

Abstract
Activation of angiotensin II type 2 receptors (AT2R) causes the release of kinins, which have beneficial effects on the cardiovascular system. However, it is not clear how AT2R interact with the kallikrein-kinin system to generate kinins. Prolylcarboxypeptidase is an endothelial membrane-bound plasma prekallikrein activator that converts plasma prekallikrein to kallikrein, leading to generation of bradykinin from high-molecular-weight kininogen. We hypothesized that AT2R-induced bradykinin release is at least in part mediated by activation of prolylcarboxypeptidase. Cultures of mouse coronary artery endothelial cells were transfected with an adenoviral vector containing the AT2R gene (Ad-AT2R) or green fluorescent protein only (Ad-GFP) as control. We found that overexpression of AT2R increased prolylcarboxypeptidase mRNA by 1.7 -fold and protein 2.5 -fold compared to Ad-GFP controls. AT2R overexpression had no effect on AT1 receptor mRNA. Bradykinin release was increased 2.2 -fold in AT2R-transfected cells. Activation of AT2R by CGP42112A, a specific AT2R agonist, increased bradykinin further in AT2R-transfected cells. These effects were diminished or abolished by AT2R blockade or a plasma kallikrein inhibitor. Furthermore, blocking prolylcarboxypeptidase with a siRNA partially but significantly reduced bradykinin release by transfected AT2R cells either at the basal condition or when stimulated by the AT2R agonist CGP42112A. These findings suggest that overexpression of AT2R in mouse coronary artery endothelial cells increases expression of prolylcarboxypeptidase, which may contribute to kinin release.
Keywords: prolylcarboxypeptidase, angiotensin II type 2 receptor, bradykinin, coronary artery endothelial cells, plasma kallikrein-kinin system
INTRODUCTION
Angiotensin II (Ang II) exerts important biological functions through two main subtypes of receptors: type 1 (AT1R) and type 2 (AT2R). The AT1R is ubiquitous and abundant in adult tissues, whereas the expression of AT2R is high in the fetus but low in adult tissues.1 AT2R appears to be upregulated in pathological conditions such as myocardial infarction (MI)2 and vascular injury.3 It is well known that AT1R mediates the effects of Ang II on blood pressure, water and sodium intake, renal sodium retention, secretion of vasopressin and aldosterone and cell growth and proliferation.4 However, the role of AT2R is less clear. Although it is currently thought that the actions of AT2R oppose those of AT1R,5 the exact mechanisms by which the AT2R elicits its cardioprotective effects are not well understood.
There is evidence that some beneficial effects of AT2R are mediated by the bradykinin/nitric oxide (NO) system.6,7 Tsutsumi et al8 demonstrated that AT2R overexpression in the vasculature stimulates the kinin system and causes dilatation, presumably by activating kininogenase. In addition, cardiac AT2R overexpression attenuates Ang II- or MI-induced fibrotic responses, which are reportedly mediated via a kinin/NO dependent mechanism.9,10
Kinins are released from high- and low-molecular-weight kininogen by two key kininogenases, plasma kallikrein and tissue kallikrein.11 Prolylcarboxypeptidase (PRCP, also called angiotensinase C) was initially described as an Ang II-inactivating enzyme.12,13 More recently it has been recognized as a plasma prekallikrein activator in endothelial cells (ECs)14–17 and is important for maintenance of EC function.18 When the complex of high-molecular-weight kininogen and plasma prekallikrein binds to the endothelial cell membrane, plasma prekallikrein is rapidly converted to kallikrein by PRCP.19 Plasma kallikrein then cleaves high-molecular-weight kininogen to liberate kinins, which act on constitutive B2 and inducible B1 receptors to stimulate the release of NO and prostacyclin. Shariat-Madar et al20 and Zhao et al21 have shown that PRCP overexpression enhances plasma prekallikrein activation and release of kinins and NO by cultured Chinese hamster ovary cells and ECs, and that these effects can be blocked by a small interfering RNA (siRNA) to PRCP, confirming the role of PRCP in kinin release. In addition, upregulation of PRCP expression in endothelial cells causes increased kallikrein generation and sustained production of bradykinin in lipopolysaccharide-induced inflammation.18 The AT1R antagonist losartan reportedly increased PRCP expression in hypertensive rats.22 We have demonstrated that the cardioprotective effect of AT1R antagonists is mediated in part by activation of AT2R.23 Also, in B2 receptor knockout mice the therapeutic effect of AT1R antagonists was diminished, indicating a link between AT2R and kinins.24 However, the precise mechanism by which AT2R mediates kinin release is not yet known and the role of PRCP in AT2R- stimulated kinins release has not been explored.
In the present study, we used an adenoviral vector system to overexpress AT2R in mouse coronary artery endothelial cells. Using this system, we tested the hypothesis that AT2R-induced bradykinin release in ECs is mediated by PRCP.
MATERIALS AND METHODS
Endothelial Cell Cultures
Coronary artery endothelial cells (ECs) were isolated from 10-week-old male C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME) as described previously.25 The study was approved by the Henry Ford Health System Institutional Animal Care and Use Committee in accord with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. ECs were maintained in medium 199-F-12 (1:1), 1% penicillin/streptomycin (Invitrogen), 100 μg/ml endothelial cell growth supplement (ECGS, Sigma) and 10% fetal bovine serum (FBS) (HyClone). All experiments were conducted with cells between passages 3 and 5.
Identification of ECs
ECs were recognized by their uptake of acetylated low-density lipoprotein labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (Dil-Ac-LDL)26 (Biomedical Technologies) (Fig. 1A) and immunostaining with an antibody to von Willebrand factor (vWF, a specific marker for ECs) (Fig. 1B)27 (for details see Materials and Methods in the online Data Supplement at http://hyper.ahajournals.org).
Fig. 1.

Characterization of mouse coronary artery endothelial cells (ECs). A: Uptake of Dil-labeled acetylated low-density lipoprotein (Ac-LDL; red fluorescence); magnification 200x. B: Immunostaining with an antibody to von Willebrand factor (vWF; green fluorescence). 4′,6-diamidino-2-phenylindole (DAPI) was used to indicate the nuclei (blue fluorescence); magnification 400x.
AT2R Gene Transfer to ECs
An adenoviral vector containing genomic AT2R DNA and a green fluorescent protein gene (Ad-AT2R-GFP) driven by a cytomegalovirus promoter (CMV) was used to induce overexpression of AT2R in ECs.28 Cells were transfected with three doses of Ad-AT2R at 40, 80 and 160 multiplicity of infection (MOI) for 24 hrs. The number of cells expressing green fluorescent protein was used to determine efficiency of Ad-AT2R transduction. ECs transfected with an adenoviral vector containing only GFP (Ad-GFP) was served as a viral control.
Reverse-Transcription Polymerase Chain Reaction
Total RNA was extracted from GFP control- or AT2R-transfected ECs using an RNeasy kit (Qiagen). RT-PCR was performed using a two-step protocol. RNA (0.2 μg) was reverse-transcribed into cDNA using an Omniscript Kit (Qiagen), and mRNA levels of AT2R, AT1R and PRCP were determined by real-time PCR using ABI 7500. Primer sequences and the detailed protocol were presented in the Materials and Methods in the online Data Supplement at http://hyper.ahajournals.org.
Small Interfering RNA
The PRCP siRNA and control scrambled siRNA were purchased from Santa Cruz (cat# sc-60171) and Qiagen (cat# 1027310), respectively. The siRNA was transfected into ECs in 6-well plates using 7.5 μl lipofectamine 2000 (Invitrogen). It was diluted in 250 μl Opti-MEM I and the mixture was incubated for 5 min at room temperature. During the incubation period, 15 μl siRNA (10 μM) was mixed with 250 μl Opti-MEM I and incubated for 25 min at room temperature to allow complex to form, after which 500 μl of the siRNA-lipofectamine was added to each well.
Western Blot
Cells were harvested in cell lysis buffer with protease inhibitors as described previously.29 Lysates were centrifuged at 14,000g for 10 min to remove insoluble material. Antibodies and the detailed protocol were presented in Materials and Methods in the online Data Supplement at http://hyper.ahajournals.org.
Measurement of Bradykinin
Bradykinin production was assessed by measuring the amount of bradykinin released into the incubation medium by ECs using EIA kit (details see Materials and Methods in the online Data Supplement at http://hyper.ahajournals.org). Bradykinin concentration was calculated according to a calibration curve, normalized to total protein and expressed as pg/μg protein.
Experimental Protocols
Cells were incubated overnight in 6-well plates with medium 199-F-12 (1:1) containing 0.5% FBS and switched to the same medium containing 10 μM captopril for 2 hrs before the experiments. Experimental protocols were:
To determine the effect of AT2R overexpression on basal bradykinin release, Ad-AT2R cells were either left untreated or treated with the AT2R antagonist PD123319 (100 μM) or the AT1R antagonist valsartan (1 μM) for 2.5 hrs. Ad-GFP cells were treated with culture medium alone and served as controls. 1 ml of the medium was collected in a test tube containing the peptidase inhibitor cocktail for bradykinin measurement.
To determine the effect of AT2R activation on bradykinin release, Ad-AT2R cells were treated with the AT2R agonist CGP42112A (0.1 μM) for 2 hrs. To determine the effect of blockade of AT2R on AT2R activation-induced bradykinin release, Ad-AT2R cells were pretreated with AT2R antagonist PD123319 (100 μM) for 30 minutes and then stimulated with CGP42112A (0.1 μM) for additional 2 hrs. Medium was collected for bradykinin measurement as described above.
To determine whether AT2R-induced bradykinin release is mediated via PRCP-dependent prekallikrein activation, Ad-AT2R cells were treated with 1 μM soybean trypsin inhibitor (SBTI, a plasma kallikrein inhibitor) for 30 minutes and then either left untreated or treated with the AT2R agonist CGP42112A (0.1 μM) for 2 hrs. Medium was then collected for bradykinin measurement.
To determine the effect of PRCP blockade on AT2R-stimulated bradykinin release, Ad-AT2R cells were treated with either a scrambled siRNA or PRCP siRNA for 48 hrs as described above and then subjected to the same experiments as in protocol 2.
Statistical Analysis
Results are expressed as mean ± SEM. Student’s two-sample t-test was used to compare the differences between two groups. Bonferroni’s adjustment was applied in multiple comparisons. A difference was considered significant if the adjusted p-value is less than 0.05.
RESULTS
Adenovirus-Mediated Overexpression of AT2R
Since the Ad-AT2R vector contains the GFP gene, the fact that cells expressed green fluorescence indicated that AT2R was successfully introduced. Fig. 2A illustrated AT2R expression following Ad-AT2R transfection at MOIs of 40, 80 and 160. Dose-dependent AT2R mRNA expression was determined by real-time RT-PCR (Fig. 2B) using GAPDH as an internal control. Endogenous AT2R was undetectable in Ad-GFP-transfected cells at a cycle of 21 but could be seen when the cycle was increased to 35 and conversely, in Ad-AT2R-transfected cells AT2R transgene mRNA was detected at a cycle of 21 and expression increased at higher MOIs: compared to MOI 40, AT2R mRNA increased 3.53 ± 0.16-fold at MOI 80 and 6.95 ± 0.49-fold at MOI 160 (Fig. 2B).
Fig. 2.
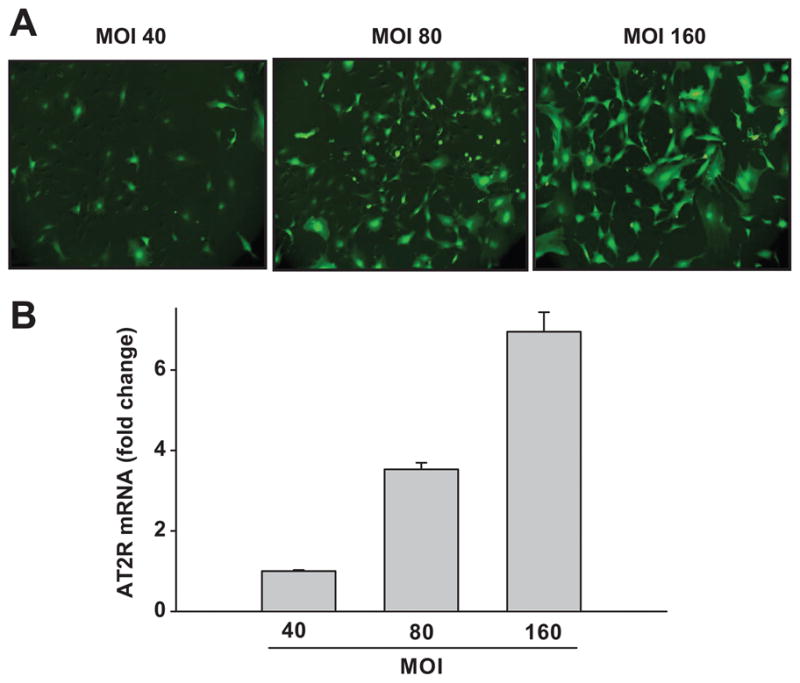
Adenovirus-induced angiotensin II type 2 receptor (Ad-AT2R) transfection in ECs. A: Representative images taken 24 hrs after transfection of Ad-AT2R (green fluorescence) at a multiplicity of infection (MOI) of 40, 80 or 160 (magnification 100x). B: Quantitative evaluation of AT2R mRNA expression in Ad-AT2R-transfected cells by real-time PCR. GAPDH was used as an internal control. Data are shown as fold change relative to MOI 40, n = 3.
To find out whether AT2R overexpression affects AT1R gene expression, we compared AT1R mRNA expression in Ad-GFP- and Ad-AT2R-transfected cells. We found that at 40 or 80 MOI there was no significant difference in AT1R mRNA expression between Ad-AT2R-and Ad-GFP-transfected cells (Fig. 3), suggesting that AT2R overexpression at these titers does not influence endogenous AT1R expression. However, AT1R mRNA was elevated at the highest MOI (160), and for this reason we used MOI 80 in subsequent experiments to study the role of AT2R without the confounding effects of the AT1R.
Fig. 3.

Effect of Ad-AT2R transfection on AT1R mRNA expression. Cells were transfected with Ad-GFP or Ad-AT2R at MOI 40, 80 or 160 for 24 hrs. mRNA levels were evaluated by real-time PCR and normalized to GAPDH. Data are shown as fold change relative to Ad-GFP-transfected cells at MOI 40; n = 3.
Effect of AT2R Overexpression on PRCP mRNA and Protein Expression
Compared with Ad-GFP, Ad-AT2R transfection increased PRCP protein expression 2.5 ± 0.32-fold (Fig. 4A and B). In addition, Ad-AT2R cells had a 1.7 ± 0.16-fold increase in PRCP mRNA compared with Ad-GFP cells (Fig. 4C). Both PRCP mRNA and protein levels were normalized to GAPDH, which did not differ significantly between Ad-GFP- and Ad-AT2R-transfected cells. These findings indicate that PRCP is upregulated by AT2R overexpression.
Fig. 4.

Effect of AT2R overexpression on PRCP protein and mRNA expression. Cells were transfected with Ad-GFP or Ad-AT2R at MOI 80 for 24 hrs. A: Representative Western blots showing the increase in PRCP protein expression induced by Ad-AT2R transfection. B. Quantitative evaluation of PRCP protein expression normalized to GAPDH. C. PRCP mRNA expression evaluated by real-time PCR and normalized to GAPDH. Data are shown as fold change relative to Ad-GFP-transfected cells, n = 3–4.
Effect of AT2R Overexpression on Bradykinin Release
Basal bradykinin release in GFP-transfected cells was 2.1 ± 0.2 pg/μg; overexpression of AT2R increased it to 4.6 ± 0.3 pg/μg, which was 2.2-fold higher than GFP-cells (Fig.5). Blocking AT2R with PD123319 reduced bradykinin levels to 2.9 ± 0.3 pg/μg, but blocking AT1R with valsartan had no effect on bradykinin release in Ad-AT2 cells (4.8 ± 0.4 pg/μg).
Fig. 5.

Effect of AT2R overexpression on bradykinin release. Ad-AT2R- transfected cells (MOI 80) were either left untreated or treated with the AT2R antagonist PD123319 (PD, 100 μM) or the AT1R antagonist valsartan (Val, 1 μM). Ad-GFP cells (MOI 80) served as controls. Bradykinin concentration was normalized to total protein and expressed as pg/μg protein. n = 5–18. NS: no statistical significance.
We next studied the effect of AT2R activation on bradykinin release. Stimulation with the AT2R agonist CGP42112A increased bradykinin levels to 6.7 ± 0.5 pg/μg in AT2R cells (Fig. 6), and this increase was significantly blocked by the AT2R antagonist PD123319 (3.4 ± 0.4 pg/μg). Taken together, these data provide strong evidence that AT2R overexpression induces bradykinin release from ECs and this increase is AT2R-specific, since the AT2R agonist CGP42112A enhanced bradykinin release, whereas the AT2R antagonist PD123319 suppressed it.
Fig. 6.

Effect of the AT2R agonist CGP42112A (CGP) on bradykinin release. Ad-AT2R-transfected cells (MOI 80) were treated with CGP (0.1 μM) with or without the AT2R antagonist PD123319 (PD, 100 μM). Ad-GFP cells (MOI 80) served as controls. n = 9–18.
Effect of Plasma Kallikrein Inhibition on Bradykinin Release
Soybean trypsin inhibitor (SBTI) has been reported to block plasma kallikrein.30 In GFP-transfected cells, SBTI reduced bradykinin to an undetectable level (data not shown). In Ad-AT2R cells SBTI significantly reduced bradykinin release from 4.6 ± 0.3 to 1.1 ± 0.5 pg/μg. Activation of AT2R with CGP42112A enhanced bradykinin release, which was reduced from 6.7 ± 0.5 to 1.8 ± 1.2 pg/μg by SBTI (Fig. 7). These data indicate that cleavage of high-molecular-weight kininogen by plasma kallikrein contributes to AT2R-induced bradykinin release by ECs.
Fig. 7.

Effect of soybean trypsin inhibitor (SBTI) on bradykinin release. Ad-AT2R- transfected cells (MOI 80) were treated with SBTI (1 μM) with or without the AT2R agonist CGP42112A (CGP, 0.1 μM). Ad-GFP cells (MOI 80) served as controls. n = 4–18.
Effect of Blocking PRCP on Bradykinin Release
Currently no PRCP-specific inhibitor is available. We therefore used a siRNA to investigate whether blockade of PRCP in AT2R-transfected cells affects bradykinin release. We found that PRCP-siRNA reduced PRCP protein expression by 84% compared with scrambled-siRNA controls (Fig. 8A). Furthermore, blockade of PRCP using a siRNA reduced basal bradykinin release by 35% and CGP42112A-stimulated bradykinin release by 49% in AT2R-transfected cells compared with scrambled siRNA controls (Fig. 8B). These data suggest that AT2R-induced bradykinin release is mediated at least in part by PRCP.
Fig. 8.

Effect of prolylcarboxypeptidase (PRCP) blockade by siRNA on bradykinin release in Ad-AT2R-transfected ECs. A: Top panel: Representative Western blot showing PRCP protein expression. Lower panel: Quantitative data. B: fold change in bradykinin release relative to Ad-GFP-transfected cells. n = 4.
DISCUSSION
We found that overexpression of AT2R in mouse coronary artery ECs increased bradykinin generation and that activation of AT2R with a specific agonist increased bradykinin levels further. These effects were diminished or abolished by AT2R blockade or a plasma kallikrein inhibitor. Overexpression of AT2R also upregulated PRCP mRNA and protein expression. Importantly, blockade of PRCP using a siRNA diminished AT2R-induced bradykinin release. We believe these results provide the first evidence that AT2R induced bradykinin release is mediated at least in part by a PRCP-dependent mechanism.
Activation of AT2R has been considered cardioprotective, partially due to stimulation of kinins.6,7 In the present study, we transfected the AT2R gene into mouse coronary artery ECs to see if its overexpression would heighten bradykinin release. We observed that the amount of bradykinin in the medium of Ad-AT2R-transfected cells was significantly higher in the presence of 0.5% FBS and that when FBS was removed bradykinin levels were undetectable. In the absence of an exogenous AT2R agonist, AT2R-mediated bradykinin release could be due to the presence of 0.5% FBS, which is known to contain Ang II and/or Ang II fragments that could activate AT2R and thereby stimulate bradykinin release. However, when we measured Ang II in medium containing 0.5% FBS Ang II was undetectable (< 15 pmol/L). Since the concentration of Ang II needed in order to induce biological effects in cultured cells generally involves nmol/L concentration, it is thus unlikely that the increased basal bradykinin release from Ad-AT2R cells is due to the presence of Ang II or its fragments in 0.5% FBS culture medium. Another possible explanation for increased basal bradykinin release in AT2R cells is ligand-independent activation AT2R. Jin et al31 reported that transfection of AT2R increased bradykinin and iNOS protein expression in vascular smooth muscle cells independent of Ang II. In addition, Li et al32 demonstrated that AT2R overexpression induced-apoptosis in prostate cancer cells is Ang II independent and AT2R itself has constitutive activity to cause apoptosis. Miura et al 33 further demonstrated that constitutive activation of AT2R induces cell signaling in a ligand-independent manner. Our data agree with these findings and demonstrate that overexpression of AT2R is able to stimulate bradykinin release independent of a specific ligand. Moreover, this bradykinin-stimulated action is AT2R-specific, since 1) this effect was blocked by the AT2R antagonist PD123319 but unaffected by AT1R blockade, and 2) activation of AT2R by a specific agonist or Ang II in the presence of valsartan (Fig. S1 in the online Data Supplement at http://hyper.ahajournals.org) increased bradykinin release further in AT2R-transfected ECs and this effect was blocked by the AT2R antagonist PD123319. We also demonstrated that overexpression of AT2R increased NO release from cultured ECs. This increase was further enhanced by CGP 42112A but blocked by PD123319 or bradykinin B2 receptor antagonist HOE-140 (Fig. S2 in the online Data Supplement at http://hyper.ahajournals.org), indicating that the effect of AT2R may be mediated by the bradykinin/NO pathway.
The precise mechanism by which AT2R increases kinin release is not known. Tsutsumi et al8 reported that in the mouse aorta AT2R activation lowered cellular pH associated with increased kininogenase activity, suggesting that an acid-optimal kininogenase may be responsible for vascular kinin release. However, it is questionable whether this enzyme is tissue kallikrein, since the optimum pH for tissue kallikrein is around 8.5. It is possible that AT2R activates an acidic protease(s) that converts tissue prekallikrein to active tissue kallikrein, or perhaps it is able to activate acidic kininogenases. Interestingly, PRCP enzyme activity appears to occur at acidic pH levels.15 However, the role of PRCP in AT2R induced kinin release has not been explored to our knowledge.
Prekallikrein is one of the physiological substrates of PRCP. Recently, PRCP has been described as a novel plasma prekallikrein activator that is responsible for kinin release from high-molecular-weight kininogen in the endothelium.16 In the present study we found that overexpression of AT2R increased PRCP mRNA 1.7-fold and protein expression 2.5-fold in mouse coronary artery ECs, coupled with increased bradykinin release. Importantly, blockade of PRCP with a siRNA significantly diminished the basal and AT2R-stimulated bradykinin release in AT2R cells, indicating that activation of PRCP, which activates plasma prekallikrein, might play a crucial role in AT2R-induced bradykinin release. To confirm this, we incubated cells in serum-free medium with or without exogenous high-molecular-weight kininogen and prekallikrein. We found that in the absence of high-molecular-weight kininogen and prekallikrein, bradykinin levels were undetectable; whereas adding them to serum-free medium, bradykinin release was significantly increased in GFP- or even more so in AT2R-transfected cells (Fig. S3 in the online Data Supplement at http://hyper.ahajournals.org). Furthermore, inhibition of plasma kallikrein with SBTI abolished the basal or AT2R agonist-stimulated bradykinin release. Taken together, our data support the hypothesis that AT2R-induced bradykinin release is mediated at least in part by activation of plasma prekallikrein via PRCP and cleavage of high-molecular-weight kininogen by kallikrein.
There is conflicting evidence regarding the effects of overexpression of AT2R on endogenous AT1R. While Jin et al reported that overexpression of AT2R downregulated the AT1R in vascular smooth muscle cells,31 others have stated that AT2R overexpression has no effect on AT1R expression in the heart or blood vessels.34 In the present study we examined the effect of overexpression of AT2R on AT1R mRNA in coronary artery ECs and found that low titers of Ad-AT2R transfection that increased bradykinin release did not affect endogenous AT1R mRNA, whereas at higher titers overexpression of AT2R increased AT1R expression. Thus our data suggest that whether or not AT2R overexpression affects AT1R expression is largely dependent on the level of AT2R gene transfection.
Limitations
The present study has some limitations. First, we demonstrated that overexpression of AT2R increased PRCP protein expression and bradykinin release. Activation of AT2R with an agonist increased bradykinin release further but did not alter PRCP protein levels (data not shown). Although this may argue against a role for PRCP in AT2R-induced bradykinin release, it is possible that 2hr-stimulation with an AT2R agonist is not long enough to alter protein expression. In addition, we can not exclude the possibility that PRCP activity or its translocation to the membrane is increased by AT2R stimulation. Furthermore, it is possible that another unknown mechanism of enzymatic activation of PRCP is involved in AT2R stimulated bradykinin release. Second, the signaling and molecular mechanism(s) by which AT2R activates PRCP have not been explored, although this is not the focus of our current study. Third, SBTI is not a specific inhibitor for plasma kallikrein. Other proteases such as tryptase and elastase are also able to cleave high-molecular-weight kininogen and liberate kinins,35 and these enzymes could also be inhibited by SBTI. Thus we cannot exclude the possibility that other proteases also contribute to AT2R-simulated bradykinin release. All of these limitations warrant further investigation. Nevertheless, these limitations do not negate the evidence that PRCP plays an important role in AT2R-induced bradykinin release from ECs.
In summary, we have shown that overexpression of AT2R increases bradykinin release from mouse coronary artery ECs. This effect was blocked by the AT2R antagonist PD123319 but was not influenced by the AT1R antagonist valsartan. Activation of AT2R by a specific agonist increased bradykinin release further. PRCP expression was increased by AT2R overexpression, while downregulation of PRCP using a siRNA reduced bradykinin release in AT2R-transfected cells. Therefore, we conclude that overexpression of AT2R increases expression of PRCP, which may contribute to AT2R-induced bradykinin release.
Perspectives
Cardiovascular disease is the leading cause of death in the USA. Angiotensin receptor blockers (ARB) reduce morbidity and mortality in patients with cardiovascular disease. The effects of ARB are mediated in part by activation of the AT2R, which leads to the release of kinins. However, the precise mechanism(s) by which AT2R stimulates kinin release are not fully understood. We believe our findings provide the first evidence that AT2R-induced bradykinin release is mediated in part by a prolylcarboxypeptidase-dependent mechanism in mouse coronary artery endothelial cells. These data will enhance our understanding of the role of the AT2R and how it interacts with the kallikrein-kinin system to provide cardioprotection; Moreover, we believe they will facilitate the development of better therapeutic targets for hypertension and cardiovascular disease.
Supplementary Material
Acknowledgments
Sources of Funding: This work was supported by NIH grant HL-28982, PPG Project II (XPY).
Footnotes
Disclosures: None.
References
- 1.Schulman IH, Raij L. The angiotensin II type 2 receptor: what is its clinical significance? Curr Hypertens Rep. 2008;10:188–193. doi: 10.1007/s11906-008-0036-8. [DOI] [PubMed] [Google Scholar]
- 2.Wharton J, Morgan K, Rutherford RAD, Catravas JD, Chester A, Whitehead BF, De Leval MR, Yacoub MH, Polak JM. Differential distribution of angiotensin AT2 receptors in the normal and failing human heart. J Pharmacol Exp Ther. 1998;284:323–336. [PubMed] [Google Scholar]
- 3.Hutchinson HG, Hein L, Fujinaga M, Pratt RE. Modulation of vascular development and injury by angiotensin II. Cardiovasc Res. 1999;41:689–700. doi: 10.1016/s0008-6363(98)00267-3. [DOI] [PubMed] [Google Scholar]
- 4.Gallinat S, Busche S, Raizada MK, Sumners C. The angiotensin II type 2 receptor: an enigma with multiple variations. Am J Physiol Endocrinol Metab. 2000;278:E357–E374. doi: 10.1152/ajpendo.2000.278.3.E357. [DOI] [PubMed] [Google Scholar]
- 5.Anavekar NS, Solomon SD. Angiotensin II receptor blockade and ventricular remodelling. J Renin Angiotensin Aldosterone Syst. 2005;6:43–48. doi: 10.3317/jraas.2005.006. [DOI] [PubMed] [Google Scholar]
- 6.Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E, Carretero OA. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J Clin Invest. 1997;99:1926–1935. doi: 10.1172/JCI119360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gohlke P, Pees C, Unger T. AT2 receptor stimulation increases aortic cyclic GMP in SHRSP by a kinin-dependent mechanism. Hypertension. 1998;31:349–355. doi: 10.1161/01.hyp.31.1.349. [DOI] [PubMed] [Google Scholar]
- 8.Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, Miwa T, Kawada N, Mori Y, Shibasaki Y, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A, Takahashi H, Iwasaka T. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Invest. 1999;104:925–935. doi: 10.1172/JCI7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, Sugino H, Matsuura H, Chayama K, Teranishi Y, Iba O, Amano K, Matsubara H. Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension. 2003;41:99–107. doi: 10.1161/01.hyp.0000050101.90932.14. [DOI] [PubMed] [Google Scholar]
- 10.Bove CM, Yang Z, Gilson WD, Epstein FH, French BA, Berr SS, Bishop SP, Matsubara H, Carey RM, Kramer CM. Nitric oxide mediates benefits of angiotensin II type 2 receptor overexpression during post-infarct remodeling. Hypertension. 2004;43:680–685. doi: 10.1161/01.HYP.0000115924.94236.91. [DOI] [PubMed] [Google Scholar]
- 11.Carretero OA, Scicli AG. The kallikrein-kinin system as a regulator of cardiovascular and renal function. In: Laragh JH, Brenner BM, editors. Hypertension: Physiology, Diagnosis, and Management. 2. New York: Raven Press; 1995. pp. 983–99. [Google Scholar]
- 12.Yang HYT, Erdös EG, Chiang TS. New enzymatic route for the inactivation of angiotensin. Nature. 1968;218:1224–1226. doi: 10.1038/2181224a0. [DOI] [PubMed] [Google Scholar]
- 13.Kumamoto K, Stewart TA, Johnson AR, Erdos EG. Prolylcarboxypeptidase (angiotensinase C) in human lung and cultured cells. J Clin Invest. 1981;67:210–215. doi: 10.1172/JCI110015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moreira CR, Schmaier AH, Mahdi F, da Motta G, Nader HB, Shariat-Madar Z. Identification of prolylcarboxypeptidase as the cell matrix-associated prekallikrein activator. FEBS Lett. 2002;523:167–170. doi: 10.1016/s0014-5793(02)02980-0. [DOI] [PubMed] [Google Scholar]
- 15.Odya CE, Marinkovic DV, Hammon KJ, Stewart TA, Erdös EG. Purification and properties of prolylcarboxypeptidase (angiotensinase C) from human kidney. J Biol Chem. 1978;253:5927–5931. [PubMed] [Google Scholar]
- 16.Shariat-Madar Z, Mahdi F, Schmaier AH. Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J Biol Chem. 2002;277:17962–17969. doi: 10.1074/jbc.M106101200. [DOI] [PubMed] [Google Scholar]
- 17.Shariat-Madar Z, Mahdi F, Schmaier AH. Recombinant prolylcarboxypeptidase activates plasma prekallikrein. Blood. 2004;103:4554–4561. doi: 10.1182/blood-2003-07-2510. [DOI] [PubMed] [Google Scholar]
- 18.Ngo ML, Mahdi F, Kolte D, Shariat-Madar Z. Upregulation of prolylcarboxypeptidase (PRCP) in lipopolysaccharide (LPS) treated endothelium promotes inflammation. J Inflamm (Lond) 2009;6:3. doi: 10.1186/1476-9255-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Motta G, Rojkjaer R, Hasan AAK, Cines DB, Schmaier AH. High molecular weight kininogen regulates prekallikrein assembly and activation on endothelial cells: a novel mechanism for contact activation. Blood. 1998;91:516–528. [PubMed] [Google Scholar]
- 20.Shariat-Madar Z, Rahimy E, Mahdi F, Schmaier AH. Overexpression of prolylcarboxypeptidase enhances plasma prekallikrein activation on Chinese hamster ovary cells. Am J Physiol Heart Circ Physiol. 2005;289:H2697–H2703. doi: 10.1152/ajpheart.00715.2005. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, Qiu Q, Mahdi F, Shariat-Madar Z, Røjkjaer R, Schmaier AH. Assembly and activation of HK-PK complex on endothelial cells results in bradykinin liberation and NO formation. Am J Physiol Heart Circ Physiol. 2001;280:H1821–H1829. doi: 10.1152/ajpheart.2001.280.4.H1821. [DOI] [PubMed] [Google Scholar]
- 22.Qin XP, Zeng SY, Tian HH, Deng SX, Ren JF, Zheng YB, Li D, Li YJ, Liao DF, Chen SY. Involvement of prolylcarboxypeptidase in the effect of rutaecarpine on the regression of mesenteric artery hypertrophy in renovascular hypertensive rats. Clin Exp Pharmacol Physiol. 2009;36:319–324. doi: 10.1111/j.1440-1681.2008.05079.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu J, Carretero OA, Liu Y-H, Shesely EG, Yang F, Kapke A, Yang X-P. Role of AT2 receptors in the cardioprotective effect of AT1 antagonists in mice. Hypertension. 2002;40:244–250. doi: 10.1161/01.hyp.0000029095.23198.ad. [DOI] [PubMed] [Google Scholar]
- 24.Yang X-P, Liu Y-H, Mehta D, Cavasin MA, Shesely E, Xu J, Liu F, Carretero OA. Diminished cardioprotective response to inhibition of angiotensin-converting enzyme and angiotensin II type 1 receptor in B2 kinin receptor gene knockout mice. Circ Res. 2001;88:1072–1079. doi: 10.1161/hh1001.090759. [DOI] [PubMed] [Google Scholar]
- 25.Teng B, Ansari HR, Oldenburg PJ, Schnermann J, Mustafa SJ. Isolation and characterization of coronary endothelial and smooth muscle cells from A1 adenosine receptor-knockout mice. Am J Physiol Heart Circ Physiol. 2006;290:H1713–H1720. doi: 10.1152/ajpheart.00826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voyta JC, Via DP, Butterfield CE, Zetter BR. Identification and isolation of endothelial cells based on their increased uptake of acetylated-low density lipoprotein. J Cell Biol. 1984;99:2034–2040. doi: 10.1083/jcb.99.6.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dame MK, Yu X, Garrido R, Bobrowski W, McDuffie JE, Murphy HS, Albassam M, Varani J. A stepwise method for the isolation of endothelial cells and smooth muscle cells from individual canine coronary arteries. In Vitro Cell Dev Biol Anim. 2003;39:402–406. doi: 10.1290/1543-706X(2003)039<0402:ASMFTI>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Gao Y, Grobe JL, Raizada MK, Katovich MJ, Sumners C. Potentiation of the antihypertensive action of losartan by peripheral overexpression of the ANG II type 2 receptor. Am J Physiol Heart Circ Physiol. 2007;292:H727–H735. doi: 10.1152/ajpheart.00938.2006. [DOI] [PubMed] [Google Scholar]
- 29.Wang F, He Q, Sun Y, Dai X, Yang XP. Female adult mouse cardiomyocytes are protected against oxidative stress. Hypertension. 2010;55:1172–1178. doi: 10.1161/HYPERTENSIONAHA.110.150839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Motta G, Shariat-Madar Z, Mahdi F, Sampaio CA, Schmaier AH. Assembly of high molecular weight kininogen and activation of prekallikrein on cell matrix. Thromb Haemost. 2001;86:840–847. [PubMed] [Google Scholar]
- 31.Jin X-Q, Fukuda N, Su J-Z, Lai Y-M, Suzuki R, Tahira Y, Takagi H, Ikeda Y, Kanmatsuse K, Miyazaki H. Angiotensin II type 2 receptor gene transfer downregulates angiotensin II type 1a receptor in vascular smooth muscle cells. Hypertension. 2002;39:1021–1027. doi: 10.1161/01.hyp.0000016179.52601.b4. [DOI] [PubMed] [Google Scholar]
- 32.Li H, Qi Y, Li C, Braseth LN, Gao Y, Shabashvili AE, Katovich MJ, Sumners C. Angiotensin type 2 receptor-mediated apoptosis of human prostate cancer cells. Mol Cancer Ther. 2009;8:3255–3265. doi: 10.1158/1535-7163.MCT-09-0237. [DOI] [PubMed] [Google Scholar]
- 33.Miura S, Karnik SS, Saku K. Constitutively active homo-oligomeric angiotensin II type 2 receptor induces cell signaling independent of receptor conformation and ligand stimulation. J Biol Chem. 2005;280:18237–18244. doi: 10.1074/jbc.M500639200. [DOI] [PubMed] [Google Scholar]
- 34.Metcalfe BL, Huentelman MJ, Parilak LD, Taylor DG, Katovich MJ, Knot HJ, Sumners C, Raizada MK. Prevention of cardiac hypertrophy by angiotensin II type-2 receptor gene transfer. Hypertension. 2004;43:1233–1238. doi: 10.1161/01.HYP.0000127563.14064.FD. [DOI] [PubMed] [Google Scholar]
- 35.Coffman LG, Brown JC, Johnson DA, Parthasarathy N, D’Agostino RB, Jr, Lively MO, Hua X, Tilley SL, Muller-Esterl W, Willingham MC, Torti FM, Torti SV. Cleavage of high-molecular-weight kininogen by elastase and tryptase is inhibited by ferritin. Am J Physiol Lung Cell Mol Physiol. 2008;294:L505–L515. doi: 10.1152/ajplung.00347.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.