

Abstract
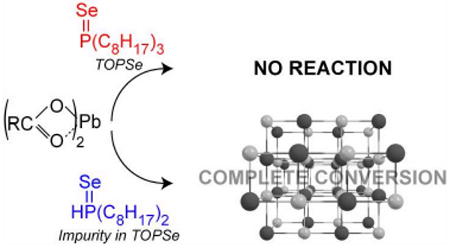
We have investigated the reaction mechanism responsible for QD nucleation using optical absorption and nuclear magnetic resonance spectroscopies. For typical II-VI and IV-VI quantum dot (QD) syntheses, pure tertiary phosphine selenide sources (e.g. trioctylphosphine selenide (TOPSe)) were surprisingly found to be unreactive with metal carboxylates and incapable of yielding QDs. Rather, small quantities of secondary phosphines, which are impurities in tertiary phosphines, are entirely responsible for the nucleation of QDs; their low concentrations account for poor synthetic conversion yields. QD yields increase to nearly quantitative levels when replacing TOPSe with a stoiciometric amount of a secondary phosphine chalcogenide such as diphenylphosphine selenide. Based on our observations, we have proposed potential monomer identities, reaction pathways and transition states, and believe this mechanism to be universal to all II-VI and IV-VI QDs synthesized using phosphine based methods.
The significant potential of semiconductor nanocrystal quantum dots (QDs) for photovoltaics, bio-labeling/imaging, and light-emitting diodes has been well-reported.1 Unfortunately, realizing this potential has yet to occur, in part due to a synthetic procedure that lacks scalability and high conversion yield. Current syntheses can produce high-quality QDs with respect to photoluminscent quantum yield (QY >50%) and size distribution (± 5%). However, growth temperatures are high (∼300 °C for CdSe), conversion yields are poor (< 2%) and the method suffers from well-known irreproducibilities.2,3 We suggest that the lack of a rational synthetic mechanism has stalled advancements in QD synthetic design.
The essential components in the synthesis of high-quality II-VI and IV-VI QDs have remained largely unchanged for 20 years; typically employing tertiary phosphine chalcogenides and metal salts as the reactive precursors.4 Notable advances include removing the need for pyrophoric compounds,5 and designing ligands to obtain a greater control over QD size, size distribution, and shape.3 Recently, secondary phosphines were recognized as useful additives to phosphine based syntheses demonstrating improvements in conversion yield.2,6 Here, we identify a reaction pathway consistent with previous synthetic observations and illuminate the role of precursor reagents in the formation of QDs. Specifically, we have identified that secondary phosphine chalcogenides are the reactive species directly responsible for the formation of QD nuclei.
We studied reactions performed between high purity tertiary phosphine chalcogenides (e.g. triethylphosphine selenide, triisopropylphosphine selenide and triphenylphosphine selenide) and Pb(oleate)2 heated to 120 °C, which were monitored by both optical absorption spectroscopy and 31P NMR. In all casers, no reactivity was observed even after five hours of heating, in stark contrast to commercially obtained trioctylphosphine selenide (TOPSe), which reacts with Pb(oleate)2 in minutes, forming a dark product. Upon addition of a secondary phosphine (diisopropylphosphine) to high purity tertiary phosphine chalcogenide precursors, reaction rates in all cases accelerated rapidly yielding PbSe QDs. Typical tertiary phosphine sources (tributylphosphine and trioctylphosphine (TOP)) used for QD syntheses are highly impure, and all contain measurable quantities of secondary phosphines (dibutylphosphine (31P δ -69.5 ppm) and dioctylphosphine (31P δ −69.7 ppm) respectively), as shown in the supporting information figures 1 and 2. The importance of secondary phosphines is further emphasized by Figure 1, which shows that growth kinetics and yield for PbSe magic-sized clusters (MSCs)7 are dependent upon dioctylphosphine concentration (i.e. [(DOP)]) rather than [TOP] or [TOPSe]. In fact, dioctylphosphine selenide (DOPSe) can be observed as an impurity in neat TOPSe (31P δ 4.7 ppm, 725 Hz 1J(31P – 77Se)), but disappears immediately upon combination with a metal carboxylate at 300K suggesting that it plays a critical part in QD nucleation.
Figure 1.
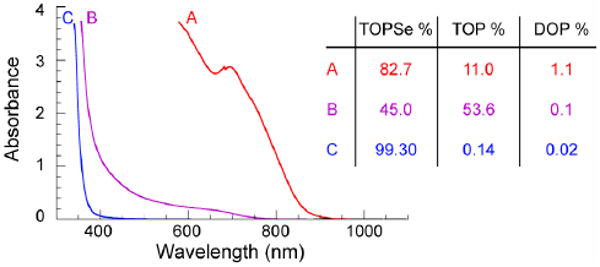
Optical absorption of PbSe MSCs after 40 minutes at 40 °C for the reaction between Pb(oleate)2 and TOPSe with varying dioctylphosphine concentrations (see SI for complete growth series). Note the negligible reactivity for the purest TOPSe source.
In order to further elucidate the role of secondary phosphines in the formation of chalcogenide QDs we studied the reaction between diphenylphosphine selenide (DPPSe) and Pb(oleate)2 via NMR spectroscopy utilizing 1H, 13C, and 31P nuclei. The combination of toluene solutions of DPPSe and Pb(oleate)2 results in an immediate reaction producing PbSe MSCs10 with complete conversion of DPPSe (verified through 31P NMR) at room temperature (See SI for experimental details).Over the course of the reaction, we identified eight reaction products in addition to the PbSe species (See Table I and NMR spectra in supporting information). Monitoring the temporal progression of the various reactants and products over the course of the reaction allowed for the postulation of a hypothetical mechanism for monomer formation consistent with our observations (Scheme 1). Significantly, we also found the organic byproducts were identical for both the CdSe and PbSe QD syntheses, leading us to conclude that this mechanism is universal to all phosphine-based syntheses of both II-VI and IV-VI semiconductor QDs.
Table I.
Observed compounds resulting from the reaction between diphenylphophine selenide and metal carboxylates.
| δ (ppm)a |
δ (ppm)a |
||
|---|---|---|---|
| 2 RCOOH | 12.2 - 12.5b,e | 7 Ph2PH | - 40.2c,e 215 Hz 1J(31P - 1H) |
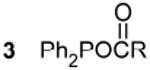 |
98.9c,g | 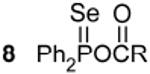 |
77.1c,h 875 Hz 1J(31P - 77Se) |
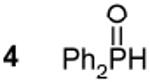 |
22.0c,e 441 Hz 1J(31P - 1H) | 12 Ph2PPPh2 | - 14.5c,e,f |
 |
168.9d,e | 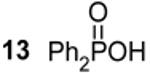 |
25.7c,e,f |
| R=C17H33 | |||
All spectra were recorded in toluene-d8
1H NMR
31P NMR
13C NMR.
Verified by addition of authentic sample.
Isolated diffraction quality single crystals from the reaction.
Chemiker Zeitung 1982, 391-395.
Chem. Commun., 2005, 2692-2694.
Scheme 1.
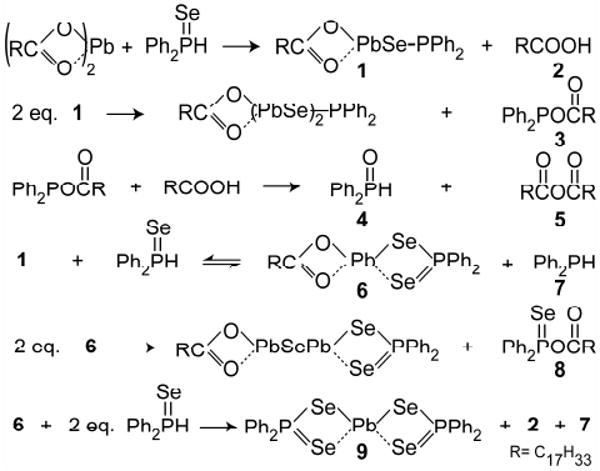
Possible reaction mechanism for metal-chalcogenide bond formation.
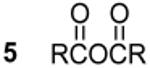
A QD monomer species is represented by structure 1 and is produced through a ligand disproportionation yielding oleic acid. Intermolecular reactions between multiple monomers (1) produce 3 while enlarging the PbSe unit. While 1 could not be isolated due to its predicted self-reactivity, 3 was observed in good quantity and shown to produce both 4 and 5 by reaction with oleic acid (See SI). An interesting parallel can be made between intermediates in condensation polymerizations (e.g. nylon) and 1 whereby asymmetrically terminated monomers combine to lengthen the repeat unit. 4 and 5 have been observed in prior mechanistic studies using the standard TOPSe QD synthesis2,8 and likely arise from the presence of secondary phosphine impurities (i.e DOPSe and DOP).
We varied the ratio of DPPSe: Cd(oleate)2 from 10:1 to 1:2 and observed that the number and relative abundance of inorganic products depended heavily on the initial proportion of reagents. Reaction conditions with a large excess (>5:1) of phosphine chalcogenide (DPPSe) yield X-ray quality crystals of 9 without production of any larger PbSe species such as MSCs. In addition to 9, several other P-containing species are observed to predominate only under DPPSe rich conditions. Conversely, reactions performed at 300 K with an excess of Pb(oleate)2 (also observed for Cd(oleate)2) rapidly yield large quantities of PbSe (CdSe) MSCs7 and oleic acid with only two major P-containing species (3 and 12). After 24 hrs at room temperature both 3 and oleic acid disappear in favor of diphenylphosphine oxide (4) and oleic anhydride (5) without significant changes to the size of the PbSe (CdSe) MSC product. This reaction appears to be an organic side-reaction independent of monomer formation and growth. Altogether, the reagent ratio dependence illustrates that at least two concurrent reaction pathways compete for the formation of product: (1) reactions rich in metal carboxylates produce monomers (1 and/or 6) that polymerize yielding MSe MSCs and eventually QDs, (2) reactions rich in DPPSe induce termination pathways yielding a stable metal-selenide complex and minimal MSe product.
Previously proposed roles for secondary phosphines do not fit with our observations (Scheme 2). Applying heat (140 °C) to a mixture of diphenylphosphine and Pb(oleate)2 did result in Pb0, but the reaction took hours, much longer than the reaction for a typical QD synthesis, which takes several minutes at equivalent temperatures. We also found that the metal product of the above reaction, Pb0, is inert to commonly used selenium sources such as TOPSe and thus is likely uninvolved in QD formation. Therefore, we suggest that the role of secondary phosphines can not simply be reduction of metal carboxylates to metal, but rather to promote the dissociation of oleate and formation of a metal-phosphine complex (10, 11).9
Scheme 2.
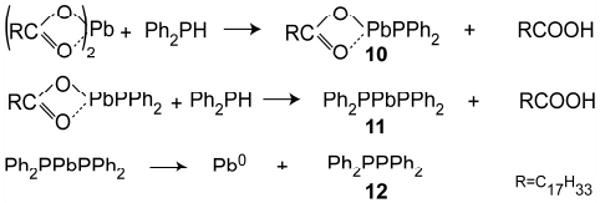
Reaction of secondary phosphines with metal carboxylates.
To test our mechanism under QD reaction conditions typically employed in the literature, we studied the evolution of product species in a reaction between Pb(oleate)2 and TOPSe when adding 15% (mol,mol) of DPP. Prior to combination of reagents, DOP (δ -69.1 ppm), DOPSe (δ 4.3 ppm) and DPPSe (δ 5.9 ppm) are observed by 31P NMR, in addition to the many impurities normally found in TOP (Figure 2A). However, in agreement with our studies of QD nucleation with pure secondary phosphine species, these species also disappear immediately after combination with Pb(oleate)2 which supports our hypothesis that secondary phosphine impurities are entirely responsible for QD growth (Figure 2B). Additional peaks are observed after combination of reagents and are assigned to tetraphenyldiphosphine (δ -14.0 ppm, 12) and 9-Octadecenoxydiphenylphosphine (δ 98.9 ppm, 3) which directly corresponds to the products of our model reactions between DPPSe and M(oleate)2.
Figure 2.
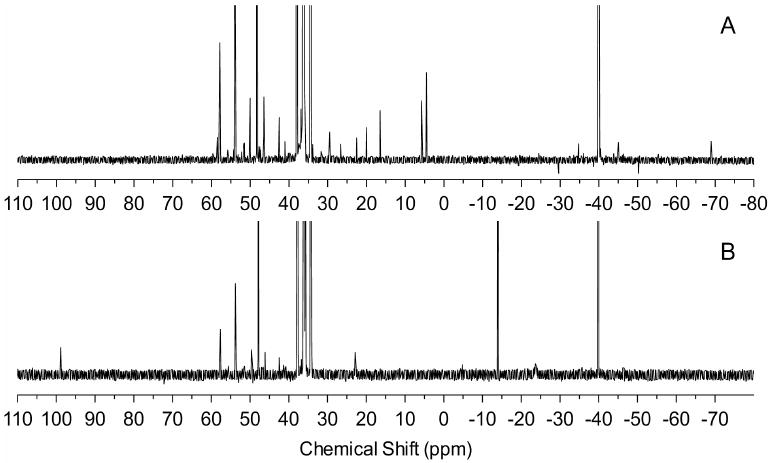
31P{1H} NMR for A.) neat TOPSe with 15% (mol,mol) DPP and B.) 30 minutes after combining with Pb(oleate)2 at 40 °C. The major products from this reaction (3 (98.9 ppm) and 12 (-14.5 ppm)) are consistent with a reaction between sec-phosphine chalcogenides and metal carboxylates.
Even in QD syntheses solely based upon TOPSe we observe byproducts indicative of the reaction between secondary phosphine chalcogenides (i.e. DOPSe, DPPSe) and metal carboxylates. Considering that pure tertiary phosphine selenides are inert to metal carboxylates, and the observation of identical reaction products for reaction with pure secondary phosphine selenide precursors, we suggest that TOPSe's role in nucleation may simply be as a homogeneous source for selenium. In support of this hypothesis we note that selenium liberated from TOPSe can exchange to either secondary phosphines or to intermediate metal phosphine complexes (10,11).
In order to generate a II-VI or IV-VI QD monomer species several pieces of evidence suggest that Se exchange is indeed occurring between an unobserved metal-phosphine complex (10,11) and TOPSe. Chalcogenide exchange is a well known and useful tool for phosphine protection strategies in organic synthesis. Rapid equilibration of Se substitution is often observed in mixtures of phosphines with the more basic phosphorus nuclei representing the preferred substitution.11 Thus, literature precedent suggests that TOPSe represents a soluble organic Se source capable of efficient Se exchange to more reactive species during a QD synthesis. Indeed, we have observed direct Se exchange between TOPSe and impurity quantities of dioctylphosphine via NMR spectroscopy; in this case, the exchange equilibrium is weighted towards TOPSe but nonetheless results in an observable exchange. Altogether, the strong effect of secondary phosphines on QD synthetic yield, the importance of the DOPSe impurity to reactivity, and the selenium exchange equilibrium present during the reaction strongly suggests that DOPSe represents the reactive species responsible for QD formation.
Reactions between DPPSe and metal carboxylates, at room temperature, completely consume both reagents rapidly yielding very small MSe MSCs. Increasing the temperature for the above reaction (to 80 °C for PbSe and 200 °C for CdSe) produced PbSe or CdSe QDs, respectively, with near-quantitative conversion yields (yield >90% based upon literature extinction coefficients10). QDs made from the above process can often possess optical properties and size-distributions comparable to the best literature methods without the use of post-synthetic processing (examples are shown in the SI). Further, the addition of excess ligand was not necessary for growth but its presence limited interparticle aggregation which became evident after several hours. On the other hand, we found QD size control using pure secondary phosphine selenide is not well understood and depends upon a complicated set of interrelated parameters including reaction temperature, surface capping ligand, and reagent stoichiometry. A detailed study is underway evaluating these many parameters and their individual roles in producing monodisperse QDs with a tunable size.
In summary, we have developed a rational synthetic mechanism for QD monomer formation that accounts for previous observations of several organic species and identifies the likely structure of the reactive intermediate. The extremely high conversion yield of QD reactions based on secondary phosphine chalcogenides, and at a reduced temperature, stresses the important role of small quantities of DOPSe impurities in the conventional synthesis. Further, our results may explain the low yields in TOPSe based procedures (<2%) and the notorious inconsistencies and irreproducibilities often seen in QD syntheses. In addition to formation of MSe QD species, we anticipate similar reactivity for secondary phosphine sulfide and telluride reagents operating under an identical nucleation mechanism as detailed above with equivalent improvements to conversion yield. A complete kinetic analysis is necessary to prove this particular mechanism; however, our study represents an important step towards understanding the complex chemistry associated with both nucleation and growth. Our expectation is that a better understanding of the QD reaction mechanism should allow for future use of cheaper and more environmentally benign reagents in the synthesis of a wide array of QD materials.
Supplementary Material
Acknowledgments
Funding was provided by the Camille and Henry Dreyfus Foundation, the Rochester Human Immunology Center supported by the National Institutes of Health, and the National Science Foundation. We also thank William Jones, Robert Boeckman, Mary Lenczewski and Pu Luo for helpful discussions. We acknowledge assistance from William Brennessel at the X-ray Crystallographic Facility of the Department of Chemistry at the University of Rochester.
Footnotes
Supporting Information Available: Experimental details regarding QD synthesis, table of observed reaction products, NMR spectra, and the X-ray crystal structure of species 9. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.(a) Nozik AJ. Annu Rev Phys Chem. 2001;52:193. doi: 10.1146/annurev.physchem.52.1.193. [DOI] [PubMed] [Google Scholar]; (b) Colvin VL, Schlamp MC, Alivisatos AP. Nature. 1996;370:354. [Google Scholar]; (c) Alivisatos P. Nat Biotechnol. 2004;22(1):47. doi: 10.1038/nbt927. [DOI] [PubMed] [Google Scholar]
- 2.Steckel JS, Yen BKH, Oertel DC, Bawendi MG. J Am Chem Soc. 2006;128(40):13032. doi: 10.1021/ja062626g. [DOI] [PubMed] [Google Scholar]
- 3.Peng ZA, Peng X. J Am Chem Soc. 2002;124(13):3343. doi: 10.1021/ja0173167. [DOI] [PubMed] [Google Scholar]
- 4.Murray CB, Norris DJ, Bawendi MG. J Am Chem Soc. 1993;115(19):8706. [Google Scholar]
- 5.Qu L, Peng A, Peng X. Nano Lett. 2001;1(6):333. [Google Scholar]
- 6.Joo J, Pietryga JM, McGuire JA, Jeon SH, Williams DJ, Wang HS, Klimov VI. J Am Chem Soc. 2009;131(30):10620. doi: 10.1021/ja903445f. [DOI] [PubMed] [Google Scholar]
- 7.Evans CM, Guo L, Peterson JJ, Maccagnano-Zacher S, Krauss TD. Nano Lett. 2008;8(9):2896. doi: 10.1021/nl801685a. [DOI] [PubMed] [Google Scholar]
- 8.Liu H, Owen JS, Alivisatos AP. J Am Chem Soc. 2007;129(2):305. doi: 10.1021/ja0656696. [DOI] [PubMed] [Google Scholar]
- 9.Driess M, Janoschek R, Pritzkow H, Rell S, Winkler U. Angew Chem Int Ed Engl. 1995;34(15):1614. [Google Scholar]
- 10.Dai Q, Wang Y, Li X, Zhang Y, Pelligrino DJ, Zhao M, Zou B, Seo J, Wang Y, Yu WW. ACS Nano. 2009;3(6):1518. doi: 10.1021/nn9001616. [DOI] [PubMed] [Google Scholar]
- 11.Brown DH, Cross RJ, Keat R. JCS Dalton. 1980;6:871. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.