

Abstract
In single-particle reconstruction methods, projections of macromolecules at random orientations are collected. Often, several classes of conformations or binding states coexist in a biological sample, which requires classification, so that each conformation can be reconstructed separately. In this work, we examine bootstrap techniques for classifying the projection data. When these techniques are applied to variance estimation, the projection images (particles) are randomly sampled with replacement from the data set and a bootstrap volume is reconstructed from each sample. In a recent extension of the bootstrap technique to classification, each particle is assigned to a volume in the space spanned by the bootstrap volumes, such that the projection of the assigned volume best matches the particle. In this work we explain the rationale of these techniques by discussing the nature of the bootstrap volumes and provide some statistical analyses.
Keywords: classification, electron microscopy, bootstrapping, single particle, variance estimation
1. INTRODUCTION
In single-particle reconstruction methods [1], projections of macromolecules at randomly unknown orientations are collected by a transmission electron microscope. Often, several classes of conformations or binding states coexist in a sample. To obtain structures with high accuracy, it is required to separate the classes before reconstruction of the macromolecule takes place. In this work, we take a close look at bootstrap techniques for classifying the projection data. In the bootstrap techniques for variance estimation [2], the projection images (or particles) are randomly sampled with replacement from the data set and a bootstrap volume is reconstructed from each sample, assuming the orientations to be known. In a recent extension of the bootstrap technique to classification [3], each particle is assigned to a volume in the space spanned by the bootstrap volumes, such that the projection (in the same orientation as the particle) of the assigned volume best matches the particle. Then, a clustering algorithm applied to the assigned volumes determines the class to which the particle belongs. In this work we explain the rationale of these techniques by discussing the nature of the bootstrap volumes: i.e., how they relate to the underlying structural classes. Furthermore, several statistical analyses should become easy to apply in our framework. Finally, the way the particles are assigned to volumes in the space spanned by the bootstrap volumes is closely examined, and our proposed solution differs from that given in [3].
In Section 2 we discuss the nature of the bootstrap volumes and the effect of noise, as well as the classification method based on the analysis of the bootstrap volumes (‘bootstrap classification’). Section 3 shows the results obtained by bootstrap classification for simulated and experimental data and a comparison of the bootstrap method with a maximum likelihood classification approach [4]. Finally, discussion and conclusions are provided in Section 4.
2. BOOTSTRAPPING IN THE SINGLE PARTICLE METHOD
The aim of the bootstrapping technique [6] is to estimate the sampling distribution of an estimator by sampling with replacement from a given sample. It is a general-purpose approach to statistical inference, which circumvents the problem posed by the unavailability of large sample-size data.
2.1. Variability of classes via the bootstrap method
If we repeatedly sample, with replacement, the projection data and reconstruct a 3D volume from each sample (assumed the corresponding orientations are known), we obtain an estimate of the probability distribution that reflects the “variability” in the data. This variability, which is estimated as variance of the bootstrap volumes, is not only due to the presence of different conformational or binding states, which is the goal in 3D variance estimation [2], but it also comes from imperfections in the data collection such as instrument shot noise, “background” noise, reconstruction artifacts, contrast transfer function effect [1], alignment error, etc. Two major sources of variance are those due to the coexistence of different conformational or binding states and instrument noise. Unlike the latter, which is characteristics for 2D projection data, the former is three-dimensional in nature. Therefore, care must be taken when relating the two. Here we attempt to establish such a relation, by describing how the bootstrap volumes relate to the underlying true structures of the classes.
2.2. Bootstrap volumes and the class structures
We show that the bootstrap volumes are in fact approximations to convex combinations of the true structures. For simplicity in the discussions, we consider the case where the projection data come from a molecule occurring in M = 2 conformations. The analysis for more than two conformations follows straightforwardly. Assuming a discrete model, given the data , the least-squares estimator is a popular criterion1 for finding the true volume :
| (1) |
where R is the discrete Radon transform and ∥∥ is the Euclidean norm. Assuming that R’R is invertible (R’ denotes the transpose of R), the solution to (1) has a closed form
| (2) |
In bootstrapping, data come from the two structures. Let and be the respective sampled projection data, where Hi is the number of projection images taken from class i = 1, 2 and I is the number of pixels in a projection image. Let and be the corresponding Radon transforms; i.e., if are the true volumes, then in the absence of imperfections in the data
| (3) |
for i = 1, 2. Without loss of generality, we can set the sampled data to be and . Substituting the values of y and R in (2), we obtain an expression for the reconstructed bootstrap volume, based on the least-square criterion,
| (4) |
Taking in to account (3) and the fact that , the bootstrap volume can be viewed as a sum of linear transformations of the true volumes x1 and x2
| (5) |
whose linear transformations , sum up to the identity matrix of appropriate size.
We have proved that a bootstrap volume is a sum of linear transformations of the true classes. In fact, this sum is an approximation to a convex combination of the classes. To see this, we note that the effect of , is essentially a blurring with a kernel that goes like 1/r (r is the radial distance; [8]) multiplied by a factor that is proportional to the number of projection images Hi taken from class i = 1, 2. Thus, the effect of the linear transformations is basically a constant multiplication by factor Hi/H, where H is the total number of projections in a bootstrap sample; i. e.,
| (6) |
2.3. Profile of the distribution of the bootstrap volumes
It is easy to see that the right hand side of (6) corresponds to summing volumes from Bernoulli trials with support {x1, x2}, probability p (whose realization is H1/H in this case), and dividing the sum by the total number of projections H:
| (7) |
That is, most of the bootstrap volumes are located near the center of the convex hull with vertices x1 and x2. A concentration near the vertices would be more desirable from the point of view of estimating the convex hull.
2.4. Imperfections in the projection data
Imperfections in the data come from the electron optics (astigmatism, spherical aberration, etc.), background noise, shot noise, alignment error, etc. Let us assume an additive noise model for the 2D projection data, such that it can be “back-projected,” leading to an additive noise model for the 3D volumes. That is, in (3) we have that, for i = 1, 2, yi =Ri (xi + gi)=Rixi + hi, for some gi and hi = Rigi, which are respectively the 3D and 2D noise component. This is realizable if, for instance, hi consists of uniform independent Gaussians at the pixels, the reconstruction region is spherical, and gi are independent Gaussians at the voxels, with lower variance close the center of the volume than away from the center. Accordingly, (7) becomes . The variance of xBS is thus composed of two terms: one that is due to the class difference, which is the signal part (computed as , where dj is the difference between x1 and x2 at voxel j, for j = 1, …, J) and the other term that is due to noise. Since both terms are scaled by factor H, the signal-to-noise ratio (SNR) for detecting class difference will not be improved by increasing H.
2.5. Classification using bootstrap method
An immediate consequence of the fact that the bootstrap volumes are convex combinations of the class volumes is that the space spanned by the bootstrap volumes approximates that spanned by the class volumes. Hence, for each projection image we can restrict ourselves in that space to estimate the volume that generated that projection. Ideally, these estimated volumes cluster around the true class volumes.
We now proceed to consider the case of M ≥ 2 classes. Suppose we have generated a sufficient number of bootstrap volumes and H is large enough, so that their principal directions are close to the principal directions of the space of the class volumes. Let be the resulting eigen-volumes and z0 the average volume. Given a projection image yP, we wish to find an element z (α) in that space (reconstitution problem); i.e., , such that the discrepancy between its projection (in the same orientation as that of yP) and yP is minimized. For simplicity, in this paper we choose the discrepancy to be the Euclidean distance
| (8) |
To avoid shift and scale variabilities, yP and Pzn, 0 ≤ n ≤ N, are replaced by their normalized (to zero mean and unit variance) version, prior to computing α in (8).
2.6. Algorithm for classification based on bootstrapping
Algorithm 1 summarizes our proposed approach to classification using bootstrapping, which is the same as the existing algorithm [3], except for the way in which the coefficients are determined. In [3], apparently the n are set to be the inner product between Pzn and yP, 1 ≤ n ≤ N. We stress that, due to the space limitation, the description of our algorithm as described here is rather sketchy. We will treat in a separate communication such issues as the dependence of the results on the number of bootstrap volumes and particles used, filtering of the bootstrap volumes, criteria for estimating α, etc., following our framework; though several useful statistical analyses are already dealt in [9] for variance estimation.
Algorithm 1.
Bootstrap Classification
| 1. Sample with replacement the projection data |
| 2. Reconstruct bootstrap volume from each sample |
| 3. Compute the eigenvolumes of the set of samples |
| 4. For each particle: |
| 4a. Project the eigenvolumes in the orientation of the particle |
| 4b. Compute α in (8) |
| 6. Classify the particles by clustering α, for all the particles |
3. RESULTS
We tested our proposed algorithm on experimental and simulated data. The experimental data set consists of ten thousand 130 × 130 particles randomly chosen from a larger data set, on which the maximum likelihood (ML) classification method (a popular alternative to ours) [5] was previously tested, giving rise to two main structures: the 70S E. coli ribosome in the classical and hybrid state (see Fig. 1). For the simulated data set, we used these two states as phantoms and generated ten thousand 130 × 130 noisy projections in the exact same manner as described in [10]; i.e., the SNR was 0.06 and the CTF was applied. To gain computational speed, we decimated the particles to size 652 in both data sets, aligned them to a library of reference projections (on a ten-degree angular grid) of a common 3D reference (the density map of the ribosome in one of the two states), and used SPIDER [11] to generate forty thousand bootstrap volumes in each case. It was necessary to filter the volumes, and for that we used a low-pass filter with cut-off 0.1, which was limited to the first lobe of the CTF [9] (though this value was not optimized). We relied on SPARX [12] to perform the eigen-decompositions. The clustering of the coefficient vectors α was performed via the k-means algorithm. To assess the classification performance, we also tested the ML method on the simulated data set.
Fig. 1.
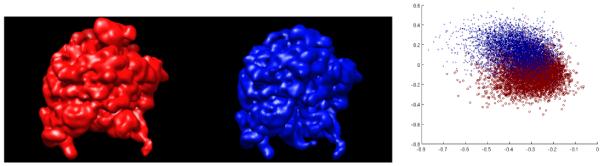
70S E. coli ribosome in classical (left) and hybrid (center) states [5], and our classification (right) of noisy mixed projections of the two structures.
3.1. Experimental data
Fig. 2 shows the result of classification using our version of the bootstrapping classification method. We used five eigen-volumes (N = 5) and looked for two classes (M = 2). One can immediately recognize the differences of the two structures in the presence/absence of the EF-G and the position of the L1 stalk. Not visible is the presence/absence of the A-site tRNA, which is another difference that the algorithm was able to pull out.
Fig. 2.
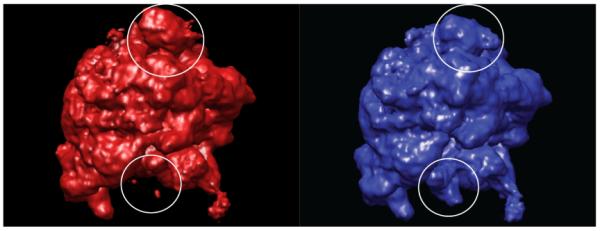
Reconstructions from ten thousand classified particles from the experimental data set, showing the two classes corresponding to the classical and hybrid states. Differences in the position of the L1 stalk and the presence/absence of the EF-G are highlighted.
3.2. Simulated data
As measured by a classification error score whose minimum is 0% (perfect classification) and the maximum is 50% (random guess), the bootstrapping method (with N = M = 2) yields 16%±0.2% (see Fig. 1) versus 34%±10% for the ML approach [4] (refinement angle of ten degrees, two classes, 20 iterations). The confidence interval of the classification error was obtained by running the respective algorithms ten times. The large dispersion of the figure in the ML method is likely due to the presence of local maxima (which is not an issue in the bootstrapping approach, except for the k-means algorithm) and the relatively small data size.
4. DISCUSSION AND CONCLUSIONS
We have explained the rationale of bootstrapping in the context of classification and proposed an algorithm the differs from the one initially proposed in an important detail. Through repeated reconstructions from bootstrap samples, we can estimate the space spanned by the underlying class structures. By searching in this space a volume whose projection best matches a given particle helps us decide on the class to which the particle belongs. We show that the bootstrapping approach offers a competitive alternative to current popular methods, such as the ML approach: the former does not suffer from local maxima effect (except for the clustering algorithm, if k-means is used). It is noted that in the experiments, the angular assignment of the projection data was done only once, at the beginning, and with respect to one reference volume. An iterative process, in which the angles are refined with respect to the current reconstructed class volumes, should provide even better results. Further improvement may also come from alternative ways of finding the coefficient vector α, since the Euclidean distance in (8) is sensitive to outliers. Finally, it should be noted that classification becomes more challenging as the variability of the structure classes competes with noise in the data, both of which are scaled down by the number of particles used in the sampling. Hence, to reduce the noise, it is necessary to find ways other than increasing the number of particles; for that, if filtering is used, the loss of high frequency information can be detrimental for the classification.
5. ACKNOWLEDGMENT
We are grateful to Zhi-Quan (Tom) Luo for help with optimization.
Footnotes
For instance, the well known SIRT algorithm can be viewed as a gradient descent algorithm for finding xLSQ [7].
6. REFERENCES
- [1].Frank J. Three-Dimensional Electron Microscopy of Macromolecular Assemblies. Oxford University Press; New York: 2006. [Google Scholar]
- [2].Penczek PA, Yang C, Frank J, Spahn CMT. Estimation of variance in single-particle reconstruction using the bootstrap technique. J. Struc. Biol. 2006;154:168–183. doi: 10.1016/j.jsb.2006.01.003. [DOI] [PubMed] [Google Scholar]
- [3].Spahn CMT, Penczek PA. Exploring conformational modes of macromolecular assemblies by mutli-particle cryo-EM. Current Opinion in Structural Biology. 2009;19:623–631. doi: 10.1016/j.sbi.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Scheres SHW, Valle M, Grob P, Nogales E, Carazo JM. Maximum likelihood refinement of electron microscopy data with normalization errors. J. Struc. Biol. 2009;166:234–240. doi: 10.1016/j.jsb.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Scheres SHW, Gao H, Valle M, Herman GT, Eggermont PPB, Frank J, Carazo JM. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat. Methods. 2007;4:27–29. doi: 10.1038/nmeth992. [DOI] [PubMed] [Google Scholar]
- [6].Efron B. Bootstrap methods: Another look at the jack-knife. The Annals of Statistics. 1979;1:1–26. [Google Scholar]
- [7].Herman GT. Image Reconstruction from Projections: The Fundamentals of Computerized Tomography. Academic Press; New York: 1980. [Google Scholar]
- [8].Deans SR. The Radon transform and some of its applications. John Wiley & Sons; New York: 2006. [Google Scholar]
- [9].Zhang W, Kimmel M, Spahn CM, Penczek PA. Heterogeneity of large macromolecular complexes revealed by 3D cryo-EM variance analysis. Structure. 2008;16:1770–1776. doi: 10.1016/j.str.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Baxter WT, Grassucci RA, Gao H, Frank J. Determination of signal-to-noise ratios and spectral SNRs in cryo-em low-dose imaging of molecules. J. Struc. Biol. 2009;166(2):126–132. doi: 10.1016/j.jsb.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shaikh TR, Gao H, Baxter W, Asturias FJ, Boisset N, Leith A, Frank J. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat. Protoc. 2008;3:1941–1974. doi: 10.1038/nprot.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hohn M, Tang G, Goodyear G, Baldwin PR, Huang Z, Penczek PA, Yang Ch., Glaeser RM, Adams P, Ludtke SJ. SPARX, a new environment for cryo-em image processing. J. Struct. Biol. 2007;157:47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]