

Most Kunitz proteins like BPTI and α-dendrotoxin are stabilized by three disulfide bonds. The crystal structure shows how subtle repacking of non-covalent interactions may compensate for disulfide bond loss in a naturally occurring two-disulfide variant, conkunitzin-S1, the first discovered member of a new conotoxin family.
Keywords: conotoxin, BPTI, α-dendroxin, native chemical ligation, conus
Abstract
Cone snails (Conus) are predatory marine mollusks that immobilize prey with venom containing 50–200 neurotoxic polypeptides. Most of these polypeptides are small disulfide-rich conotoxins that can be classified into families according to their respective ion-channel targets and patterns of cysteine–cysteine disulfides. Conkunitzin-S1, a potassium-channel pore-blocking toxin isolated from C. striatus venom, is a member of a newly defined conotoxin family with sequence homology to Kunitz-fold proteins such as α-dendrotoxin and bovine pancreatic trypsin inhibitor (BPTI). While conkunitzin-S1 and α-dendrotoxin are 42% identical in amino-acid sequence, conkunitzin-S1 has only four of the six cysteines normally found in Kunitz proteins. Here, the crystal structure of conkunitzin-S1 is reported. Conkunitzin-S1 adopts the canonical 310–β–β–α Kunitz fold complete with additional distinguishing structural features including two completely buried water molecules. The crystal structure, although completely consistent with previously reported NMR distance restraints, provides a greater degree of precision for atomic coordinates, especially for S atoms and buried solvent molecules. The region normally cross-linked by cysteines II and IV in other Kunitz proteins retains a network of hydrogen bonds and van der Waals interactions comparable to those found in α-dendrotoxin and BPTI. In conkunitzin-S1, glycine occupies the sequence position normally reserved for cysteine II and the special steric properties of glycine allow additional van der Waals contacts with the glutamine residue substituting for cysteine IV. Evolution has thus defrayed the cost of losing a disulfide bond by augmenting and optimizing weaker yet nonetheless effective non-covalent interactions.
1. Introduction
Cone snails of the genus Conus are predatory marine molluscs that include vermivorous, molluscivorous and piscivorous species (Terlau & Olivera, 2004 ▶). Evolution of fish-hunting behavior in snails is a remarkable feat considering that snails move slowly and do not swim. Cone snails rapidly immobilize their prey by injection with venom containing approximately 50–200 neurotoxic polypeptides. A majority of these polypeptides are small disulfide-rich conotoxins. Each conotoxin is synthesized as a precursor protein which matures through proteolysis and chemical modifications including C-terminal amidation and oxidation of cysteines. The number and pattern of cysteine–cysteine cross-links are features that define a given conotoxin family (Terlau & Olivera, 2004 ▶). Conotoxin families are further defined by their molecular targets. For instance, α-conotoxins recognize, bind and inhibit nicotinic acetylcholine receptors (McIntosh et al., 1999 ▶) and ω-conotoxins are specific for Ca2+ channels (Olivera et al., 1994 ▶).
The discovery of conkunitzin-S1, a potassium-channel inhibitor isolated from the venom of C. striatus, brought to light a new conotoxin family (J. S. Imperial, unpublished results; Bayrhuber et al., 2005 ▶). Members of this conkunitzin family are homologous to Kunitz proteins such as α-dendrotoxin and bovine pancreatic trypsin inhibitor (BPTI; Fig. 1 ▶). Conkunitzin-S1 shares 42% sequence identity with α-dendrotoxin and has all of the highly conserved amino acids such as Gly14, Phe35, Gly39, Asn45 and Phe47 found in Kunitz-folded proteins1. Most Kunitz proteins are stabilized by specific intramolecular disulfide bridges. Indeed, the six cysteines involved in these disulfide cross-links and the spacing between cysteine residues are defining characteristics of Kunitz proteins. A few Kunitz proteins, however, make do with only four cysteines and such is the case for conkunitzin-S1. In conkunitzin-S1, glycine and glutamine occupy sequence positions normally reserved for CysII and CysIV, posing interesting questions related to the structure and evolution of the conkunitzin family.
Figure 1.

Amino-acid sequence alignment of conkunitizin-S1 and other Kunitz proteins and domains. Highly conserved positions are shaded. Disulfide connections are shown with either a solid line or, in the case of the CysII–CysIV disulfide, a broken line. Conkunitzin-S1, the focus of this work, is a two-disulfide Kunitz-fold conotoxin isolated from C. striatus venom. Two-disulfide variants selected for comparison are cow TK-3 (amino acids 122–172 of trophoblast Kunitz domain protein-3 from cow) and Ixolaris (amino acids 101–151 of a tissue-factor pathway inhibitor from the tick Ixodes scapularis). Selected Kunitz folds with three disulfides that were superimposed to make the ensemble search model are as follows: α-dendrotoxin (a K+-channel-blocking toxin from the green mamba Dendroaspis angusticeps), collagen type VI (the C5 domain of human type VI collagen α-3 chain), TFPI-2 (amino acids 121–178 of the human tissue-factor pathway inhibitor) and BPTI (bovine pancreatic trypsin inhibitor, the first described Kunitz protein; Kunitz & Northrop, 1936 ▶).
In an effort to address these questions, we have characterized intermediates encountered during oxidative folding and elucidated the crystal structure of conkunitzin-S1. Total chemical synthesis of conkunitzin-S1 was achieved through native chemical ligation. Oxidative folding of conkunitzin-S1 proceeded efficiently to give a native and functional protein in high yield. The crystal structure of conkunitzin-S1 was determined in space group P1 to a 2.45 Å resolution limit. This X-ray crystal structure complements a solution NMR structure of conkunitzin-S1 (Bayrhuber et al., 2005 ▶). The crystal structure is more precise, however, and provides a clear view of the positions of disulfide-bonded S atoms and solvent molecules, structural details that are not readily measurable by NMR. As expected from sequence comparisons and in agreement with the NMR-determined model, crystalline conkunitzin-S1 folds as a canonical 310–β–β–α Kunitz protein. Considering the prominent role disulfides have on protein folding a less obvious result is that conkunitzin-S1 retains several conserved attractive interactions including hydrogen bonds and van der Waals contacts in the region normally cross-linked by a disulfide bond between CysII and CysIV, even though these cysteines are not present in conkunitzin-S1 (Fig. 1 ▶). Replacement of CysII with glycine, a residue with unique steric properties, allows additional van der Waals contacts as a result of modest repacking, thereby apparently recouping some of the energetic cost incurred by loss of a disulfide bond.
2. Materials and methods
2.1. Total chemical synthesis and oxidative folding of conkunitzin-S1
Two peptide fragments comprising Lys1–Gln31 (fragment 1) and Cys32–Thr60 (fragment 2) of conkunitzin-S1 were synthesized separately using standard Fmoc chemistry. Fragment 1 was synthesized on a 4-sulfamylbutyryl AM resin to produce upon cleavage a thioester linking the carboxylic acid of Gln31 with an S-CH2-COOC2H5 group (Shin et al., 1999 ▶; Ingenito et al., 1999 ▶). Fragment 2 was synthesized on a Wang resin to produce upon cleavage an amino-terminal cysteine and a free C-terminal carboxylic acid. The peptides were purified by reverse-phase HPLC on a C18 column (Vydac) using linear gradients of buffer A (0.1% trifluoroacetic acid; TFA) and buffer B (90% acetonitrile in 0.1% TFA). Experimental details were similar to those previously described for the synthesis of δ-conotoxin–PVIIA with its propeptide through native chemical ligation (Buczek et al., 2005 ▶). During chemical ligation a cysteine residue at the N-terminus of fragment 2 exchanges with the C-terminal thioester of fragment 1 and the resulting intermediate rearranges to yield a native peptide bond linking Gln31 and Cys32. Progress of the ligation reaction was monitored by HPLC (Fig. 2 ▶ a). Fractions were analyzed by MALDI–TOF on a Bruker Daltonics OmniFLEX mass spectrometer using α-cyano-4-hydroxycinnamic acid as matrix and the product-containing fractions were combined, lyophilized and used for folding reactions. The resulting linear polypeptide has a measured average weight of 6934.43 Da, which corresponds closely to the predicted weight (MH+) of 6934.66 Da.
Figure 2.

Synthesis, oxidative folding and disulfide mapping of conkunitzin-S1. (a) HPLC analysis of polypeptide fragments before chemical ligation (top trace) and after 10 h of chemical ligation (bottom trace). As ligation proceeded, peptides corresponding to amino acids 1–31 (aa 1–31) and 32–60 (aa 32–60) were covalently linked by a peptide bond to form the linear conkunitzin-S1 polypeptide (aa 1–60). The species labeled aa 1–31* is an intermediate encountered after transthioesterification of aa 1–31 with thiophenol. (b) HPLC analysis of intermediates encountered during oxidative folding of conkunitzin-S1. The fully reduced polypeptide (Linear, top trace) folded via single-disulfide (IA, IB, IC, ID) and non-native double-disulfide (IIA, IIB) intermediates to form one predominant fully oxidized product (Native, bottom trace). (c) Disulfide mapping confirms I–VI and III–V disulfide linkages in natively folded conkunitzin-S1. Peptide fragments were identified by mass spectrometry (Table 1 ▶). Peptides in peak b are related to peptides in peak a through the removal of two residues (NS) from the NSARKQCLRF peptide. Peptides in peak d are related to peptides in peak e through the removal of six N-terminal residues (KDRPSL).
Folding reactions were carried out in folding buffer (0.1 M Tris–HCl pH 8.7, 1 mM EDTA, 0.5 mM reduced glutathione and 0.25 mM oxidized glutathione). Analytical folding reactions were initiated by adding 10 µl 100 µM reduced linear peptide dissolved in 0.01% TFA to 40 µl folding buffer. Folding reactions were quenched by the addition of 5 µl formic acid and were analyzed by reverse-phase HPLC with a linear 40 min gradient from 20 to 24% buffer B at 318 K. Peaks were collected and analyzed by MALDI–TOF MS. Mass spectrometry indicated the loss of two or four protons upon formation of one-disulfide and non-native two-disulfide species. Species predominant in early folding steps correspond to the peaks labeled IA, IB, IC and ID in Fig. 2 ▶(b). The measured average weight of each species was observed to be 6932.17, 6932.07, 6932.53 and 6932.31 Da, all of which are within experimental uncertainty of the weight of 6932.66 Da (MH+) expected for a one-disulfide species. Two additional species were detected later in folding reactions (peaks labeled IIA and IIB in Fig. 2 ▶ b) and the measured average weights of these species were 6930.80 and 6930.12 Da, consistent with non-native two-disulfide species. Folding intermediates rapidly converted to one major product: the natively folded two-disulfide linked protein. Natively folded conkunitizin-S1 had a measured weight of 6930.41 Da, consistent with the loss of four protons relative to the reduced unfolded peptide.
2.2. Verification of disulfide connectivity and bioactivity
Verification of disulfide-bond connectivity in oxidized conkunitzin-S1 was accomplished by proteolytic cleavage and analysis using reverse-phase HPLC and MALDI–TOF mass spectrometry. In the first step, 10 nmol conkunitzin-S1 was digested with immobilized pepsin (Pierce Biotechnology) in digest buffer (20 mM sodium acetate pH 3.5) for 1 h at 310 K, yielding small peptide fragments and one species which retained both disulfide bonds (peak e of Fig. 2 ▶ c). The molecular weight of the two-disulfide proteolysis product was 4549.74 Da and corresponded to that expected for three peptide fragments linked by two disulfides. These three fragments were amino acids 1–19, 26–35 and 48–58, with a total calculated mono-protonated molecular weight of 4549.23 Da (see also Table 1 ▶). In the second step, to obtain species with single disulfide linkages the product corresponding to peak e of Fig. 2 ▶(c) was collected and treated with pepsin for an additional 6 h at 310 K. Additional species were identified upon separation by HPLC and mass spectrometry (Fig. 2 ▶ c and Table 1 ▶). Three of these corresponded to species with two fragments joined by a single disulfide bond either linking CysIII and CysV (peaks a and b) or linking CysI and CysVI (peak c). These results confirm that the two disulfides of conkunitzin-S1 bridge CysI and CysVI and CysIII and CysV exactly as seen in canonical Kunitz proteins.
Table 1. Molecular weight of conkunitzin-S1 pepsin-digestion products.
Conkunitzin-S1 is neurotoxic (J. S. Imperial, unpublished results; Bayrhuber et al., 2005 ▶). To test whether synthetic conkunitzin-S1 exhibits neurotoxic activity, synthesized and oxidatively folded conkunitzin-S1 was administered to male CF-1 mice (26–35 g, Charles River Laboratories). Conkunitzin-S1 dissolved in normal saline (0.9% NaCl), when administered to mice by intracerebroventricular injection, produced spastic running followed by tonic extension seizures. Five doses (0.1, 0.3, 1, 2 and 3 nmol per mouse) of conkunitzin-S1 with 3–5 mice per dose group were used for ED50 determination. Seizure activity was dose-dependent, with ED50 = 1 nmol per mouse [95% confidence limits using probit analysis (Litchfield, 1949 ▶) were 0.3–2 nmol].
2.3. Crystallization
In preparation for crystallization, conkunitzin-S1 was washed repeatedly with 0.1% acetic acid and lyophilized, followed by repeated washing with water to remove TFA carried over with HPLC solvents. Washed conkunitzin-S1 was dissolved in water to a concentration of 15 mg ml−1. Crystallization experiments were carried out at 277 and 291 K using the hanging-drop vapor-diffusion method. Each drop contained equal volumes of protein and reservoir solution. Reservoir solution contained 0.1 M acetate buffered to pH 4–5, 2% PEG 400, 0.02%(w/v) sodium azide and (NH4)2SO4 at various concentrations between 1.5 and 3.0 M. Crystals appeared within a period of several weeks. Most of the crystalline material consisted of clusters of needles or clusters of plates, but isolated plate-shaped crystals were obtained in some reactions. Crystals were briefly soaked in a cryoprotectant solution containing 30%(w/v) ethylene glycol before flash-freezing in liquid propane at 100 K.
2.4. X-ray data collection and processing
X-ray data extending to the 2.45 Å resolution limit were collected at 100 K from a single crystal using a rotating copper-anode X-ray source and a CCD detector (Nonius). Data were indexed, scaled and processed using DENZO and SCALEPACK as implemented in the HKL-2000 interface (Otwinowski & Minor, 1997 ▶). The data indexed in space group P1 and Matthews coefficient probability distributions (Matthews, 1968 ▶; Kantardjieff & Rupp, 2003 ▶) indicated that six molecules of conkunitzin-S1 are likely to have occupied the asymmetric unit. Alternative space groups, including a C2 space group and a different P1 space group with a smaller volume, were considered and eliminated on the basis of merging statistics or failure at subsequent stages of refinement. Data-collection and processing statistics are listed in Table 2 ▶.
Table 2. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | P1 |
| Unit-cell parameters (Å, °) | a = 50.76, b = 51.54, c = 51.60, α = 119.9, β = 107.5, γ = 91.1 |
| Resolution limits (Å) | 20–2.45 (2.54–2.45) |
| Rcryst† (%) | 9.8 (36.5) |
| I/σ(I) | 9.1 (3.8) |
| Completeness (%) | 99.0 (97.8) |
| Total reflections | 60461 |
| Independent reflections | 15238 |
| Refinement | |
| R‡ (%) | 22.1 |
| Rfree‡ (%) | 25.5 |
| No. of atoms | 2821 |
| No. of waters | 52 |
| No. of sulfates | 13 |
| Average B values (Å2) | 23.9 |
| Protein atoms | 23.5 |
| Solvent atoms | 20.7 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.0080 |
| Bond angles (°) | 1.3 |
| Ramachandran statistics | |
| Most favored (%) | 88.3 |
| Additionally favored (%) | 11.7 |
| Generously allowed (%) | 0.0 |
| Disallowed (%) | 0.0 |
R
cryst = 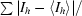
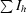 .
.
R and R
free = 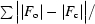
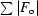 , where F
o and F
c are the observed and calculated structure-factor amplitudes. R
free was calculated with 8% of reflections not used in maximum-likelihood refinement.
, where F
o and F
c are the observed and calculated structure-factor amplitudes. R
free was calculated with 8% of reflections not used in maximum-likelihood refinement.
2.5. Structure determination and refinement
The coordinates for the crystal structure of conkunitzin-S1 (PDB code 1y62) were submitted ahead of the NMR solution structure (PDB code 1yl2). The NMR structure was neither used as a search model during molecular replacement nor at any time during refinement of the crystal structure reported here.
The structure of conkunitzin-S1 was determined by molecular replacement using an ensemble of four previously determined Kunitz-fold crystal structures as a search model. Single-protein search models consisted of the Kunitz-fold proteins or domains from PDB codes 1dtx, 1tfx, 2knt and 2ptc, with N-terminal residues preceding the first cysteine and C-terminal residues following the final cysteine omitted. Cross-rotation and translation functions were implemented in CNS (Brünger et al., 1998 ▶) using data between 20 and 4 Å resolution. Initial B values were estimated on the basis of Wilson scaling. Peaks in the rotation function were sorted by correlation coefficient and directed multiple rounds of translation. Intermediate results and final solutions were evaluated by packing quality, correlation coefficient, R and R free values.
Refinement was carried out with cycles of energy minimization, torsion-angle simulated annealing and temperature-factor optimization using maximum-likelihood target functions implemented in CNS (Brünger et al., 1998 ▶). Each round of refinement alternated with a round of model building using the program O (Jones et al., 1991 ▶). A simulated-annealing 2|F o| − |F c| OMIT map was generated by removing atoms corresponding to 8% of the asymmetric unit, subjecting the resulting structure to torsion-angle simulated annealing, using the resulting phases to calculate a map for the omitted volume and repeating this process to sample the entire asymmetric unit. As phases improved, ordered solvent molecules were added, including waters and sulfate ions. The final structure included six molecules of conkunitzin-S1 refined without non-crystallographic symmetry (NCS) restraints. Two N-terminal residues and two C-terminal residues from each conkunitzin-S1 molecule were omitted owing to lack of electron density. R and R free values converged to values of 22.1 and 25.5%, respectively (Table 2 ▶).
2.6. Comparison of X-ray and NMR structures
R.m.s. deviations for atomic coordinates of main-chain atoms (N, CA and C) were calculated for pairwise comparisons to the average structure or to a representative structure using CNS (Brünger et al., 1998 ▶). Energies related to NOE distance restraints deposited with the NMR structure (PDB code 1yl2) were computed with CNS using a biharmonic potential, T = 300 K and an inverse sixth-power distance weighting scheme.
H atoms were added using the atom-building algorithm of CNS for the purpose of computing NOE restraint energies. NOE restraints were not used during the atom-building procedure. Of the original 659 NOE restraints, 624 restraints that met the following two criteria were included in the energy analysis: (i) the restraint is among residues 3–58 included in both NMR and crystal structures and (ii) the restraint does not refer to stereochemically ambiguous H atoms. Including the ambiguous restraints (e.g. methyl H atoms) did not change the conclusion of the analysis.
2.7. Sequence comparisons
Amino-acid sequences corresponding to the Kunitz domain found in trophoblast Kunitz proteins and the two domains from selected serine protease inhibitor proteins were compared with CLUSTAL-X (Thompson et al., 1997 ▶). Phylogenetic trees were constructed and tested by neighbor-joining, most-parsimonious and maximum-likelihood methods implemented in PHYLIP (Felsenstein, 2004 ▶).
3. Results
3.1. Overall structure of conkunitzin-S1
The crystal structure reveals conkunitzin-S1 folded with 310–β–β–α secondary-structure elements (Fig. 3 ▶ a) as seen in other Kunitz protein structures. The two disulfides of conkunitzin-S1 were readily visible, with 5σ peaks in the simulated-annealing 2|F o| − |F c| OMIT maps (Fig. 4 ▶ a). These electron-density peaks are consistent with the disulfide connectivity expected for a Kunitz fold (Fig. 1 ▶) and as verified in this work by proteolytic cleavage and mass spectrometry (Fig. 2 ▶ c, Table 1 ▶). Conkunitzin-S1 is most similar to the Kunitz fold of α-dendrotoxin, with a main-chain r.m.s.d. of 0.80 Å.
Figure 3.

Conkunitzin-S1 crystal structure overview and comparison with NMR-determined models. (a) Conkunitzin-S1 folds into the 310–β–β–α fold characteristic of Kunitz domains. S atoms in each of two disulfide bonds are shown as orange spheres. Two water molecules, shown as green spheres, are completely buried inside conkunitzin-S1. The secondary-structure elements are colored with the 310-helix blue, β-strands cyan and α-helix red. (b) Cα superposition of the six conkunitzin-S1 molecules found in the asymmetric unit. (c) Cα superposition of the six unique NMR-ensemble members currently available (PDB code 1yl2). In (b) and (c) disulfide bonds are shown with orange lines, selected residues are labeled with Arabic numerals and cysteines are labeled with Roman numerals. (d) R.m.s. deviations of main-chain atoms (N, CA and C) are plotted as a function of residue number for crystal (top panel) and NMR models (bottom panel). (e) Energies computed for individual NOE distance restraints are plotted as a bar graph, with blue and orange bars indicating the energies computed for representative X-ray-determined and NMR-determined structures. (a), (b) and (c) were generated using PyMOL (DeLano, 2002 ▶).
Figure 4.

Electron-density maps. (a) Electron density defining positions of disulfide-bonded S atoms and buried solvent molecules is shown contoured at 4.6σ for sulfur and 2.2σ for water. (b) Electron density surrounding loops II and IV of conkunitzin-S1 is shown in stereo. The map is a simulated-annealed 2|F o| − |F c| OMIT map calculated to the 2.45 Å resolution limit and contoured at 1.2σ. N atoms are blue, O atoms are red and C atoms are gray. This figure was produced using Bobscript (Kraulis, 1991 ▶; Esnouf, 1999 ▶) and Raster3D (Merritt & Murphy, 1994 ▶).
Since conkunitzin-S1 has only two of the three disulfides normally stabilizing Kunitz proteins, we were especially interested to know if the structure adopts several conformations as a consequence of missing a disulfide bond. Ideally, questions regarding protein dynamics and conformational exchange are best addressed by NMR relaxation experiments; however, refined B-value parameters should be approximate indicators of conformational heterogeneity if it exists in the crystal structure. The average B value for all conkunitzin-S1 protein atoms is 24 Å2 (Table 2 ▶), with values ranging above 35 Å2 only for Arg3, the first N-terminal residue visible in electron-density maps. Profiles of B values with respect to sequence position (not shown) indicate that residues 4–57, including those regions cross-linked by a third disulfide bond in other Kunitz proteins, assume well determined positions in conkunitzin-S1. On the basis of these low B values, the structure of conkunitzin-S1 does not appear to be prone to conformational heterogeneity despite having an incomplete complement of disulfide bonds.
3.2. Ensemble search model for molecular replacement
The structure of conkunitzin-S1 was solved using molecular replacement despite several challenges. Six molecules of conkunitzin-S1 occupied the P1 asymmetric unit, meaning that a correctly placed and completely accurate model would represent only 16% of the scattering mass. The self-rotation function was uninformative. Compounding this lack of internal symmetry, previously determined Kunitz protein structures exhibit greater than 1 Å positional variation in certain regions. Various single Kunitz proteins failed as search models in molecular-replacement trials. A similar difficulty was noted for α-dendrotoxin, where attempts to solve the structure via molecular replacement failed (Skarzynski, 1992 ▶).
Molecular replacement was successful when the search model consisted of an ensemble of four previously determined Kunitz protein crystal structures. Difficult molecular-replacement cases such as that encountered here have previously been solved with X-ray-derived (Kleywegt et al., 1994 ▶) or NMR-derived ensembles (Chen & Clore, 2000 ▶; Pauptit et al., 2001 ▶).
For the ensemble search model, correlation coefficients, unit-cell packing and R values improved in a stepwise manner for each of six translation searches, but this was not the case for single-protein search models. A structure consisting of six translated ensembles yielded R and R free values of 49.3 and 48.0%, respectively, whereas structures consisting of translated single-protein search models did not achieve R values below 51%. Although these are small differences, crystal packing corroborated the ensemble-generated model and excluded the single-protein-generated models on the basis of steric overlap.
The most convincing evidence that the ensemble search model had succeeded came from 2|F o| − |F c| electron-density maps. Residues omitted from the ensemble search model were clearly seen in these initial electron-density maps. Model bias is a concern whenever solving structures with molecular replacement (Adams et al., 1999 ▶; Terwilliger, 2004 ▶); however, we are convinced of the structure’s correctness on the basis of excellent refinement statistics (Table 2 ▶), the emergence of new structural information in 2|F o| − |F c| electron-density maps during the course of refinement and the high quality of simulated-annealing 2|F o| − |F c| OMIT maps (e.g. Fig. 4 ▶).
3.3. Comparison of X-ray and NMR structures
The overall fold determined by X-ray crystallography agrees with the conclusion of an NMR solution structure study that showed conkunzin-S1 to be a two-disulfide variant of the Kunitz fold (Bayrhuber et al., 2005 ▶). The NMR-ensemble model members are similar to but not exactly the same as the structure refined using X-ray diffraction data. The r.m.s. deviations comparing NMR-ensemble members superimposed with one representative example of crystalline conkunitzin-S1 range from 0.8 to 1.5 Å, comparable to values reported for other NMR and X-ray structure comparisons at similar experimental resolution (Garbuzynskiy et al., 2005 ▶).
Figs. 3 ▶(b) and 3 ▶(c) show Cα traces for superimposed crystal structures and NMR solution ensemble members, respectively. The crystal’s asymmetric unit comprised six molecules, which were independently refined without NCS restraints. The average r.m.s.d. for main-chain atoms among the six conkunitzin-S1 molecules is 0.2 Å, a value close to the Luzzati coordinate error of 0.3 Å. The NMR-determined structure consists of an ensemble of six energy-minimized models (PDB code 1yl2) with an average r.m.s.d. for main-chain atoms among ensemble members of 0.6 Å. Thus, atomic coordinates are somewhat more precisely determined by X-ray crystallography than by NMR distance restraints (Fig. 3 ▶ d). This has important implications for the structural details described in the following sections. For instance, the crystal structure reveals conserved buried water molecules. In the NMR structure these waters are absent and the corresponding volume is occupied by Asn43. Additionally, the conformations of disulfide-linked cysteine residues (compare Figs. 3 ▶ b and 3 ▶ c) are more reliably determined by X-ray diffraction data.
Differences in atomic coordinate precision could in theory arise from real differences in structure and atomic flexibility for solution and crystalline phases. If crystallization perturbs the structure or range of accessible protein conformations, then the crystal structure would be expected to show systematic inconsistencies with NMR-determined distance restraints. To test this idea, H atoms were added to the crystal structure and the distances between these H atoms were compared with NOE distance restraints. Interestingly, the distances computed for the crystal structure were on average characterized by similar or lower NOE distance restraint energies relative to those of NMR-ensemble members (Fig. 3 ▶ e). On the basis of these energy comparisons, we conclude that the structure of conkunitzin-S1 is not greatly perturbed by crystallization and that apparent positional variation among NMR-ensemble members reflects uncertainty in position arising from mathematical treatment of the data.
3.4. Buried waters
The structure of conkunitzin-S1 includes two buried waters (Fig. 4 ▶ a). Buried waters at analogous positions are also seen in α-dendrotoxin and BPTI. In the case of BPTI, which has been extensively studied by NMR, these buried waters exchange with bulk solvent on the microsecond time scale coupled with local unfolding and refolding events (Denisov et al., 1995 ▶, 1996 ▶). In conkunitzin-S1 the absence of a direct route from the outside to the positions of these waters indicates that the waters associate with their hydrogen-bonding partners during the process of folding. The residues bridged through hydrogen bonds with the buried waters have some of the lowest r.m.s. deviations (Fig. 3 ▶ d) and B values. The average B value of the buried waters (13 Å2) is also remarkably low. In other Kunitz proteins, these buried waters and their hydrogen-bonding partners are likewise associated with lower than average B values. The conserved nature of these waters and their characteristically low B values suggest that buried waters are integral structural elements that stabilize conkunitzin-S1.
3.5. Structural consequences of disulfide-bond ‘loss’
Gly16 and Glu40 occupy positions normally reserved for CysII and CysIV in canonical Kunitz proteins (Fig. 1 ▶). To understand how these residues are accommodated in conkunitzin-S1, the loop regions containing Gly16 and Gln40 were explored in greater detail (Figs. 4 ▶ b and 5 ▶ a). For simplicity, we will refer to the loop comprising residues 13–17 in conkunitzin-S1 and α-dendrotoxin as loop II and the loop comprising residues 37–41 as loop IV.
Figure 5.

Hydrogen-bonding network of loops II and IV is conserved. (a) Despite the lack of a third disulfide in conkunitzin-S1, hydrogen bonding (indicated by yellow dashes) is strikingly similar to that observed in α-dendrotoxin (b) and BPTI (c), two proteins that retain the disulfide (orange). A buried water molecule (red sphere) helps to organize the hydrogen-bonding network in BPTI (c).
Comparison of loop II and loop IV in conkuntizin-S1 with those in α-dendrotoxin and BPTI (Fig. 5 ▶) show that attractive interactions involving a hydrogen-bonding network are preserved in conkunitzin-S1 despite the lack of a disulfide. The hydrogen-bonding network in conkunitizin-S1 consists of hydrogen bonds between backbone atoms in loops II and IV and hydrogen bonds between the hydroxyl group of Thr38 and backbone atoms of loop II. Packing interactions involving a π–π stack and van der Waals contacts are also preserved (Fig. 4 ▶ b). At position 37 a conserved aromatic residue, often tyrosine but sometimes tryptophan (Fig. 1 ▶), makes a shelf that supports the peptide linking residues 39 and 40 of loop IV (Fig. 4 ▶ b). In conkunitzin-S1, the β carbon of Thr38 packs closely with the α carbon of Gly16, similar to what is seen in α-dendrotoxin, where the β carbon of Ser38 packs closely with the α carbon of Cys16.
Compared with the situation in previously determined Kunitz crystal structures, loops II and IV of conkunitzin-S1 are slightly closer to each other as a consequence of modest repacking made possible by the special steric qualities of glycine at position 16 and the lack of distance restraints imposed by a covalent disulfide bond. The α carbon of Gly16 is sandwiched between Thr38 and Gln40, making van der Waals contacts with the β carbons of these two residues. NOE distance restraints and the NMR-ensemble model (Bayrhuber et al., 2005 ▶) corroborate these van der Waals contacts deduced independently from the crystal structure. A larger amino acid would likely not fit into this closely spaced environment. In canonical Kunitz proteins, such close-packing interactions are prevented since the disulfide bond linking CysII and CysIV hold these residues slightly farther apart. From the observed closely packed structure of loops II and IV, we speculate that non-covalent interactions are optimized in conkunitzin-S1 to partially compensate for the lack of a covalent bond.
4. Discussion
4.1. Pore-blocking strategy
Conkunitzin-S1 is a potassium ion-channel pore-blocking toxin, as demonstrated by electrophysiology experiments (Bayrhuber et al., 2005 ▶), and the crystal structure provides clues as to which residues are important for activity. Pore-blocking activity was retained for a conkunitzin-S1 variant with a CysII–CysIV disulfide engineered through substitution at Gly16 and Gln40 (Bayrhuber et al., 2005 ▶), meaning that loss of the CysII–CysIV disulfide is not an innovation necessary for biological function. Other potent K+-channel pore-blocking toxins generally occlude the K+ ion pore by inserting a particular lysine residue, which is often in close proximity to a hydrophobic residue, a motif termed the ‘functional dyad’ (Dauplais et al., 1997 ▶; Mouhat et al., 2004 ▶). A dyad pore-blocking strategy is used in the context of several different protein folds such as the snake Kunitz protein α-dendrotoxin (Gasparini et al., 1998 ▶), Conus κ-PVIIA (Savarin et al., 1998 ▶; Scanlon et al., 1997 ▶), the scorpion toxin charybdotoxin (Goldstein & Miller, 1993 ▶; Goldstein et al., 1994 ▶), the spider toxin cobatoxin-1 (Jouirou et al., 2004 ▶) and the sea anemone toxin BgK (Gilquin et al., 2002 ▶). In the case of the K+-channel blocker conotoxin κM-RIIIK from C. radiatus, however, several positively charged groups presented by two arginines, a lysine and the amino-terminus work cooperatively to block the channel without a lysine–hydrophobe dyad (Al-Sabi et al., 2004 ▶; Verdier et al., 2005 ▶). In the crystal structure of conkunitzin-S1, positive electrostatic potential is concentrated on one side of the molecule (not shown) and there is apparently no dyad-like lysine–hydrophobe pair. Based on these structural observations, conkunitzin-S1 is likely to operate similarly to κM-RIIIK using a combination of positively charged residues to block K+ channels.
4.2. Disulfide bonds in the Kunitz family
The canonical Kunitz fold is stabilized by three highly conserved disulfides (Fig. 1 ▶). However, two-disulfide variants similar to conkunitzin-S1 exist and these are widely distributed among many animals including worms, ticks, midges, mosquitoes, cows, sheep and Conus marine snails. Examination of the protein-family sequence database (Bateman et al., 2004 ▶) reveals that the most commonly substituted disulfide involves CysII and CysIV which connect residues 16 and 40 in α-dendrotoxin (Fig. 5 ▶ b) and which are missing in conkunitzin-S1 (Fig. 3 ▶ and Fig. 5 ▶ a).
Disulfides stabilize a protein fold by reducing the chain entropy of the unfolded state and the removal of a disulfide generally impacts on protein folding and stability. Of the three disulfides encountered in Kunitz proteins, the CysI–CysVI and CysIII–CysV disulfides are buried within the protein core while the CysII–CysIV disulfide is relatively solvent-exposed (Wlodawer, Deisenhofer et al., 1987 ▶; Wlodawer, Nachman et al., 1987 ▶; Skarzynski, 1992 ▶). This solvent-accessible II–IV disulfide forms last in the preferred pathway for oxidative folding of BPTI (Creighton & Goldenberg, 1984 ▶). The II–IV disulfide of BPTI is reduced by mild agents such as sodium borohydride and the resulting two-disulfide form retains trypsin inhibition activity (Kress & Laskowski, 1967 ▶), indicating an intact native-like structure. Variants of BPTI engineered with alternate residues substituting for CysII and CysIV can fold properly in vitro (Marks et al., 1987 ▶), albeit with altered rate constants under mildly oxidizing conditions (Goldenberg, 1988 ▶). Consistent with the idea that the II–IV disulfide is dispensable for Kunitz protein folding and stability, conkunitzin-S1 folds efficiently and cooperatively with only transient accumulation of single-disulfide and non-native two-disulfide intermediates (Fig. 2 ▶ b).
Loss of the II–IV disulfide in Kunitz-folded proteins seems to have occurred several times in the course of evolution, with examples in cows and sheep (MacLean et al., 2004 ▶), blood-sucking insects (Francischetti et al., 2002 ▶; Monteiro et al., 2005 ▶; Francischetti et al., 2004 ▶), nematodes and Conus snails. A phylogenetic tree constructed for the trophoblast Kunitz domain proteins (Fig. 6 ▶), a set that includes three members from cows and sheep missing CysII and CysIV, suggests that an ancestral protein had three disulfides. Independent lineages experiencing loss of CysII and CysIV can also be inferred through sequence comparisons of the numerous proteins found in nematodes and ticks (data not shown).
Figure 6.

Phylogenetic analysis showing relations among selected Kunitz domains. The domains are derived from the two-Kunitz-domain serine protease inhibitors (SPI), which all retain three disulfides, and from the trophoblast Kunitz (TK) domain proteins found in cow and sheep. Proteins from sea anemone and from the egg white of a marine turtle provide outgroups. (a) Alignment of the 22 Kunitz domains with conserved sites shaded. Proteins with a bullet point appended to their names have only two disulfides. (b) Dendrogram showing the consensus tree derived from neighbor-joining, most-parsimonious and maximum-likelihood analyses. Numbers indicate bootstrap percentages from 1000 resampled data sets analyzed by maximum parsimony or, in parentheses, by the neighbor-joining method. Nodes preceding the two-disulfide variants are consistently associated with ancestral three-disulfide proteins at the 95% confidence limit.
Of naturally occurring two-disulfide variants, most have glycine at the position normally reserved for CysII. This pattern of CysII-to-Gly substitution was also observed in genetic screens selecting for folding competent revertants of a mutant BPTI with only one disulfide from either random (Hagihara & Kim, 2002 ▶) or site-directed libraries (Hagihara et al., 2002 ▶). Glycine is not thought of as a conservative substitute for cysteine and certainly cannot form a covalent disulfide bond. Our work provides a structural rationale for why glycine is the best substitution for CysII. Because glycine is the smallest and most conformationally unrestrained amino acid, glycine can maximize hydrogen bonds and attractive van der Waals contacts in the loop II–loop IV region of conkunitzin-S1. Thus, evolution has devised a strategy to substitute a covalent disulfide bond with weaker but nevertheless effective non-covalent interactions.
A solution structure of conkunitzin-S1 determined by NMR confirms that conkunitzin-S1 is a well folded rigid protein with excellent chemical shift dispersion (Bayrhuber et al., 2005 ▶), a surprising result given the profound effect that disulfide elimination has on small disulfide-rich conotoxins (Price-Carter et al., 1998 ▶; Kaerner & Rabenstein, 1999 ▶; Sun et al., 2002 ▶; Flinn et al., 1999 ▶). On the basis of these conotoxin folding studies and prior to publication of the NMR conkunitzin-S1 structure and determination of its crystal structure, we had anticipated that loops II and IV of conkunitzin-S1 would either be disordered or stabilized by several compensatory attractive interactions not seen in three-disulfide Kunitz proteins. The crystal structure shows that neither is the case; loops II and IV are only slightly different owing to modest repacking of this region. The consequences following loss of a disulfide bond are apparently context-dependent. Studies of BPTI stability and folding (Creighton & Goldenberg, 1984 ▶; Creighton, 1986 ▶) are consistent with this finding. Preservation of structure in the face of disulfide-bond loss is expected at solvent-exposed sites, such as the II–IV disulfide of Kunitz proteins, where the disulfide bond works in concert with weak but numerous non-covalent interactions that are collectively sufficient to define protein structure.
Supplementary Material
PDB reference: conkunitzin-S1, 1y62, r1y62sf
Acknowledgments
We thank B. M. Olivera for his generous support of this work, D. P. Goldenberg, J. Seger and F. R. Adler for helpful discussions and encouragement, I. Ota, R. S. Orr and C. P. Hill for critical reading of the manuscript during preparation and R. T. Layer for performing the bioassays. The work was funded through University of Utah startup funds to MPH and an NIH grant (GM48677, Conus Peptides and Their Receptor Targets) to B. M. Olivera.
Footnotes
Residues are numbered according to the sequence of α-dendrotoxin and conkunitzin-S1. BPTI is shorter by two residues at the amino-terminus, meaning that corresponding positions in BPTI can be derived by subtracting 2.
References
- Adams, P. D., Pannu, N. S., Read, R. J. & Brunger, A. T. (1999). Acta Cryst. D55, 181–190. [DOI] [PubMed] [Google Scholar]
- Al-Sabi, A., Lennartz, D., Ferber, M., Gulyas, J., Rivier, J. E., Olivera, B. M., Carlomagno, T. & Terlau, H. (2004). Biochemistry, 43, 8625–8635. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R. D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E. L., Studholme, D. J., Yeats, C. & Eddy, S. R. (2004). Nucleic Acids Res.32, D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayrhuber, M., Vijayan, V., Ferber, M., Graf, R., Korukottu, J., Imperial, J., Garrett, J. E., Olivera, B. M., Terlau, H., Zweckstetter, M. & Becker, S. (2005). J. Biol. Chem.280, 23766–23770. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Buczek, P., Buczek, O. & Bulaj, G. (2005). Biopolymers, 80, 50–57. [DOI] [PubMed] [Google Scholar]
- Chen, Y. W. & Clore, G. M. (2000). Acta Cryst. D56, 1535–1540. [DOI] [PubMed] [Google Scholar]
- Creighton, T. E. (1986). Methods Enzymol.131, 83–106. [DOI] [PubMed] [Google Scholar]
- Creighton, T. E. & Goldenberg, D. P. (1984). J. Mol. Biol.179, 497–526. [DOI] [PubMed] [Google Scholar]
- Dauplais, M., Lecoq, A., Song, J., Cotton, J., Jamin, N., Gilquin, B., Roumestand, C., Vita, C., de Medeiros, C. L., Rowan, E. G., Harvey, A. L. & Menez, A. (1997). J. Biol. Chem.272, 4302–4309. [DOI] [PubMed] [Google Scholar]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific.
- Denisov, V. P., Halle, B., Peters, J. & Horlein, H. D. (1995). Biochemistry, 34, 9046–9051. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., Peters, J., Horlein, H. D. & Halle, B. (1996). Nature Struct. Biol.3, 505–509. [DOI] [PubMed] [Google Scholar]
- Esnouf, R. M. (1999). Acta Cryst. D55, 938–940. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J. (2004). PHYLIP: Phylogeny Inference Package v. 3.6. http://evolution.genetics.washington.edu/phylip/phylipweb.html.
- Flinn, J. P., Pallaghy, P. K., Lew, M. J., Murphy, R., Angus, J. A. & Norton, R. S. (1999). Biochim. Biophys. Acta, 1434, 177–190. [DOI] [PubMed] [Google Scholar]
- Francischetti, I. M., Mather, T. N. & Ribeiro, J. M. (2004). Thromb. Haemost.91, 886–898. [DOI] [PubMed] [Google Scholar]
- Francischetti, I. M., Valenzuela, J. G., Andersen, J. F., Mather, T. N. & Ribeiro, J. M. (2002). Blood, 99, 3602–3612. [DOI] [PubMed] [Google Scholar]
- Garbuzynskiy, S. O., Melnik, B. S., Lobanov, M. Y., Finkelstein, A. V. & Galzitskaya, O. V. (2005). Proteins, 60, 139–147. [DOI] [PubMed] [Google Scholar]
- Gasparini, S., Danse, J. M., Lecoq, A., Pinkasfeld, S., Zinn-Justin, S., Young, L. C., de Medeiros, C. C., Rowan, E. G., Harvey, A. L. & Menez, A. (1998). J. Biol. Chem.273, 25393–25403. [DOI] [PubMed] [Google Scholar]
- Gilquin, B., Racape, J., Wrisch, A., Visan, V., Lecoq, A., Grissmer, S., Menez, A. & Gasparini, S. (2002). J. Biol. Chem.277, 37406–37413. [DOI] [PubMed] [Google Scholar]
- Goldenberg, D. P. (1988). Biochemistry, 27, 2481–2489. [DOI] [PubMed] [Google Scholar]
- Goldstein, S. A. & Miller, C. (1993). Biophys. J.65, 1613–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, S. A., Pheasant, D. J. & Miller, C. (1994). Neuron, 12, 1377–1388. [DOI] [PubMed] [Google Scholar]
- Hagihara, Y. & Kim, P. S. (2002). Proc. Natl Acad. Sci. USA, 99, 6619–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagihara, Y., Shiraki, K., Nakamura, T., Uegaki, K., Takagi, M., Imanaka, T. & Yumoto, N. (2002). J. Biol. Chem.277, 51043–51048. [DOI] [PubMed] [Google Scholar]
- Ingenito, R., Bianchi, E., Fattori, D. & Pessi, A. (1999). J. Am. Chem. Soc.121, 11369–11374.
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Jouirou, B., Mosbah, A., Visan, V., Grissmer, S., M’Barek, S., Fajloun, Z., Van Rietschoten, J., Devaux, C., Rochat, H., Lippens, G., El Ayeb, M., De Waard, M., Mabrouk, K. & Sabatier, J. M. (2004). Biochem. J.377, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaerner, A. & Rabenstein, D. L. (1999). Biochemistry, 38, 5459–5470. [DOI] [PubMed] [Google Scholar]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci.12, 1865–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt, G. J., Bergfors, T., Senn, H., Le Motte, P., Gsell, B., Shudo, K. & Jones, T. A. (1994). Structure, 2, 1241–1258. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. (1991). J. Appl. Cryst.24, 946–950. [Google Scholar]
- Kress, L. F. & Laskowski, M. Sr (1967). J. Biol. Chem.242, 4925–4929. [PubMed] [Google Scholar]
- Kunitz, M. & Northrop, J. H. (1936). J. Gen. Physiol.19, 991–1007. [DOI] [PMC free article] [PubMed]
- Litchfield, J. T. Jr (1949). J. Pharmacol. Exp. Ther.97, 399–408. [PubMed] [Google Scholar]
- McIntosh, J. M., Santos, A. D. & Olivera, B. M. (1999). Annu. Rev. Biochem.68, 59–88. [DOI] [PubMed] [Google Scholar]
- MacLean, J. A. II, Roberts, R. M. & Green, J. A. (2004). Biol. Reprod.71, 455–463. [DOI] [PubMed] [Google Scholar]
- Marks, C. B., Naderi, H., Kosen, P. A., Kuntz, I. D. & Anderson, S. (1987). Science, 235, 1370–1373. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Merritt, E. A. & Murphy, M. E. (1994). Acta Cryst. D50, 869–873. [DOI] [PubMed] [Google Scholar]
- Monteiro, R. Q., Rezaie, A. R., Ribeiro, J. M. & Francischetti, I. M. (2005). Biochem. J.387, 871–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouhat, S., Jouirou, B., Mosbah, A., De Waard, M. & Sabatier, J. M. (2004). Biochem. J.378, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera, B. M., Miljanich, G. P., Ramachandran, J. & Adams, M. E. (1994). Annu. Rev. Biochem.63, 823–867. [DOI] [PubMed] [Google Scholar]
- Otwinowski, A. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pauptit, R. A., Dennis, C. A., Derbyshire, D. J., Breeze, A. L., Weston, S. A., Rowsell, S. & Murshudov, G. N. (2001). Acta Cryst. D57, 1397–1404. [DOI] [PubMed] [Google Scholar]
- Price-Carter, M., Hull, M. S. & Goldenberg, D. P. (1998). Biochemistry, 37, 9851–9861. [DOI] [PubMed] [Google Scholar]
- Savarin, P., Guenneugues, M., Gilquin, B., Lamthanh, H., Gasparini, S., Zinn-Justin, S. & Menez, A. (1998). Biochemistry, 37, 5407–5416. [DOI] [PubMed] [Google Scholar]
- Scanlon, M. J., Naranjo, D., Thomas, L., Alewood, P. F., Lewis, R. J. & Craik, D. J. (1997). Structure, 5, 1585–1597. [DOI] [PubMed] [Google Scholar]
- Shin, Y., Winans, K. A., Backes, B. J., Kent, S. B. H., Ellman, J. A. & Bertozzi, C. R. (1999). J. Am. Chem. Soc.121, 11684–11689.
- Skarzynski, T. (1992). J. Mol. Biol.224, 671–683. [DOI] [PubMed] [Google Scholar]
- Sun, Y. M., Liu, W., Zhu, R. H., Goudet, C., Tytgat, J. & Wang, D. C. (2002). J. Pept. Res.60, 247–256. [DOI] [PubMed] [Google Scholar]
- Terlau, H. & Olivera, B. M. (2004). Physiol. Rev.84, 41–68. [DOI] [PubMed] [Google Scholar]
- Terwilliger, T. C. (2004). Acta Cryst. D60, 2144–2149. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. (1997). Nucleic Acids Res.25, 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdier, L., Al-Sabi, A., Rivier, J. E., Olivera, B. M., Terlau, H. & Carlomagno, T. (2005). J. Biol. Chem.280, 21246–21255. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A., Deisenhofer, J. & Huber, R. (1987). J. Mol. Biol.193, 145–156. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A., Nachman, J., Gilliland, G. L., Gallagher, W. & Woodward, C. (1987). J. Mol. Biol.198, 469–480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: conkunitzin-S1, 1y62, r1y62sf