

Abstract
The process of protein synthesis must be sufficiently rapid and sufficiently accurate to support continued cellular growth. Failure in speed or accuracy can have dire consequences, including disease in humans. Most estimates of the accuracy come from studies of bacterial systems, principally Escherichia coli, and have involved incomplete analysis of possible errors. We recently used a highly quantitative system to measure the frequency of all types of misreading errors by a single tRNA in E. coli. That study found a wide variation in error frequencies among codons; a major factor causing that variation is competition between the correct (cognate) and incorrect (near-cognate) aminoacyl-tRNAs for the mutant codon. Here we extend that analysis to measure the frequency of missense errors by two tRNAs in a eukaryote, the yeast Saccharomyces cerevisiae. The data show that in yeast errors vary by codon from a low of 4 × 10−5 to a high of 6.9 × 10−4 per codon and that error frequency is in general about threefold lower than in E. coli, which may suggest that yeast has additional mechanisms that reduce missense errors. Error rate again is strongly influenced by tRNA competition. Surprisingly, missense errors involving wobble position mispairing were much less frequent in S. cerevisiae than in E. coli. Furthermore, the error-inducing aminoglycoside antibiotic, paromomycin, which stimulates errors on all error-prone codons in E. coli, has a more codon-specific effect in yeast.
Keywords: mistranslation, cognate, near-cognate, dual luciferase, aminoglycoside, eukaryotic translation
INTRODUCTION
Cells express their genetic information mostly by translating it into encoded protein products. Translational accuracy critically influences cell viability since inaccurately translated proteins can interfere with processes essential to viability. On the other hand, cells must balance the demands of accurate synthesis with competing demands for rapid synthesis (for review, see Parker 1989). The protein synthetic system has therefore evolved to minimize errors without adversely affecting protein output, using many mechanisms to block a variety of misincorporation events. For example, error correction by editing during aminoacylation of some tRNAs (Hendrickson and Schimmel 2003) can reduce misacylation to as few as once in 106 events (Soll 1990; Schulman 1991). Aminoacylation of other tRNAs lacks editing, resulting in much higher levels of error, as high as one in 104 events (Ibba and Soll 2000).
Errors during other steps in protein synthesis appear to be more frequent. Missense errors during protein synthesis result from incorrect codon recognition. Correct decoding requires the tRNA to form three base pairs with the mRNA codon. The intrinsic stability of correct (cognate) tRNAs compared with near-cognate tRNAs, those making a single mismatch, is insufficient to explain the accuracy of protein synthesis (Uhlenbeck et al. 1971; Thompson and Stone 1977; Grosjean et al. 1978). An energy difference of 3 kcal/mol caused by the loss of 1 base pair (bp) would predict an error frequency of 10−2 (Ogle and Ramakrishnan 2005), but translation is much more accurate (Parker 1989; Kurland et al. 1996). The long accepted explanation of this discrepancy is the kinetic proofreading model (Hopfield 1974; Ninio 1975), which proposes that repeated discrimination in a process involving two 100-fold discriminating steps that are separated by an irreversible step could provide 10,000-fold selectivity. Recruitment of an aminoacyl-tRNA (aa-tRNA) occurs in complex with an elongation factor, EF-Tu, bound to GTP; recruitment activates an intrinsic GTPase in EF-Tu so aa-tRNA binding requires GTP cleavage. Under the kinetic proofreading model, the ribosome discriminates in favor of cognate tRNAs both before and after irreversible GTP hydrolysis on EF-Tu (Thompson 1988); discrimination would depend on intrinsic differences in aa-tRNA stability between cognate and near-cognate complexes. Recent kinetic analysis has suggested that accuracy actually mainly derives not from differences in stability but from different rates of activation of the EF-Tu GTPase (for review, see Rodnina et al. 2005). GTPase activation is almost three orders of magnitude faster for cognate than for near-cognate complexes. The ribosome fails to maximize discrimination based on differential stability because selection proceeds too rapidly for stability differences to exert their full effect (Gromadski et al. 2006). Structural analysis of the ribosome suggests that different rates of GTPase activation result from the fact that cognate, but not near-cognate, tRNA occupying the ribosomal decoding site (the A site) induces a large-scale conformational change thought to cause GTPase activation (Ogle et al. 2002; Ogle and Ramakrishnan 2005). Recently, another mode of error correction has been suggested in which errant peptidyl-tRNAs are released from the ribosome in a quality control mechanism (Zaher and Green 2009). The relative contribution to translational accuracy of this quality control mechanism compared with events before peptide transfer is unknown.
There have been surprisingly few estimates of the frequency of misreading errors in vivo (for review, see Parker 1992). Most have been measured in Escherichia coli and individual estimates of misreading error frequency have varied widely (see Kramer and Farabaugh 2007, and references therein). The errors analyzed in these studies differed by type of base-pair mismatches, position of the codon•anticodon complex involved, and analytical method used. The fact that each of these factors might affect the observed frequency of errors at a codon could explain variation in error estimates. To better understand the true in vivo range of misreading errors, we developed an assay system that measured the frequency of every near-cognate misreading error by E. coli 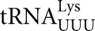 (Kramer and Farabaugh 2007). The system uses mutant forms of a firefly luciferase (Fluc) reporter gene with inactivating mutations in the essential active-site lysine-529 (Branchini et al. 2000). The proteins expressed from these mutant genes have low but measurable activity, which could be explained in two ways. The “functional replacement model” predicts that the mutant protein is not completely inactive but that the mutant amino acid might partially perform the function of the wild-type amino acid, providing a low level of activity. We have shown in bacteria that the activity of some lysine-529 mutants actually results from occasional mistranslation of the mutant transcript, leading to misincorporation of the wild-type lysine at a mutant codon. This mistake generates a wild-type protein with full enzymatic activity. Because this mistake occurs only rarely, most encoded proteins remain inactive. The frequency of misincorporation is equal to the fractional activity caused by the rare production of a wild-type protein (Kramer and Farabaugh 2007). Using this system, we found that the error rates in E. coli do in fact vary widely, from ∼4 × 10−3 to ∼3 × 10−4 per codon, and that a major factor contributing to error is competition for decoding at the mutant codons between cognate and near-cognate aa-tRNAs; higher concentrations of the competing cognate tRNA reduce error rates and lower concentrations allow higher error rates.
(Kramer and Farabaugh 2007). The system uses mutant forms of a firefly luciferase (Fluc) reporter gene with inactivating mutations in the essential active-site lysine-529 (Branchini et al. 2000). The proteins expressed from these mutant genes have low but measurable activity, which could be explained in two ways. The “functional replacement model” predicts that the mutant protein is not completely inactive but that the mutant amino acid might partially perform the function of the wild-type amino acid, providing a low level of activity. We have shown in bacteria that the activity of some lysine-529 mutants actually results from occasional mistranslation of the mutant transcript, leading to misincorporation of the wild-type lysine at a mutant codon. This mistake generates a wild-type protein with full enzymatic activity. Because this mistake occurs only rarely, most encoded proteins remain inactive. The frequency of misincorporation is equal to the fractional activity caused by the rare production of a wild-type protein (Kramer and Farabaugh 2007). Using this system, we found that the error rates in E. coli do in fact vary widely, from ∼4 × 10−3 to ∼3 × 10−4 per codon, and that a major factor contributing to error is competition for decoding at the mutant codons between cognate and near-cognate aa-tRNAs; higher concentrations of the competing cognate tRNA reduce error rates and lower concentrations allow higher error rates.
Even fewer estimates of misreading rates are available for eukaryotic organisms. The few studies that have been done in vivo in the yeast Saccharomyces cerevisiae reported error frequencies ranging from ∼10−3 to 10−5 per codon (Stansfield et al. 1998; Rakwalska and Rospert 2004; Salas-Marco and Bedwell 2005; Plant et al. 2007). Again, most of these studies measured one or a few distinct errors at different codons, involving different mismatches, and at all three positions of the codon•anticodon complex. For example, Salas-Marco and Bedwell (2005) tested half of the potential errors that can occur at each anticodon position by reading codon 529 of Fluc and by 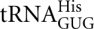 reading codon 245. They reported a range of error frequencies from 1 to 5 × 10−4 per codon for and a much higher level of error by , up to 2 × 10−3 per codon (Salas-Marco and Bedwell 2005). The highest activities they recorded they ascribed to replacement of the wild-type histidine by the encoded mutant amino acid. Using an extension of their system, we have attempted to analyze the full range of misreading errors by these tRNAs in S. cerevisiae. We confirm that the highest enzymatic activities reported at codon 245 by Salas-Marco and Bedwell (2005) reflect functional replacement rather than translational error but that half of the other mutants appear to have been incorrectly identified as error prone. For misreading by at codon 529 we show that, as in E. coli, error frequency is not solely determined by codon•anticodon pairing but rather is influenced by competition for decoding between cognate and near-cognate tRNAs. Additionally, we show that, instead of inducing error in general, the aminoglycoside antibiotic paromomycin induces misreading of only a subset of error-prone codons in yeast.
reading codon 245. They reported a range of error frequencies from 1 to 5 × 10−4 per codon for and a much higher level of error by , up to 2 × 10−3 per codon (Salas-Marco and Bedwell 2005). The highest activities they recorded they ascribed to replacement of the wild-type histidine by the encoded mutant amino acid. Using an extension of their system, we have attempted to analyze the full range of misreading errors by these tRNAs in S. cerevisiae. We confirm that the highest enzymatic activities reported at codon 245 by Salas-Marco and Bedwell (2005) reflect functional replacement rather than translational error but that half of the other mutants appear to have been incorrectly identified as error prone. For misreading by at codon 529 we show that, as in E. coli, error frequency is not solely determined by codon•anticodon pairing but rather is influenced by competition for decoding between cognate and near-cognate tRNAs. Additionally, we show that, instead of inducing error in general, the aminoglycoside antibiotic paromomycin induces misreading of only a subset of error-prone codons in yeast.
RESULTS
An assay system for measuring the frequency of every possible misreading error by in yeast
We used a previously described dual luciferase reporter construct, pDB688 (Salas-Marco and Bedwell 2005), to measure all potential translational misreading errors by in the yeast S. cerevisiae. The reporter consists of a translational fusion of the gene encoding Renilla reniformis (sea pansy) luciferase (Rluc) to a downstream gene encoding the Photinus pyralis (firefly) luciferase (Fluc) gene under the control of the S. cerevisiae PGK promoter (Fig. 1). Fluc is the misreading reporter; Rluc serves as an internal control for protein abundance. Lysine-529 (K529) of Fluc is highly conserved among firefly luciferases (Ye et al. 1997) and even among the more highly diverged superfamily of adenylate-forming enzymes (Branchini et al. 2000). Point mutations in K529 reduce enzymatic activity up to 1600-fold in bacteria (Branchini et al. 2000; Kramer and Farabaugh 2007). We have shown in E. coli that the residual activity of several K529 mutants results from the mutant codon being misread by , producing a small fraction of translation products that have wild-type activity (Kramer and Farabaugh 2007). The ratio of activity of such a mutant to that of the wild type is a measure of the frequency of misreading by .
FIGURE 1.

Map of the dual luciferase reporter plasmid pDB688. The plasmid pDB688 includes a single open reading frame encoding a fusion of the Renilla (Rluc), and firefly (Fluc) luciferase is shown flanked by the promoter of the constitutively expressed PGK gene and the poly(A) addition site of the CYH2 genes, both from S. cerevisiae. The location of a segment of the 2 μ circle that confers autonomous replication in S. cerevisiae and the URA3+ selectable gene are also indicated.
To estimate the range of misreading frequencies by we measured the residual Fluc activity of a set of dual luciferase constructs individually bearing all possible single nucleotide substitutions in the K529 codon (see Fig. 2). These mutant codons represent every possible near-cognate substrate for . As a negative control for misreading, we measured the residual Fluc activity of a mutant with a UUU codon at position 529. This codon can make no Watson-Crick base pairs with the UUU anticodon of and should therefore minimally misread the codon as lysine; when assayed in bacteria, it produced background activity (Kramer and Farabaugh 2007).
FIGURE 2.

Near- and noncognate mutations for lysine-529 (K529). The two Lys codons are shown in italics in this standard genetic code table. Near-cognate codons, differing at one codon position, are shown in reverse on black. Noncognate codons differ from Lys codons in more than one codon position; noncognate codons synonymous with tested near-cognate codons are shown in black on gray. The control UUU codon is shown in boldface.
The Fluc activities of K529 near-cognate mutants varied by a factor of ∼9 (Fig. 3). The activity of several of these mutants was similar to the UUU control, with an average Fluc activity of 1.6 × 10−4 in wild type, about twofold lower than the activity seen in bacteria. Two of the near-cognate codons, however, had activity significantly higher than the others (P <0.05, determined by ANOVA with Tukey's post-hoc analyses), a termination codon (UAG) and an Arg codon (AGG). The activity of the UAG nonsense codon should reflect misreading since truncation of the protein to 528 amino acids would eliminate Fluc activity. The increased activity of the AGG mutant could result from misreading but could also result from the mutant Arg functionally replacing the wild-type Lys. A test of the functional replacement model is to measure the activity of synonymous mutants, either near-cognate mutants, requiring to form one mismatch during codon recognition, or noncognate mutants, requiring more than one mismatch. If the activities of these mutant proteins result from functional replacement the activity of the synonymous mutants should be nearly identical. The activity, however, may result from misreading of the mutant codon by . In that case, the noncognate synonymous codons should be very poorly recognized by the tRNA and should have much less activity than the near-cognate codon mutants.
FIGURE 3.

Variation in Fluc activity of K529 mutants. The graph shows the Fluc expression of the indicated constructs expressed as a fraction of the expression of wild-type Fluc. The activities for mutants that according to ANOVA are not significantly different from each other or the negative control are shown in black and the two activities that are significantly higher (P <0.05) are shown in white; the negative control UUU mutant is shown in gray. Indicated below each codon is the amino acid it encodes. Error bars are standard error of the mean.
As shown in Figure 4, synonymous near-cognate codon mutants (UAG/UAA and AGA/AGG) had significantly different Fluc activities in both cases (P <0.05). The functional replacement model would predict that synonymous codons would produce proteins with the same activity, so this result is inconsistent with that model. The data suggest that misreading is influenced by wobble pairing since codons ending in A produced less activity than those ending in G. To test further the functional replacement model, we replaced the K529 codon with five synonymous noncognate mutant codons—the termination codon UGA and the Arg codons CGU, CGC, CGA, and CGG. All of these codons would require two or three mismatches during decoding by and are expected to be minimally misread as Lys. As shown in Figure 4, the UAG mutant had much greater activity than UGA. Among the Arg mutants, the AGG mutant had more activity than the five other near- and noncognate mutants, and the four noncognate mutants were not distinguishable from each other (P <0.001). These data are also inconsistent with the functional replacement model and suggest that the increased activity of the UAG and AGG mutants reflects a higher level of missense errors than on the other codons.
FIGURE 4.

High residual activity of two K529 mutants results from near-cognate decoding. A comparison of the Fluc activity relative to wild type for synonymous near- and noncognate codon mutants are shown for the three nonsense mutants, UAA, UAG, and UGA, and six Arg mutants, AGA, AGG, CGU, CGC, CGA, and CGG. The data are represented as in Figure 3.
The Asn mutants, AAU and AAC, have the least activity among the near-cognate mutants tested (∼7 × 10−5 times wild type). This result is particularly striking because would misread these codons by a wobble position pyrimidine–pyrimidine mismatch. That error was among the most frequent in E. coli. We will discuss possible reasons for this difference below.
Competition for decoding between cognate and near-cognate tRNA species affects misreading rates in yeast
The Arg codon AGG had the highest activity among the mutants tested, from two- to ninefold higher than the remaining near-cognate 529 codons. This codon is rarely used in yeast genes and is decoded by the low-abundance cognate 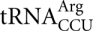 (Ikemura 1982; Percudani et al. 1997). We suspected that frequent errors at AGG are caused by poor decoding by the rare cognate tRNA; we have demonstrated such a competition effect in E. coli (Kramer and Farabaugh 2007). If the high error frequency at AGG is in fact a result of slow recognition by low-abundance , then further reducing the rate of recognition of AGG should increase misreading. In a strain lacking , near-cognate decodes AGG more slowly than would the cognate tRNA (Kawakami et al. 1993). In the strain, the activity of the AGG mutant increased about fivefold to ∼2 × 10−3 but there was no effect on the activity of AGA or UUU mutants (Fig. 5). This result is further proof that increased activity of the AGG mutant results from misreading by and that, as in E. coli, a factor influencing misreading rates in yeast is competition for decoding between cognate and near-cognate tRNA species.
(Ikemura 1982; Percudani et al. 1997). We suspected that frequent errors at AGG are caused by poor decoding by the rare cognate tRNA; we have demonstrated such a competition effect in E. coli (Kramer and Farabaugh 2007). If the high error frequency at AGG is in fact a result of slow recognition by low-abundance , then further reducing the rate of recognition of AGG should increase misreading. In a strain lacking , near-cognate decodes AGG more slowly than would the cognate tRNA (Kawakami et al. 1993). In the strain, the activity of the AGG mutant increased about fivefold to ∼2 × 10−3 but there was no effect on the activity of AGA or UUU mutants (Fig. 5). This result is further proof that increased activity of the AGG mutant results from misreading by and that, as in E. coli, a factor influencing misreading rates in yeast is competition for decoding between cognate and near-cognate tRNA species.
FIGURE 5.

Deletion of the single gene encoding the AGG-decoding tRNA drastically increases error at the AGG codon. The Fluc activity relative to wild type is shown for the negative control (UUU, Phe) and the two Arg codons, AGA and AGG. The black columns represent the activity of the three mutants in the wild-type background and the white columns represent the activity in the background deleted for the tRNA gene. The data are represented as in Figure 3.
Histidine-245 of firefly luciferase is not suitable for measuring misreading rates by in yeast
We wanted to extend our analysis and to estimate the full range of misreading error rates by a second yeast tRNA species. Luciferase is an acyl-adenylate-forming enzyme (for review, see Deluca 1976); it adenylates a carboxyl group on its substrate, luciferin. Oxidation of the acyl-adenylate produces oxyluciferin in an excited state, which relaxes with emission of light. A second active site residue, histidine at position 245 (H245), is proposed to coordinate the position of the carboxyl side group of the substrate luciferin to facilitate its adenylation (Branchini et al. 2000). H245 regulates the color of the emitted light apparently by influencing the tautomeric form of the luciferin substrate (Branchini et al. 1999). A H245D mutant, replacing the His with Asp, has extremely low activity, suggesting that the residue might also be required for the rate of the adenylation reaction; other mutants had substantially more activity that could result either from functional replacement of the His activity or from misreading errors (Branchini et al. 1998). A recent report by Salas-Marco and Bedwell (2005) argued for error as the source of variation for some mutants. They based this identification of error-prone codons on their being stimulated to higher activity by paromomycin. We decided to perform a more extensive test of the role of misreading errors at this codon.
We measured the Fluc activity of reporters with all possible single nucleotide substitutions in the H245 codon, all of which are near-cognate substrates for (Fig. 6). As a negative control for error we also measured the Fluc activity of a mutant introducing a GUG Val codon noncognate for . Like the K529F(UUU) mutation, the inability of to base pair with GUG should allow minimal misreading of this codon. The H245V(GUG) mutation did indeed have low activity, 1.7 × 10−3 times the activity of wild type; this activity, however, is about an order of magnitude above that of the least active of the K529 mutations, suggesting that H245 may be substantially less important to Fluc activity.
FIGURE 6.

Near- and noncognate mutations for histidine-245 (H245). The wild-type, near-cognate, and synonymous noncognate mutant codons for H245 are represented as in Figure 2.
The luciferase activities of the near-cognate H245 codon mutants varied >2000-fold (Fig. 7; Table 1). For most of the tested mutants, synonymous codon mutants had indistinguishable activities; we cannot conclude that the level of activity of any of these mutants results from error. Among these mutants most have activities averaging 2 × 10−3 times wild type, although the His-to-Asp mutants, GAU and GAC, had much lower activity, 4 × 10−5 times wild type. Four mutants (UAU, UAC, AAU, and AAC) had much higher activity, ranging from 3% to 11% for wild type but, because the synonymous codons had indistinguishable activity, we cannot conclude that the elevated activity results from translational misreading. The only direct evidence for misreading is seen with the two Arg codons, CGU and CGC, which show significantly different activities (P <0.001). These data suggest that misreads the CGC codon at a rate of 1.5 × 10−3 per codon.
FIGURE 7.

Variation in Fluc activity of H245 mutants. The graph shows the variation in Fluc activity of the near-cognate H245 mutants using the format of Figure 3 except that the y-axis is logarithmic to encompass data over three orders of magnitude. The black columns represent activities that are not significantly different among synonymous codons. The single white column indicates that the activity of the CGC mutant is significantly higher than the synonymous CGU mutant.
TABLE 1.
Range of apparent misreading frequencies of His245 mutants with and without paromomycin

From these data we cannot conclude that the higher level of activity of most of the H245 mutants is due to misreading. These data are consistent with the functional replacement model. Most of the H245 mutants have activities much greater than the background activity of the K529 mutants (the activity of those near-cognate mutants that do not show any significant evidence of error). The higher activity of the H245 mutants suggests that this residue is not as essential to luciferase activity so that many mutant proteins have substantial activity. In many cases (Val, Leu, Pro, and Gln) the mutant amino acid allows a moderate level of activity, ∼2 × 10−3 for wild type. In some cases (Tyr and Asn) the mutant amino acid appears to be able to partially fulfill the role of His in coordinating the luciferin substrate. In other cases (Asp and Arg) the mutant amino acid may actually interfere with adenylation of luciferin, resulting in reduced activity. The activity of the Arg mutant was low enough that misreading by could be identified.
In order to determine if the activities of the H245 Fluc mutants are caused by functional replacement we determined the activity of the mutants in E. coli. If the activities reflect the specific activity of the mutant enzymes we expect their activities in bacteria to be similar to those in yeast. If, on the other hand, the activities reflect translational error we expect the bacterial and yeast systems to produce quite different activites, as we saw with the K529 mutants. The H245 Fluc mutants were transferred into the bacterial expression plasmid pEK4 (Kramer and Farabaugh 2007). Figure 8 presents the results of assaying these mutants in the bacterial strain Xac. Overall, the activity of the mutants is quite similar to that seen in yeast. The activities of the majority of the mutants in yeast averaged 1.9 times the activity in bacteria with the relative activities being nearly unchanged. The only exceptions were the Asp mutants GAU and GAC, which in yeast had 0.3 and 0.5 times the bacterial activity. This difference and the fact that the activity in bacteria of the GAU mutant was 1.8-fold higher than the GAC mutant suggest that the higher activity in bacteria reflect misreading at 1.5 × 10−4 per codon for GAU and 8.3 × 10−5 per codon for GAC. A second pair of synonymous codons, the Arg codons CGU and CGC, had activities 1.7 × 10−4 and 7.9 × 10−4 times wild type. The four synonymous noncognate mutants (CGA, CGG, AGA, and AGG) averaged 2 × 10−4 times wild type (data not shown), suggesting that the activity of the CGU mutant did not reflect error; the higher activity of the CGC mutant appears to result from misreading. These data strongly suggest that the activity of most H245 mutants results from functional replacement and not misreading.
FIGURE 8.

Paromomycin increases inaccuracy of only three K529 mutations in S. cerevisiae. The activity of the near-cognate mutants of K529 is shown in the absence (black) or presence (white) of 200 μg/mL paromomycin. Significant increases in activity (ANOVA, P <0.05) were seen for the two nonsense mutants, UAA and UAG, and the Asn mutant, AAU. The data are represented as in Figure 3.
The aminoglycoside antibiotic paromomycin increases a subset of near-cognate misreading errors
Paromomycin induces translational misreading errors in both E. coli and yeast. It stimulated misreading of poly(U) in yeast cell free extracts (Benveniste and Davies 1973; Palmer et al. 1979; Singh et al. 1979) and induced phenotypic suppression of both nonsense and missense mutations in yeast (Palmer et al. 1979; Singh et al. 1979). Paromomycin binds to a region of helix 44 (h44) of the small ribosomal subunit (Carter et al. 2000) and this binding is thought to increase misreading by inducing a structural change in the decoding center similar to that induced by the binding of a cognate tRNA (for review, see Ogle et al. 2003), thereby altering the kinetics of decoding to increase the probability of acceptance of near-cognate tRNAs in the A site (Pape et al. 2000). Paromomycin and other aminoglycoside antibiotics also bind to a second site in Helix 69 (H69) of the 50S ribosomal subunit (Borovinskaya et al. 2007). H69 is a highly conserved structure located between the A and P sites that is involved in several intersubunit contacts and it contacts both the aminoacyl and peptidyl-tRNAs (Borovinskaya et al. 2007). Paromomycin has recently been shown to block recognition of termination codons by bacterial peptide release factors (RFs), and that effect has been attributed to the h44 site (Youngman et al. 2007). This effect could contribute to misreading of termination codons by slowing recognition by RFs. Paromomycin also interferes with ribosome recycling but this effect appears to involve the H69 site (Borovinskaya et al. 2007). The involvement of the H69 site in misreading has not been determined. The h44 binding site for paromomycin in eukaryotes, including S. cerevisiae, differs from the bacterial site by the replacement of A1408 with G1408, a change that in bacteria causes increased resistance to aminoglycosides including paramomycin; this difference may be the basis of the prokaryotic-specific effect of the drugs (Recht et al. 1999).
In E. coli, we showed that paromomycin and a second aminoglycoside antibiotic, streptomycin, increased misreading of only some near-cognate codons, suggesting that their effect is less general than previously thought, but to increase errors at all error-prone near-cognate codons (Kramer and Farabaugh 2007). Salas-Marco and Bedwell (2005) showed in S. cerevisiae that exposure to a sublethal concentration of paromomycin significantly increased misreading by of only one near-cognate codon, AAU. The amount of paromomycin necessary to induce errors was much higher than in bacteria; Salas-Marco and Bedwell (2005) used 200 μg/mL of paromomycin, 40 times the concentration used in bacteria (Kramer and Farabaugh 2007). We do not know if only the H69 site is occupied in vivo at this higher concentration or if the increased concentration also allows binding to the G1408 variant of the h44 site. Salas-Marco and Bedwell (2005) measured only half of the potential errors by this tRNA, excluding the G-ending codons and termination codons. We wanted to determine the specific effects of paromomycin on all potential misreading events by in yeast.
As shown in Figure 8, paromomycin significantly increased the Fluc activity more than twofold of only three of the near-cognate codon 529 mutants (P <0.05). Misreading of UAA, UAG, and AAU increased from 11-fold, 7.6-fold, and 2.8-fold, respectively, with the highest relative activity of 2.7 × 10−3 at UAG. These correspond to either first position errors in competition with translation termination or, at AAU, a wobble position error. The activity of the remaining near-cognate codons showed little or no effect of paromomycin. In particular, paromomycin had no effect on errors at AGG, even though it is the most error-prone codon in the absence of the drug. These data suggest that paromomycin affects a subset of misreading events, all of which appear to be special cases. The much stronger effect on UAA/UAG can be explained if, as in E. coli (Youngman et al. 2007), paromomycin interfered with recognition by peptide release factor. Paromomycin appears to stimulate a wobble position error by at AAU. The drug also induced wobble position errors in E. coli; the AAU codon showed the greatest effect of paromomycin, 4.4-fold, compared with the 2.8-fold effect in S. cerevisiae. However, paromomycin stimulated errors in E. coli at all codons that were error-prone in the absence of the drug—UAG, AGA, AGG, and AAU—as well as at three codons that do not appear error-prone in its absence—UAA, AUA, and AAC (Kramer and Farabaugh 2007). Paromomycin has a much more restricted effect in yeast.
As shown in Figure 9 and Table 1, paromomycin significantly increased the activity in S. cerevisiae of only three near-cognate H245 codon mutants (P <0.05): GAU and GAC (Asp) and CGC (Arg). Activity was stimulated two- to threefold for the Asp codons and sixfold for CGC. The remaining near-cognate codons were unaffected by treatment with paromomycin, including the synonymous Tyr (UAU and UAC) and Asn (AAU and AAC) codons with the highest residual activity. In bacteria, as shown in Figure 10, paromomycin induced significantly higher activity in five mutants: GAU (10-fold), GAC (13-fold), CGU (6.9-fold), CGC (4.0-fold), and CAA (3.9-fold). The other mutants were not significantly increased.
FIGURE 9.

Paromomycin increases inaccuracy of only three H245 codons in S. cerevisiae. The activity of the near-cognate mutants of H245 is shown in the absence (black) or presence (white) of 200 μg/mL paromomycin. Significant increases in activity (ANOVA, P <0.05) were seen for the two Asp mutants, GAU and GAC, and the Arg mutant, CGC. The data are represented as in Figure 7.
FIGURE 10.

Paromomycin increases inaccuracy of five H245 codons in E. coli. The activity of the near-cognate mutants of H245 is shown in the absence (black) or presence (white) of 200 μg/mL paromomycin. Significant increases in activity (ANOVA, P <0.05) were seen for the two Asp mutants, GAU and GAC, two Arg mutants, CGU and CGC, and the Gln mutant, CAG. The data are represented as in Figure 7.
The fact that paromomycin increases the activity of the GAU, GAC, and CGC mutants in yeast suggests that these codons can be misread by . Misreading would require first position G•G mismatches for GAU and GAC and a second position G•U mismatch for CGC. In bacteria, the CGU and CAA codons also appear to be misread by (the Q represents the wobble base queuine, a hypermodified form of guanine) (Katze et al. 1982), requiring a second position G•U and a wobble position Q•A mismatch. The activity of the Asp mutants in both bacteria and yeast in the absence of paromomycin is about an order of magnitude lower than the background activity among the K529 mutants. This lower activity establishes a maximal level for misreading in yeast of ∼4 × 10−5 per codon. The extremely low activity of these mutants relative to the others tested probably reflects interference by the negatively charged side groups of Asp actively with the adenylation of luciferin when it replaces the partially positively charged H245. If the activity of these mutants had been as high as the activity of the K529 non-error-prone codons we would not have been able to measure the low levels of misreading shown by the Asp codons. This fact emphasizes that error-prone codons identified in this analysis provide a minimum list of error-inducing codons; many weakly misread codons would be missed because of the background activity of the mutant proteins. The functional replacement of His in most mutants of H245 would obscure even very high levels of error. For example, because of functional replacement of His by Asn in the AAU and AAC wobble position mutants we cannot determine whether paromomycin stimulates wobble errors by .
DISCUSSION
Here we report the range of all possible misreading errors by in S. cerevisiae, providing an unbiased comparison of error frequencies on 14 near-cognate codons. Previously, we used the same assay based on the Photinus pyralis (firefly) luciferase (Fluc) expressed in E. coli to estimate the frequency of errors at an essential lysine (K529), finding that misreading was elevated at a minority of near-cognate codons, meaning those that differ from an AAA or AAG Lys codon by a single change (Kramer and Farabaugh 2007). The error frequency in E. coli varied from a high of 3.6 × 10−3 (at an AGA codon) to no more than 2.0 × 10−4 (at a GAA codon) or a range of at least 18-fold. That work suggested that a major cause of increased error was the low availability of the competing cognate tRNA; codons recognized by low-abundance cognate tRNAs experience a higher rate of errors than ones recognized by high-abundance tRNAs. The exception to this rule is when misreading requires a wobble error; in that case, an abundant cognate tRNA cannot reduce error to background. We also found that error-inducing antibiotics like the aminoglycosides paromomycin and streptomycin induced higher error rates for all error-prone codons.
Our findings with errors in the yeast S. cerevisiae are similar in general but there are significant differences in detail. We began with the same set of Fluc mutations to assess error at the K529 codon. We find that at this codon misreads near-cognate codons at a frequency from a low of 8 × 10−5 (for GAA and AAU) to a high 6.9 × 10−4 (for AGG). This spectrum of errors is three- to fivefold lower overall than error frequencies measured at the same codon in E. coli (Kramer and Farabaugh 2007). Most of the near-cognate codons had Fluc activities near that of the noncognate control, UUU, with an estimated misreading frequency no higher than ∼1.6 × 10−4. Three near-cognate codons (UAA, GAA, and AAU) had significantly less activity than the UUU control, with an estimated misreading frequency as low as 8 × 10−5. The very low activity of the UAA codon probably reflects the low activity of prematurely terminated proteins. The lower activity of the Glu (GAA) and Asn (AAU) mutants may suggest that, whereas other substituted amino acids simply fail to provide the function of the wild-type lysine, these residues actually interfere with enzyme activity.
Two near-cognate codons had significantly elevated residual activities, UAG and AGG. The activities of these mutants were significantly higher than all synonymous near- and noncognate mutants, inconsistent with the functional replacement model and supporting the alternative misreading model. The activity of the UAG mutant no doubt reflects readthrough by first position misreading by in competition with its recognition by peptide release factor. Misreading at AGG by , which requires a middle position U•G mismatch, would compete with slow decoding by the rare cognate for the codon. Deletion of the gene encoding this tRNA causes a significant increase in AGG misreading. This deletion forces even slower decoding by the AGA cognate 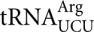 (Kawakami et al. 1993). This result is consistent with our observation that poor recognition by a cognate tRNA is a major factor in determining error frequency (Kramer and Farabaugh 2007). AGG had previously been shown to induce frameshift errors in the sequence CUU-AGG-C and frameshifting efficiency was inversely related to the availability of (Belcourt and Farabaugh 1990; Kawakami et al. 1992, 1993; Pande et al. 1995). Thus, as we previously reported for E. coli, misreading in yeast is not determined by codon•anticodon interaction alone but is also strongly influenced by competition for decoding between cognate and near-cognate tRNAs.
(Kawakami et al. 1993). This result is consistent with our observation that poor recognition by a cognate tRNA is a major factor in determining error frequency (Kramer and Farabaugh 2007). AGG had previously been shown to induce frameshift errors in the sequence CUU-AGG-C and frameshifting efficiency was inversely related to the availability of (Belcourt and Farabaugh 1990; Kawakami et al. 1992, 1993; Pande et al. 1995). Thus, as we previously reported for E. coli, misreading in yeast is not determined by codon•anticodon interaction alone but is also strongly influenced by competition for decoding between cognate and near-cognate tRNAs.
Unexpectedly, there is no evidence of misreading of the codons AAU and AAC, both of which could decode by wobble position pyrimidine–pyrimidine mispairing. In E. coli, we had observed errors at AAU and, to a lesser extent, AAC (Kramer and Farabaugh 2007). In yeast, the AAU/AAC-decoding 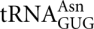 that would compete with at these codons is a major isoacceptor (Percudani et al. 1997). However, the abundance of the AAU/AAC cognate
that would compete with at these codons is a major isoacceptor (Percudani et al. 1997). However, the abundance of the AAU/AAC cognate 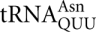 in E. coli did not block errors (Ikemura 1981), suggesting that cognate competition has little effect on wobble errors. If the difference in error rate does not reflect a difference in tRNA availability it must reflect some structural feature either of the cognate or the misreading tRNA. The most obvious difference between the Asn-decoding tRNAs is that the yeast tRNA has an unmodified guanine wobble base while the E. coli tRNA has a hypermodified wobble base, queuine [7-([(4,5-cis-dihydroxy-2-cyclopenten-1-yl)amino]methyl)-7-deazaguanosine or Q]. It may be that the presence of Q reduces recognition of the AAU/AAC bases by their cognate tRNA, allowing misreading by . The yeast tRNA may allow fewer frequent errors on AAU because the G•U wobble interaction strengthens cognate recognition of that base.
in E. coli did not block errors (Ikemura 1981), suggesting that cognate competition has little effect on wobble errors. If the difference in error rate does not reflect a difference in tRNA availability it must reflect some structural feature either of the cognate or the misreading tRNA. The most obvious difference between the Asn-decoding tRNAs is that the yeast tRNA has an unmodified guanine wobble base while the E. coli tRNA has a hypermodified wobble base, queuine [7-([(4,5-cis-dihydroxy-2-cyclopenten-1-yl)amino]methyl)-7-deazaguanosine or Q]. It may be that the presence of Q reduces recognition of the AAU/AAC bases by their cognate tRNA, allowing misreading by . The yeast tRNA may allow fewer frequent errors on AAU because the G•U wobble interaction strengthens cognate recognition of that base.
Previously work has suggested that a second Fluc codon, encoding His245, would be useful as an error reporter (Salas-Marco and Bedwell 2005). That study showed that the activity of single nucleotide mutants of the codon varied widely, from a low of 7 × 10−5 (for GAC) to a high of 7.7 × 10−2 per codon (for AAC). Because synonymous codon mutants were tested in only one case (CAA and CAG) the study provides less evidence to reject the functional replacement hypothesis as an explanation for the variation in activity. To resolve the question we measured activities of a full set of near-cognate and synonymous noncognate codon mutations at this site. Salas-Marco and Bedwell (2005) constructed only mutants one step from the His codon CAC; we constructed all possible near-cognate synonymous CAC and CAU codons as well as all possible synonymous noncognate mutants. Only the two Arg mutants, CGU and CGC, showed a significant difference in activity, with the CGC mutant having almost sevenfold higher activity. In only this case can we conclude that the difference in activity reflects missense error, with the CGC mutant being misread at a rate of 1.5 × 10−3 per codon; that rate is comparable with error rates of the error-prone K529 mutants. We found that four of the near-cognate H245 mutant codons (UAU, UAC, AAU, and AAC) had very high residual Fluc activity, over 50-fold above background, but there was no significant difference in activity between these or most other synonymous mutants, consistent with the functional replacement hypothesis, although it does not exclude that the activity derives from error. In addition, the activities produced by these mutants in yeast and in E. coli were extremely similar, consistent with the conclusion that the activity reflected functional replacement of the wild-type His by the mutant amino acids.
Aminoglycoside antibiotics such as paromomycin cause translational misreading. This effect has been attributed to the drug's inducing structural rearrangements in the ribosome that reduce discrimination between cognate and near-cognate tRNA substrates in the A site (Pape et al. 2000; Ogle et al. 2003). We have shown in E. coli that paromomycin and streptomycin have an effect on misreading that is much less general than previously thought (Kramer and Farabaugh 2007). Both antibiotics stimulated only a subset of all potential near-cognate decoding errors, but both increased error at all codons that are error prone in their absence. We wondered whether the error-inducing effect of paromomycin in yeast was similar. In the current study, paromomycin stimulated misreading of only three of the 14 possible near-cognate 529 codons by . The first position readthrough of UAA and UAG as well as the wobble position misreading of AAU were increased from three- to 11-fold in the presence of paromomycin. Significantly, paromomycin had no effect on the activity of the AGG mutant of codon 529. At codon 245, paromomycin increased the activity of three of the 14 His245 mutants: GAU, GAC, and CGC. The increases varied from 2.5- to 6.4-fold. However, the absolute increase in error varied among the affected codons with the greatest effect being on the CGC mutant of His245 (9.6 × 10−3 per codon) and the two nonsense mutants of Lys529 (1 × 10−3 at UAA and 2.7 × 10−3 for UAG). The effect on misreading is much more specific than in E. coli, where paromomycin induced errors at all error-prone codons and some that did not show error in its absence (Kramer and Farabaugh 2007). Stansfield et al. (1998) measured the effect of paromomycin on misreading by at a near cognate Tyr codon, UAC. That study found a 50-fold increase in error, a much greater effect than we have found. The fact that paromomycin does not induce errors on the AGG mutant of codon 529 is significant since the drug strongly induced errors at this codon in E. coli. These data suggest that the nature of the paromomycin effect may be strongly influenced by the details of codon•anticodon interaction or other sequence-specific effects. Alternatively, the lack of effect on the AGG mutant of codon 529 may mean that the level of activity seen at this codon does not reflect codon•anticodon misreading but rather error at another step, for example, aminoacylation of the the AGG-cognate tRNA; the lack of apparent errors at AGA could be because it is mainly recognized by a distinct tRNA isoacceptor, which might not be misacylated. This conclusion is not consistent with the fact that elimination of the AGG-decoding cognate tRNA causes a large increase in error; that result is more consistent with errors at AGG caused by misreading by tRNALys.
Although misreading frequency has been reported to be as high as 1.5 × 10−3 in yeast (Rakwalska and Rospert 2004), most studies have estimated the frequency to be more on the order of 10−5–10−4 (Stansfield et al. 1998; Salas-Marco and Bedwell 2005; Plant et al. 2007). Our findings that tRNALys1 misreads near-cognate codons at a frequency of 8 × 10−5–7 × 10−4 agree. The range of misreading frequencies of the equivalent E. coli tRNA species is 2 × 10−4–4 × 10−3 (Kramer and Farabaugh 2007), almost an order of magnitude higher. Stansfield et al. (1998) suggested that E. coli might be intrinsically more error-prone than yeast. They reported a misreading of the first codon position in yeast that was almost two orders of magnitude lower than several reports of first position errors in E. coli, although the errors compared were different. Here we measured the same errors in yeast that we have previously measured in E. coli, providing a direct comparison between the two species. Some activities were very similar in E. coli and yeast, including sites that would require to misread at the first (CAA, CAG, and GAG at ∼3 × 10−4 times wild type) or second codon position (AUG at 2–3 × 10−4). These values, however, may reflect functional replacement rather than error since they are at the background level. However, in several cases we found quite different activities in E. coli and yeast, including the first position misreading of UAG (1.4 × 10−3 in E. coli versus 3.5 × 10−4 in yeast), the second position misreading of AGA (3.6 × 10−3 in E. coli versus 1.8 × 10−4 in yeast), the second position misreading of AGG (3.1 × 10−3 in E. coli versus 7 × 10−4 in yeast), and the wobble position misreading of AAU (1.6 × 10−3 in E. coli versus 8 × 10−5 in yeast). These differences must reflect differences in error rate. The 20-fold difference in wobble position error is especially striking since errors of other types vary much less. It may be that this is a general phenomenon in yeast or that a lack of wobble-position errors at AAU reflects the competition between particular tRNAs including specific effects of their modification. The consistent observation that in yeast errors occur at approximately one-third the frequency in E. coli raises the issue of whether yeast may have evolved mechanisms to reduce misreading at low-usage codons in general. Addressing that issue will require a more extensive comparison of error rates that tests multiple competing tRNAs and multiple sequence contexts.
Because this study was performed in vivo using enzyme activity as an indication of the level of error, it is necessarily indirect. We have interpreted the errors we measure as resulting from misreading of a near-cognate codon by tRNALys or tRNAHis but, in the absence of explicit proof, the activity could result from errors at other steps in expression of the gene, for example, during transcription or during aminoacylation of the cognate tRNA for the tested codons. We have not done a study to directly test if any of the observed activity results from transcriptional errors. The frequency of transcriptional errors is estimated at ∼1 × 10−5 per nucleotide, over an order of magnitude below our observed frequency, suggesting that the errors are unlikely to be transcriptional. Further, the sensitivity of the observed errors to wobble position and enhancement by tRNA deletion are inconsistent with their being transcriptional errors. Alternatively, the errors could result from misacylation by aa-tRNA synthetases. These enzymes can have error rates in the range of 10−4 per codon, which is similar to the levels we measure here. Again, it is unclear why misacylation errors would vary in the way the errors measured here have, that is, distinguishing near- from noncognate codons, wobble errors from those at other codon positions, or being stimulated by deletion of the cognate tRNA for the tested codon. In our study of errors in E. coli we tested the effect of a hyperaccurate mutant of ribosomal protein S12 (rpS12) on our errors, finding that hyperactivity reduced errors down to background level (Kramer and Farabaugh 2007). That background level could represent misacylation errors or it could result not from error but from the activity of mutant proteins. Similar hyperaccurate mutants affecting the yeast homolog of rpS12, ribosomal protein S23, are available (Alksne et al. 1993) and are being used to test whether they reduce apparent translational errors in yeast.
MATERIALS AND METHODS
Strains and growth conditions
The strain used for assaying error frequencies in E. coli was Xac (ara Δ[lacproAB] gyrA rpoB argE[amber]). The S. cerevisiae strains used in this study were: BY4728, MATa his3Δ200 trp1Δ63 ura3Δ0 (Brachmann et al. 1998), and a congenic pair of strains that differ only in the presence or absence of the gene HSX1, the only gene encoding : KK242, MATa his3 trp1 ura3 leu2, and KK240, MATa his3 trp1 ura3 leu2 hsx1∷HIS3 (Kawakami et al. 1993). Cultures were grown at 30°C in liquid yeast nitrogen base (YNB) minimal medium, 1.7 g yeast nitrogen base (Difco), 5 g (NH4)2SO4 and 2% glucose per liter. Histidine and tryptophan were added at 20 mg/L to the growth media for strain BY4728. Histidine and tryptophan were added at 20 mg/L and leucine at 100 mg/L to the growth media for strains KK242 and KK240. The aminoglycoside antibiotic paromomycin was added to a sublethal concentration (200 μg/mL) to induce translational errors.
Plasmids
The reporter plasmid used in the studies of error frequency in E. coli was pEK4 (Kramer and Farabaugh 2007). The reporter plasmid used in this study is pDB688, shown in Figure 1 (Salas-Marco and Bedwell 2005). Each plasmid carries copies of luciferase genes derived from the sea pansy Renilla reniformis (Rluc) and the firefly Photinus pyralis (Fluc). The two genes are fused into a single open reading frame and encode a single bifunctional protein. QuikChange Site-Directed Mutagenesis (Stratagene) was used to introduce mutations in the AAA lysine codon at position 529 and the CAC histidine codon at position 245 in the firefly luciferase gene (Salas-Marco and Bedwell 2005). The identity of the codon mutation was confirmed in each case by DNA sequencing using primers adjacent to each mutant site.
Preparation of cell extracts and dual luciferase assays
Transformation, extract preparation, and dual luciferase assays in E. coli were performed as previously described (Kramer and Farabaugh 2007). Plasmids were transformed into S. cerevisiae cells as previously described (Ito et al. 1983). Individual transformants were grown at 30°C to an OD600 of 0.5–0.7 and cell extracts prepared essentially as previously described (Salas-Marco and Bedwell 2005). Cells were collected by centrifugation at 2000 rpm, washed twice, and then resuspended in 250 μL ice cold 1× phosphate buffered saline (PBS) (8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, and 0.24 g KH2PO4 per liter). Cells were lysed by vortexing with glass beads for 3 min and clarified by centrifugation at 4000 rpm at 4°C (Harger and Dinman 2003; Plant et al. 2007). Aliquots of the extracts (5 μL) were assayed for Photinus and Renilla luciferase activities and error frequencies were estimated as previously described (Kramer and Farabaugh 2007). Luminescence was measured using a Veritas Microplate luminometer (Turner Biosystems) according to the manufacturer.
ACKNOWLEDGMENTS
We thank Dr. David Bedwell for providing the yeast plasmids used in this study and Julie Wolf and Yelena Betz for technical assistance. This work was supported by a grant from the National Institute of General Medical Sciences (GM029480).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2201210.
REFERENCES
- Alksne LE, Anthony RA, Liebman SW, Warner JR 1993. An accuracy center in the ribosome conserved over 2 billion years. Proc Natl Acad Sci 90: 9538–9541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcourt MF, Farabaugh PJ 1990. Ribosomal frameshifting in the yeast retrotransposon Ty: tRNAs induce slippage on a 7 nucleotide minimal site. Cell 62: 339–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste R, Davies J 1973. Structure–activity relationships among the aminoglycoside antibiotics: Role of hydroxyl and amino groups. Antimicrob Agents Chemother 4: 402–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, Kaji H, Kaji A, Cate JH 2007. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat Struct Mol Biol 14: 727–732 [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115–132 [DOI] [PubMed] [Google Scholar]
- Branchini BR, Magyar RA, Murtiashaw MH, Anderson SM, Zimmer M 1998. Site-directed mutagenesis of histidine 245 in firefly luciferase: A proposed model of the active site. Biochemistry 37: 15311–15319 [DOI] [PubMed] [Google Scholar]
- Branchini BR, Magyar RA, Murtiashaw MH, Anderson SM, Helgerson LC, Zimmer M 1999. Site-directed mutagenesis of firefly luciferase active site amino acids: A proposed model for bioluminescence color. Biochemistry 38: 13223–13230 [DOI] [PubMed] [Google Scholar]
- Branchini BR, Murtiashaw MH, Magyar RA, Anderson SM 2000. The role of lysine 529, a conserved residue of the acyl-adenylate-forming enzyme superfamily, in firefly luciferase. Biochemistry 39: 5433–5440 [DOI] [PubMed] [Google Scholar]
- Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V 2000. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407: 340–348 [DOI] [PubMed] [Google Scholar]
- Deluca M 1976. Firefly luciferase. Adv Enzymol Relat Areas Mol Biol 44: 37–68 [DOI] [PubMed] [Google Scholar]
- Gromadski KB, Daviter T, Rodnina MV 2006. A uniform response to mismatches in codon–anticodon complexes ensures ribosomal fidelity. Mol Cell 21: 369–377 [DOI] [PubMed] [Google Scholar]
- Grosjean H, DeHenau S, Crothers DM 1978. On the physical basis for the ambiguity in genetic coding interactions. Proc Natl Acad Sci 75: 610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harger JW, Dinman JD 2003. An in vivo dual-luciferase assay system for studying translational recoding in the yeast Saccharomyces cerevisiae. RNA 9: 1019–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson T, Schimmel P 2003. Transfer RNA-dependent amino acid discrimination by aminoacyl-tRNA synthetases. In Translational mechanisms (ed. Lapointe J, Brakier-Gingras L), pp. 34–64 Kluwer Academic, New York [Google Scholar]
- Hopfield J 1974. Kinetic proofreading: A new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc Natl Acad Sci 71: 4135–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M, Soll D 2000. Aminoacyl-tRNA synthesis. Annu Rev Biochem 69: 617–650 [DOI] [PubMed] [Google Scholar]
- Ikemura T 1981. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrenceof the respective codons in its protein genes. J Mol Biol 146: 1–21 [DOI] [PubMed] [Google Scholar]
- Ikemura T 1982. Correlation between the abundance of yeast transfer RNAs and the occurrence of the respective codons in protein genes. J Mol Biol 158: 573–597 [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A 1983. Transformation of intact yeast cells treated with alkali cations. J Bacteriol 153: 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katze JR, Basile B, McCloskey JA 1982. Queuine, a modified base incorporated post-transcriptionally into eukaryotic transfer RNA: Wide distribution in nature. Science 216: 55–56 [DOI] [PubMed] [Google Scholar]
- Kawakami K, Shafer BK, Garfinkel DJ, Strathern JN, Nakamura Y 1992. Ty element-induced temperature-sensitive mutations of Saccharomyces cerevisiae. Genetics 131: 821–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Pande S, Faiola B, Moore D, Boeke J, Farabaugh P, Strathern J, Nakamura Y, Garfinkel D 1993. A rare tRNA-Arg(CCU) that regulates Ty1 element ribosomal frameshifting is essential for Ty1 retrotransposition in Saccharomyces cerevisiae. Genetics 135: 309–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer EB, Farabaugh PJ 2007. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA 13: 87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland C, Hughes D, Ehrenberg M 1996. Limitations of translational accuracy. In Escherichia coli and Salmonella: Cellular and molecular biology (ed. Neidhardt FC et al. ), Vol. 1, pp. 979–1004 ASM Press, Washington, DC [Google Scholar]
- Ninio J 1975. Kinetic amplification of enzyme discrimination. Biochimie 57: 587–595 [DOI] [PubMed] [Google Scholar]
- Ogle JM, Ramakrishnan V 2005. Structural insights into translational fidelity. Annu Rev Biochem 74: 129–177 [DOI] [PubMed] [Google Scholar]
- Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V 2002. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111: 721–732 [DOI] [PubMed] [Google Scholar]
- Ogle JM, Carter AP, Ramakrishnan V 2003. Insights into the decoding mechanism from recent ribosome structures. Trends Biochem Sci 28: 259–266 [DOI] [PubMed] [Google Scholar]
- Palmer E, Wilhelm JM, Sherman F 1979. Phenotypic suppression of nonsense mutants in yeast by aminoglycoside antibiotics. Nature 277: 148–150 [DOI] [PubMed] [Google Scholar]
- Pande S, Vimaladithan A, Zhao H, Farabaugh PJ 1995. Pulling the ribosome out of frame +1 at a programmed frameshift site by cognate binding of aminoacyl-tRNA. Mol Cell Biol 15: 298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape T, Wintermeyer W, Rodnina MV 2000. Conformational switch in the decoding region of 16S rRNA during aminoacyl-tRNA selection on the ribosome. Nat Struct Biol 7: 104–107 [DOI] [PubMed] [Google Scholar]
- Parker J 1989. Errors and alternatives in reading the universal genetic code. Microbiol Rev 53: 273–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J 1992. Variations in reading the genetic code. In Transfer RNA in protein synthesis (ed. Hatfield DL et al. ), pp. 23–85 CRC Press, Boca Raton, FL [Google Scholar]
- Percudani R, Pavesi A, Ottonello S 1997. Transfer RNA gene redundancy and translational selection in Saccharomyces cerevisiae. J Mol Biol 268: 322–330 [DOI] [PubMed] [Google Scholar]
- Plant EP, Nguyen P, Russ JR, Pittman YR, Nguyen T, Quesinberry JT, Kinzy TG, Dinman JD 2007. Differentiating between near- and noncognate codons in Saccharomyces cerevisiae. PLoS ONE 2: e517 doi: 10.1371/journal.pone.0000517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakwalska M, Rospert S 2004. The ribosome-bound chaperones RAC and Ssb1/2p are required for accurate translation in Saccharomyces cerevisiae. Mol Cell Biol 24: 9186–9197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recht MI, Douthwaite S, Puglisi JD 1999. Basis for prokaryotic specificity of action of aminoglycoside antibiotics. EMBO J 18: 3133–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnina MV, Gromadski KB, Kothe U, Wieden HJ 2005. Recognition and selection of tRNA in translation. FEBS Lett 579: 938–942 [DOI] [PubMed] [Google Scholar]
- Salas-Marco J, Bedwell DM 2005. Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J Mol Biol 348: 801–815 [DOI] [PubMed] [Google Scholar]
- Schulman LH 1991. Recognition of tRNAs by aminoacyl-tRNA synthetases. Prog Nucleic Acid Res Mol Biol 41: 23–87 [PubMed] [Google Scholar]
- Singh A, Ursic D, Davies J 1979. Phenotypic suppression and misreading in Saccharomyces cerevisiae. Nature 277: 146–148 [DOI] [PubMed] [Google Scholar]
- Soll D 1990. The accuracy of aminoacylation—ensuring the fidelity of the genetic code. Experientia 46: 1089–1096 [DOI] [PubMed] [Google Scholar]
- Stansfield I, Jones KM, Herbert P, Lewendon A, Shaw WV, Tuite MF 1998. Missense translation errors in Saccharomyces cerevisiae. J Mol Biol 282: 13–24 [DOI] [PubMed] [Google Scholar]
- Thompson R 1988. EF-Tu provides an internal kinetic standard for translational accuracy. Trends Biochem Sci 13: 91–93 [DOI] [PubMed] [Google Scholar]
- Thompson RC, Stone PJ 1977. Proofreading of the codon–anticodon interaction on ribosomes. Proc Natl Acad Sci 74: 198–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlenbeck OC, Martin FH, Doty P 1971. Self-complementary oligoribonucleotides: Effects of helix defects and guanylic acid–cytidylic acid base pairs. J Mol Biol 57: 217–229 [DOI] [PubMed] [Google Scholar]
- Ye L, Buck LM, Schaeffer HJ, Leach FR 1997. Cloning and sequencing of a cDNA for firefly luciferase from Photuris pennsylvanica. Biochim Biophys Acta 1339: 39–52 [DOI] [PubMed] [Google Scholar]
- Youngman EM, He SL, Nikstad LJ, Green R 2007. Stop codon recognition by release factors induces structural rearrangement of the ribosomal decoding center that is productive for peptide release. Mol Cell 28: 533–543 [DOI] [PubMed] [Google Scholar]
- Zaher HS, Green R 2009. Quality control by the ribosome following peptide bond formation. Nature 457: 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]