Means AR, Quiocho FA. Target enzyme recognition by calmodulin: 2.4 Å Structure of a calmodulin-peptide complex. Science. 1992;257:1251–1255. doi: 10.1126/science.1519061. [DOI] [PubMed] [Google Scholar]
- 18.van Petegem F, Chatelain FC, Minor DL., Jr Insights into voltage-gated calcium channel regulation from the structure of the Ca(V)1.2 IQ domain-Ca2+/calmodulin complex. Nat Struct Mol Biol. 2005;12:1108–1115. doi: 10.1038/nsmb1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fallon JL, Halling DB, Hamilton SL, Quiocho FA. Structure of calmodulin bound to the hydrophobic IQ domain of the cardiac Cav1.2 calcium channel. Structure. 2005;13:1881–1886. doi: 10.1016/j.str.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 20.Meador WE, Means AR, Quiocho FA. Modulation of calmodulin plasticity in molecular recognition on the basis of X-ray structures. Science. 1993;262:1718–1721. doi: 10.1126/science.8259515. [DOI] [PubMed] [Google Scholar]
- 21.Yamauchi E, Nakatsu T, Matsubara M, Kato H, Taniguchi H. Crystal structure of a MARCKS peptide containing the calmodulin-binding domain complex with Ca2+-calmodulin. Nat Struct Biol. 2003;10(3):226–231. doi: 10.1038/nsb900. [DOI] [PubMed] [Google Scholar]
- 22.Clapperton JA, Martin SR, Smerdon SJ, Gamblin SJ, Bayley PM. Structure of the Complex of Calmodulin with the Target Sequences of Calmodulin-Dependent Protein Kinase I: Studies of the Kinase Activation Mechanism. Biochemistry. 2002;41:14669–14679. doi: 10.1021/bi026660t. [DOI] [PubMed] [Google Scholar]
- 23.Ataman ZA, Gakhar L, Sorensen BR, Hell JW, Shea MA. The NMDA Receptor NR1 C1 Region Bound to Calmodulin: Structural Insights into Functional Differences between Homologous Domains. Structure. 2007;15(12):1603–1617. doi: 10.1016/j.str.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crivici A, Ikura M. Molecular and Structural Basis of Target Recognition by Calmodulin. Annu Rev Biophys Biomol Struct. 1995;24:85–116. doi: 10.1146/annurev.bb.24.060195.000505. [DOI] [PubMed] [Google Scholar]
- 25.Ishida H, Vogel HJ. Protein-peptide interaction studies demonstrate the versatility of calmodulin target protein binding. Protein Pept Lett. 2006;13(5):455–465. doi: 10.2174/092986606776819600. [DOI] [PubMed] [Google Scholar]
- 26.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 27.Hudmon A, Schulman H. Structure–function of the multifunctional Ca2+/calmodulin- dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hudmon A, Schulman H. Neuronal Ca2+/calmodulin dependent protein kinase II: The role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- 29.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11(4):409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 30.Anderson ME. Calmodulin kinase signaling in heart: an intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol Ther. 2005;106(1):39–55. doi: 10.1016/j.pharmthera.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Gaertner TR, Kolodziej SJ, Wang D, Kobayashi R, Koomen JM, Stoops JK, Waxham MN. Comparative analyses of the three-dimensional structures and enzymatic properties of α, β, γ, and δ, isoforms of Ca2+-calmodulin--dependent protein kinase II. J Biol Chem. 2004;279(13):12484–12494. doi: 10.1074/jbc.M313597200. [DOI] [PubMed] [Google Scholar]
- 32.Rosenberg OS, Deindl S, Sung R, Nairn AC, Kuriyan J. Structure of the Autoinhibited Kinase Domain of CaMKII and SAXS Analysis of the Holoenzyme. Cell. 2005;12310(1016):849–860. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 33.Waxham MN, Aronowski J, Westgate SA, Kelly PT. Mutagenesis of Thr-286 in monomeric Ca2+/calmodulin-dependent protein kinase II eliminates Ca2+/calmodulin-independent activity. Proc Natl Acad Sci U S A. 1990;87(4):1273–1277. doi: 10.1073/pnas.87.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waxham MN, Tsai A-L, Putkey JA. A mechanism for calmodulin (CaM) trapping by CaM-kinase II defined by a family of CaM-binding peptides. J Biol Chem. 1998;273(28):17579–17584. doi: 10.1074/jbc.273.28.17579. [DOI] [PubMed] [Google Scholar]
- 35.Torok K, Tzortzopoulos A, Grabarek Z, Best SL, Thorogate R. Dual effect of ATP in the activation mechanism of brain Ca2+/calmodulin-dependent protein kinase II by Ca2+/calmodulin. Biochemistry. 2001;40(49):14878–14890. doi: 10.1021/bi010920+. [DOI] [PubMed] [Google Scholar]
- 36.Tse JK, Giannetti AM, Bradshaw JM. Thermodynamics of calmodulin trapping by Ca2+/calmodulin-dependent protein kinase II: subpicomolar Kd determined using competition titration calorimetry. Biochemistry. 2007;46(13):4017–4027. doi: 10.1021/bi700013y. [DOI] [PubMed] [Google Scholar]
- 37.Shifman JM, Choi MH, Mihalas S, Mayo SL, Kennedy MB. Ca2+/calmodulin-dependent protein kinase II (CaMKII) is activated by calmodulin with two bound calciums. Proc Natl Acad Sci U S A. 2006;103(38):13968–13973. doi: 10.1073/pnas.0606433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peersen OB, Madsen TS, Falke JJ. Intermolecular tuning of calmodulin by target peptides and proteins: differential effects on Ca2+ binding and implications for kinase activation. Protein Sci. 1997;6(4):794–807. doi: 10.1002/pro.5560060406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson JD, Snyder C, Walsh M, Flynn M. Effects of myosin light chain kinase and peptides on Ca2+ exchange with the N- and C-terminal Ca2+ binding sites of calmodulin. J Biol Chem. 1996;271:761–767. doi: 10.1074/jbc.271.2.761. [DOI] [PubMed] [Google Scholar]
- 40.Newman RA, VanScyoc WS, Sorensen BR, Jaren OR, Shea MA. Interdomain coopertivity of calmodulin to melittin preferentially increases calcium affinity of sites I and II. Proteins. 2008;71(4):1792–1812. doi: 10.1002/prot.21861. [DOI] [PubMed] [Google Scholar]
- 41.Sorensen BR, Faga LA, Hultman R, Shea MA. Interdomain linker increases thermostability and decreases calcium affinity of calmodulin N-domain. Biochemistry. 2002;41(1):15–20. doi: 10.1021/bi011718+. [DOI] [PubMed] [Google Scholar]
- 42.Sorensen BR, Shea MA. Interactions between domains of apo calmodulin alter calcium binding and stability. Biochemistry. 1998;37:4244–4253. doi: 10.1021/bi9718200. [DOI] [PubMed] [Google Scholar]
- 43.Pedigo S, Shea MA. Quantitative endoproteinase GluC footprinting of cooperative Ca2+ binding to calmodulin: Proteolytic susceptibility of E31 and E87 indicates interdomain interactions. Biochemistry. 1995;34:1179–1196. doi: 10.1021/bi00004a011. [DOI] [PubMed] [Google Scholar]
- 44.Putkey JA, Slaughter GR, Means AR. Bacterial expression and characterization of proteins derived from the chicken calmodulin cDNA and a calmodulin processed gene. J Biol Chem. 1985;260(8):4704–4712. [PubMed] [Google Scholar]
- 45.Beaven GH, Holiday ER. Ultraviolet absorbtion spectra of proteins and amino acids. Adv Protein Chem. 1952;7:319–386. doi: 10.1016/s0065-3233(08)60022-4. [DOI] [PubMed] [Google Scholar]
- 46.Johnson ML, Frasier SG. Nonlinear least-squares analysis. Methods Enzymol. 1985;117:301–342. [Google Scholar]
- 47.VanScyoc WS, Shea MA. Phenylalanine fluorescence studies of calcium binding to N-Domain fragments of Paramecium calmodulin mutants show increased calcium affinity correlates with increased disorder. Protein Sci. 2001;10(9):1758–1768. doi: 10.1110/ps.11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.VanScyoc WS, Sorensen BR, Rusinova E, Laws WR, Ross JB, Shea MA. Calcium binding to calmodulin mutants monitored by domain-specific intrinsic phenylalanine and tyrosine fluorescence. Biophys J. 2002;83(5):2767–2780. doi: 10.1016/S0006-3495(02)75286-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winzor DJ, Sawyer WH. Quantitative Characterization of Ligand Binding. New York, NY: Wiley-Liss; 1995. p. 168. [Google Scholar]
- 50.Kranz JK, Lee EK, Nairn AC, Wand AJ. A direct test of the reductionist approach to structural studies of calmodulin activity: relevance of peptide models of target proteins. J Biol Chem. 2002;277:16351–16354. doi: 10.1074/jbc.C200139200. [DOI] [PubMed] [Google Scholar]
- 51.Hanley RM, Means AR, Ono T, Kemp BE, Burgin KE, Waxham N, Kelly PT. Functional analysis of a complementary DNA for the 50-kilodalton subunit of calmodulin kinase II. Science. 1987;237(4812):293–297. doi: 10.1126/science.3037704. [DOI] [PubMed] [Google Scholar]
- 52.Persechini A, McMillan K, Leakey P. Activation of myosin light chain kinase and nitric oxide synthase activities by calmodulin fragments. J Biol Chem. 1994;269:16148–16154. [PubMed] [Google Scholar]
- 53.Forest A, Swulius MT, Tse JK, Bradshaw JM, Gaertner T, Waxham MN. Role of the N-and C-Lobes of Calmodulin in the Activation of Ca(2+)/Calmodulin-Dependent Protein Kinase II. Biochemistry. 2008 doi: 10.1021/bi8007033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olwin BB, Edelman AM, Krebs EG, Storm DR. Quantitation of energy coupling between Ca2+, calmodulin, skeletal muscle myosin light chain kinase, and kinase substrates. J Biol Chem. 1984;259(17):10949–10955. [PubMed] [Google Scholar]
- 55.Olwin BB, Storm DR. Calcium Binding to Complexes of Calmodulin and Calmodulin Binding Proteins. Biochemistry. 1985;24:8081–8086. doi: 10.1021/bi00348a037. [DOI] [PubMed] [Google Scholar]
- 56.Kim J, Ghosh S, Liu H, Tateyama M, Kass RS, Pitt GS. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004;279(43):45004–45012. doi: 10.1074/jbc.M407286200. [DOI] [PubMed] [Google Scholar]
- 57.Theoharis NT, Sorensen BR, Theisen-Toupal J, Shea MA. The Neuronal Voltage-Dependent Sodium Channel Type II IQ Motif Lowers the Calcium Affinity of the C-Domain of Calmodulin. Biochemistry. 2008;47(1):112–123. doi: 10.1021/bi7013129. [DOI] [PubMed] [Google Scholar]
- 58.Evenas J, Thulin E, Malmendal A, Forsen S, Carlstrom G. NMR studies of the E140Q mutant of the carboxy-terminal domain of calmodulin reveal global conformational exchange in the Ca2+-saturated state. Biochemistry. 1997;36:3448–3457. doi: 10.1021/bi9628275. [DOI] [PubMed] [Google Scholar]
- 59.Evenas J, Malmendal A, Thulin E, Carlstrom G, Forsen S. Ca2+ Binding and Conformational Changes in a Calmdoulin Domain. Biochemistry. 1998;37(39):13744–13754. doi: 10.1021/bi9806448. [DOI] [PubMed] [Google Scholar]
- 60.Mukherjea P, Maune JF, Beckingham K. Interlobe communication in multiple calcium-binding site mutants of Drosophila calmodulin. Protein Sci. 1996;5:468–477. doi: 10.1002/pro.5560050308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beckingham K. Use of site-directed mutations in the individual Ca2+-binding sites of calmodulin to examine Ca2+-induced conformational changes. J Biol Chem. 1991;266:6027–6030. [PubMed] [Google Scholar]
- 62.Findlay WA, Martin SR, Beckingham K, Bayley PM. Recovery of native structure by calcium binding site mutants of calmodulin upon binding of sk-MLCK target peptides. Biochemistry. 1995;34:2087–2094. doi: 10.1021/bi00007a001. [DOI] [PubMed] [Google Scholar]
- 63.Haiech J, Kilhoffer M-C, Lukas TJ, Craig TA, Roberts DM, Watterson DM. Restoration of the calcium binding activity of mutant calmodulins toward normal by the presence of a calmodulin binding structure. J Biol Chem. 1991;266:3427–3431. [PubMed] [Google Scholar]
- 64.Gao ZH, Krebs J, VanBerkum MFA, Tang W-J, Maune JF, Means AR, Stull JT, Beckingham K. Activation of four enzymes by two series of calmodulin mutants with point mutations in individual Ca2+ binding sites. J Biol Chem. 1993;268:20096–20104. [PubMed] [Google Scholar]
- 65.VanScyoc WS, Newman RA, Sorensen BR, Shea MA. Calcium Binding by Calmodulin Mutants Having Domain-Specific Effects on Regulation of Ion Channels. Biochemistry. 2006;45(48):14311–14324. doi: 10.1021/bi061134d. [DOI] [PubMed] [Google Scholar]
- 66.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15(4):327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 67.Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39(6):951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- 68.Babu YS, Bugg CE, Cook WJ. Structure of calmodulin refined at 2.2 Å resolution. J Mol Biol. 1988;204:191–204. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]