

Abstract
Recombinant oncolytic viruses represent a promising alternative option for the treatment of malignant cancers. We have previously reported the safety and efficacy of recombinant vesicular stomatitis virus (VSV) vectors in a rat model of hepatocellular carcinoma (HCC). However, the full potential of VSV therapy is limited by a sudden decline in intratumoral virus replication observed early after viral administration, a phenomenon which coincides with an accumulation of inflammatory cells within infected lesions. To overcome the anti-viral function of these cells, we present a recombinant virus, rVSV-UL141, which expresses a protein from human cytomegalovirus known to downregulate the NK cell-activating ligand CD155. The modified vector resulted in an inhibition of NK cell recruitment in vitro, as well as decreased intra-tumoral accumulations of NK and NKT cells in vivo. Administration of rVSV-UL141 via hepatic artery infusion in immune-competent Buffalo rats harboring orthotopic, multifocal HCC lesions, resulted in a one-log elevation of intratumoral virus replication over a control rVSV vector, which translated to enhanced tumor necrosis and substantial prolongation of survival. Moreover, these results were achieved in the absence of apparent toxicities. The present study suggests the applicability of this strategy for the development of effective and safe oncolytic agents to treat multi-focal HCC, and potentially a multitude of other cancers, in the future.
Keywords: oncolytic virotherapy, HCC, inflammatory response, natural killer cells, recombinant VSV
INTRODUCTION
Hepatocellular carcinoma (HCC) represents a major health concern throughout the world, accounting for over one million cases annually1–3. While the incidence of HCC has more than doubled over the last two decades,4, 5 the emergence of innovative treatment options remains extremely limited. Currently, only a small percentage of patients are candidates for curative treatments, due to underlying liver disease, extent of metastasis, and limited availability of livers for transplantation.6 The remaining majority of patients are eligible only for palliative treatments, which result in modest prolongation of survival7, 8. HCC is therefore an aggressive disease, characterized by poor prognosis and limited treatment options, highlighting the critical need for the development of novel and effective therapeutic agents.
Oncolytic viruses have gained recognition as attractive alternatives to conventional cancer therapy, due to their intrinsic ability to selectively replicate and kill tumor cells in vitro and in vivo.9–12 Vesicular stomatitis virus (VSV), a member of the Rhabdoviridae family, is a particularly appealing oncolytic vector because of its short replication cycle and ability to reach high titers in many rodent and human tumor cell lines. It is an enveloped, negative-strand RNA virus with a wide host range, which replicates selectively within tumor cells due to defects in anti-viral type I interferon responses in these cells.13 While the natural hosts for VSV infection are cattle, horses and pigs, infections in humans are generally asymptomatic or result in mild febrile illness,14 indicating that it has potential for safe clinical application in the future. Moreover, VSV is not endemic to the North American population, implying that there will not be preexisting neutralizing antibodies or memory cellular immune responses to interfere with its replication potential in patients.15
We have previously described the efficacy of recombinant VSV as an oncolytic vector for treatment of orthotopic HCC in immune-competent rats.16 We demonstrated that VSV, when administered at its maximum tolerated dose (MTD) via the hepatic artery, could gain access to and selectively replicate in multi-focal HCC tumors of various sizes, resulting in tumor necrosis and prolongation of animal survival.17–19 Although encouraging, complete tumor regression was not achieved, and the remaining viable tumor ultimately relapsed, resulting in the eventual demise of the treated animals. These observations encouraged us to seek an alternate approach to improve the outcome of VSV treatment for multi-focal HCC.
Following intra-arterial infusion of the vector, robust virus replication quickly commences within infected tumor cells, reaching a peak in viral titer at approximately 24 hours post-treatment, followed by a logarithmic decline in subsequent days.17 Given that the neutralizing anti-viral antibody response in the host is not observed until later timepoints,17 we suspected that the rapid reduction in intra-tumoral virus titers after the first day is instead the result of anti-viral inflammatory responses launched at the site of infected tumor cells. Rapid recruitment and activation of cellular components of the innate immune system, such as granulocytes, natural killer (NK) cells, NKT cells and macrophages, have been observed at sites of viral infection.20 These cells participate in the anti-viral response both by direct killing of infected cells and by production of anti-viral cytokines. It was recently demonstrated that the host inflammatory response is correlated with the limited ability of Herpes Simplex Virus (HSV) to replicate within tumor cells,21 and suppression of inflammatory responses by treatment with chemotherapeutic cyclophosphamide resulted in enhanced anti-tumor efficacy of HSV.22 Based on these data, coupled with our own observations of NK and NKT cell accumulation coinciding with the logarithmic decline of intratumoral VSV titers after one day, we demonstrated in a previous report that the host inflammatory response to VSV infection plays a critical role in suppression of intratumoral VSV replication, and counteracting these responses substantially enhances VSV oncolysis and treatment efficacy23.
As the inflammatory processes of the host provide a difficult challenge to the survival of invading viruses, successful viral propagation within mammalian hosts is dependent in part on their ability to evade the anti-virus arsenal launched by the host immune system. To this end, many viruses have evolved intricate mechanisms to evade detection and subsequent destruction by various immune cells in the host.24 We have previously reported a recombinant VSV vector, which encodes a viral chemokine binding protein (vCKBP) derived from a heterologous virus, for the purpose of inhibiting anti-viral inflammatory cells in vivo. In that study, we characterized a vector expressing the secreted form of the equine herpesvirus-1 glycoprotein G (gGEHV-1), which is a known vCKBP, and demonstrated that the expression of gGEHV-1 inhibited the accumulation of NK and NKT cells within treated tumors, which resulted in an improvement in therapeutic outcome. However, given that gGEHV-1 is considered to be a broad-range vCKBP which binds C, CC, and CXC chemokines with high affinity25, in our current study we sought to employ an alternate strategy to further substantiate our hypothesis, while simultaneously restricting the specificity of the transgene to NK and NKT cells.
Here we present a novel VSV vector, rVSV-UL141, which expresses a gene cloned from human cytomegalovirus (HCMV), known to inhibit the function of NK cells by blocking the ligand of NK cell-activating receptors. In addition to the inhibitory function on NK cells, we demonstrated that UL141 also blocks the migration of NKT cells to infected tumor sites, and the combined affect resulted in greatly elevated intratumoral virus replication, leading to enhanced tumor necrosis and substantially prolonged survival of immune-competent rats bearing syngeneic and multi-focal HCC lesions in the liver.
Materials and Methods
Cell Lines
The rat HCC cell line McA-RH7777 was purchased from the American Type Culture Collection (ATCC) (Manassas, VA) and maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Mediatech, Herndon, VA) in a humidified atmosphere at 10% CO2 and 37°C. BHK-21 cells (ATCC) were maintained in DMEM in a humidified atmosphere at 5% CO2 and 37°C. All culture media were supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich, St. Louis, MO) and 100U/ml penicillin-100mg/ml streptomycin (Mediatech).
Plasmid Construction
The construction of the recombinant VSV vector expressing mutant (L289A) Newcastle Disease Virus fusion protein (rVSV-F) was previously described18. To clone a secreted form of the UL141HCMV gene, the predicted C-terminal transmembrane domain was determined by hydrophobicity plot to correspond to the first 823bp of the UL141 open reading frame, which was consistent with the findings of others26. This portion of the UL141 open reading frame, with a stop codon and appropriate restriction sites for subsequent cloning, was synthesized chemically (GenScript, Piscataway, NJ). To generate a VSV plasmid simultaneously expressing UL141HCMV and firefly Luciferase, the ubiquitously expressed ECMV internal ribosomal entry site (IRES) was introduced such that the two genes could be co-translated as a single transcriptional unit. This cassette was inserted into the full-length pVSV-XN2 plasmid, using the unique restriction sites XhoI and NheI in the untranslated region of the endogenous VSV glycoprotein. This was the same cloning scheme employed in the construction of rVSV- gGEHV-1 described in our previous publication23. Sequencing of the plasmids was conducted in the DNA Sequencing Core Facility at Mount Sinai School of Medicine.
Generation of Recombinant Viruses
To rescue the recombinant VSV vectors, an established method of reverse genetics was employed 27, 28. Briefly, BHK-21 cells were infected with vaccinia virus expressing the T7 RNA polymerase (vTF-7.3), and subsequently transfected with the full-length rVSV plasmid in addition to plasmids encoding VSV N, P and L proteins using LipofectAMINE 2000 transfection reagent (Invitrogen, Carlsbad, CA). 72 hours post-transfection, supernatants were centrifuged and filtered through a 0.22µm filter to remove the majority of vaccinia virus, and transferred onto fresh BHK-21 cells. Any remaining vaccinia virus was eliminated by plaque purification, and the titers of recombinant VSV stocks were determined by plaque assays on BHK-21 cells.
Multicycle Growth Curves
Morris (McA-RH7777) cells were plated in 24-well plates at 5 × 104 cells/well and infected at a MOI of 0.01 with rVSV-F or rVSV-UL141 in triplicate. After infection at room temperature for 30 minutes, cells were washed twice with PBS to remove any unabsorbed virus, and fresh complete medium was added. At the indicated time points after infection, 100µl of supernatant were collected and assayed for viral titer by TCID50 assays.
In Vitro Cytotoxicity Assay
McA-RH7777 cells were seeded in 24-well plates at 5 × 104 cells/well overnight and then infected with rVSV-F or rVSV-UL141 in triplicate, at a MOI of 0.01 the following day. Cell viability was measured at the indicated time points after infection using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Cell Proliferation Kit I, Roche, Indianapolis, IN). All cell viability data are expressed as a percentage of viable cells as compared to mock-infected controls at each time point.
In Vitro NK Cell Migration Assay
For preparation of rat NK cells, spleens were harvested from male Buffalo rats 24 hours following treatment with 10µg/g of Poly I:C (EMD Biosciences, La Jolla, CA) by i.p. injection. The mononuclear cells (MNCs) in splenocytes were dispersed from the spleens, followed by gradient centrifugation in Lympholyte Cell Separation Media (Cedarlane, Ontario, Canada). NK cells were enriched from the MNCs by Miltenyi magnetic separation after binding the cells with phycoerythrin (PE)-conjugated anti-rat CD161α antibody (BD Biosciences, San Diego, CA), followed by anti-PE MicroBeads (Miltenyi Biotec, Auburn, CA), according to the manufacturer’s instructions. Analysis by flow cytometry revealed that the preparations were >85% pure. The purified cells were cultured in complete DMEM medium containing 0.5% BSA (Sigma). To determine the optimal concentration of rat MIP-1α for the migration of rat NK cells, a dose response study was conducted in 24-well transwell plates (Corning INC, Corning, NY) with 5 µm pore size filters using 0, 25, 50, 100, or 200ng/ml rat MIP-1α (Peprotech, Rocky Hill, NJ) in the lower chambers followed by a 4 hour incubation at 37°C. Migration of rat NK cells (5×105/well) from the upper to lower chamber in response to the varying concentrations of chemokine was monitored. McA-RH7777 cells in serum free DMEM (Mediatech, Herndon, VA) were infected with rVSV-F or rVSV-UL141 at MOI=5. The culture media were harvested 24 hours post-infection, and filtered through 0.2µm Acrodisc syringe filters (Pall Corp., Ann Arbor, MI) after infectious virus in the filtrate was quantitatively inactivated by UV irradiation. At the defined dose of 100ng/ml of MIP-1α migration of rat NK cells was measured in the presence of the filtered and UV inactivated culture supernatants.
Animal Studies
All procedures involving animals were approved and performed according to the guidelines of the Institutional Animal Care and Use Committee of the Mount Sinai School of Medicine. Five to seven-week old male Buffalo rats weighing 200g were purchased from Charles River Labs (Wilmington, MA) and housed in a specific pathogen-free environment under standard conditions. To establish multifocal HCC lesions within the liver, 107 syngeneic McA-RH7777 rat HCC cells were infused into the portal vein in a 1ml suspension in serum-free DMEM. 21 days after tumor cell implantation, the development of multifocal hepatic tumors of 1–10 mm in diameter was confirmed, and 1 × 107 plaque-forming units (pfu) of rVSV-UL141 or rVSV-F or PBS control (in 1ml total volume) were administered via the hepatic artery. To evaluate tumor response to treatment, animals were sacrificed 3 days after infusion and tumors were subjected to histological, immunohistochemical and immunofluorescent staining, as well as TCID50 analysis of tumor extracts for quantification of VSV yield. In addition, groups of animals infused with VSV vectors or PBS were followed for survival, which was monitored daily in all animals.
Histology and Immunohistochemistry
Liver sections containing tumor were fixed overnight in 4% paraformaldehyde and then paraffin-embedded. 5µm-thin sections were subjected to either H&E staining for histological analysis or immunohistochemical staining using monoclonal antibodies against VSV-G protein (Alpha Diagnostic, TX), myeloperoxidase (MPO) (Abcam, MA). A second set of liver sections containing tumor were fixed overnight in 4% paraformaldehyde and then equilibrated in 20% sucrose in PBS overnight. 5µm-thin frozen sections were subjected to immunohistochemical staining using monoclonal antibodies against NKR-P1A (BD Pharmingen, CA), OX-52 (BD Pharmingen, CA), or ED-1 (Chemicon, CA). Semi-quantification of positively stained cells was performed using ImagePro Software (Media Cybernetics, Inc., Silver Spring, MD), and immune cell index was calculated as a ratio of positive cell number to unit tumor area (10,000 pixels as one unit tumor area).
Indirect Immunofluorescence
Tumor-containing liver samples were obtained on day 3 after virus or PBS infusion. 5µm-thin frozen sections were fixed with cold acetone and blocked with 4% goat serum, followed by staining with R-PE-conjugated mouse anti-rat CD3 monoclonal antibody (BD Pharmingen, CA) and FITC-conjugated mouse anti-rat NKR-P1A antibody (BD Pharmingen, CA). Nuclear DNA was stained with 4’,6’-diamidino-2-phenylindole (DAPI). Cover slips were mounted on glass slides using VECTASHIELD Mounting Medium (Vector Laboratories, CA).
Statistical Analyses
For comparison of individual data points, two-sided student t-test was applied to determine statistical significance. Survival curves of animals were plotted according to the Kaplan-Meier method, and statistical significance in different treatment groups was compared using the log-rank test. Results and graphs were obtained using the GraphPad Prism 3.0 program (GraphPad Software, San Diego, CA).
RESULTS
Functional Characterization of a Recombinant VSV Vector Expressing the UL141 gene from HCMV
After rescue of the recombinant rVSV-UL141 vector, via the established reverse genetics system described previously27, 28, the vector was subjected to various methods of characterization. We have shown previously that the recombinant vector rVSV-F, which expresses a genetically modified NDV fusion membrane glycoprotein, replicated to similar titers as wild-type VSV in BHK-21 and Hepatoma cells.18 To determine whether or not the expression of the UL141 transgene altered the infectivity or replication of the virus, growth curves of rVSV-UL141 were compared to that of rVSV-F in vitro. TCID50 assays were performed from supernatants collected at various time points after infection of the rat HCC cell line McA-RH7777 with each virus at an MOI of 0.01 (Fig. 1A). At each time point, the vectors replicated to similar titers, indicating that the inclusion of the UL141 gene in the recombinant vector did not significantly alter the viral life cycle or viral yield of VSV in rat HCC cells in vitro.
Figure 1. Viral replication and cell killing by rVSV-UL141 versus rVSV-F in Morris (McA-RH7777) rat hepatoma cells in vitro.
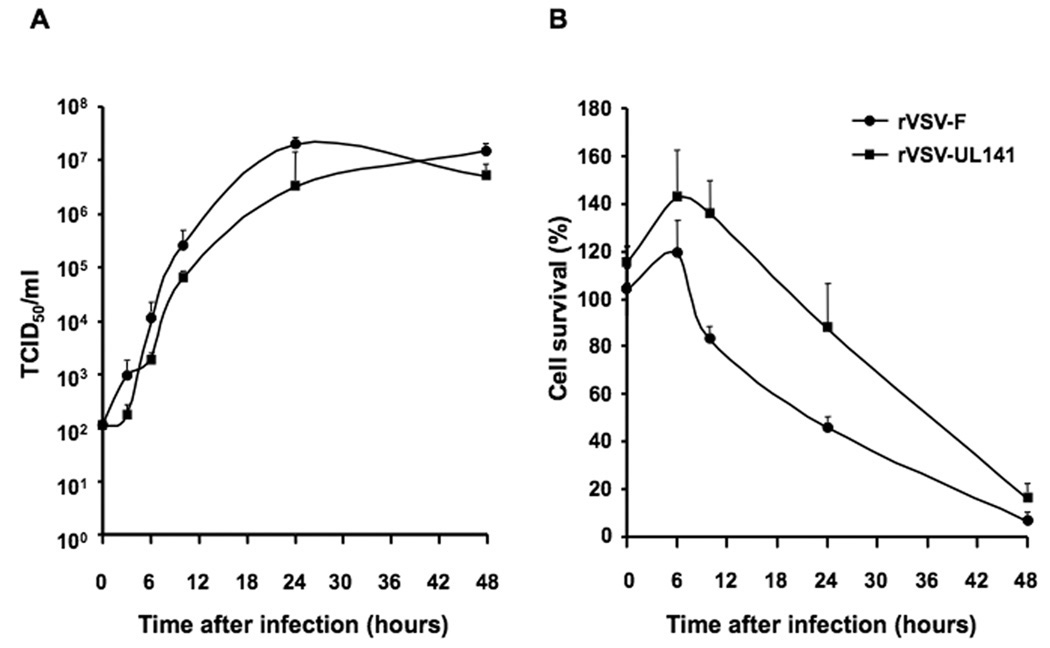
Rat hepatoma cells were infected with rVSV-F or rVSV-UL141 at MOI = 0.01. Panel A, TCID50 assay was performed on conditioned media at 0, 3, 6, 10, 24, and 48 hours post-infection; Panel B, MTT assays for cell viability were performed at 0, 3, 6, 10, 24, and 48 hours post-infection. Triplicate samples were analyzed at each time point. Data are shown as mean + standard deviation.
To compare the tumor cell killing potential of the VSV vectors in vitro, McA-RH7777 cells were infected with either rVSV-UL141 or rVSV-F at an MOI of 0.01, or mock-infected. The cytotoxicity to the cells was quantified by MTT assay, and calculated as a percentage of mock-infected cells at each time point. Although there appeared to be an initial delay in the cell killing effects observed in rVSV-UL141 infected cells as compared to rVSV-F, the kinetics were not significantly different, and by 48 hours post-infection both viruses resulted in nearly 100% cell death (Fig. 1B), demonstrating that rVSV-UL141 is an effective oncolytic agent in Morris hepatoma cells in vitro.
In Vitro Inhibition of Natural Killer (NK) Cell Migration by Conditioned Media from rVSV-UL141 Infected Rat HCC Cells
To determine if the UL141HCMV protein expressed by rVSV-UL141 infected cells is functional, assays to look at the migration of NK cells in response to macrophage inflammatory protein-1α (MIP-1α) were performed in 24-well transwell plates. To define the optimal dose of rat MIP-1α in the lower chamber for recruitment of rat NK cells, a dose response experiment was performed using increasing concentrations of MIP-1α while the number of rat NK cells added to the upper chamber remained constant. The migration of NK cells from the upper to the lower chamber was monitored, and it was determined that NK cell migration increased in a MIP-1α dose-responsive manner until saturation was reached at 100ng/ml (Fig. 2A). This dose of MIP-1α was then used in subsequent experiments to evaluate the inhibition of rat NK cell migration by conditioned media from rVSV-UL141 versus rVSV-F infected rat McA-RH7777 Hepatoma cells, which were ultrafiltered and UV-irradiated to quantitatively remove infectious viruses. As controls, media from mock-infected cells and migration in the absence of MIP-1α were used. The results showed that the number of migrating NK cells was significantly inhibited by conditioned media from rVSV-UL141 infected rat HCC cells as compared to that from rVSV-F and mock infected cells (Fig. 2B, p=0.01).
Figure 2. Inhibition of Natural Killer (NK) cell migration by conditioned media from rVSV-UL141, but not rVSV-F, infected rat HCC cells.
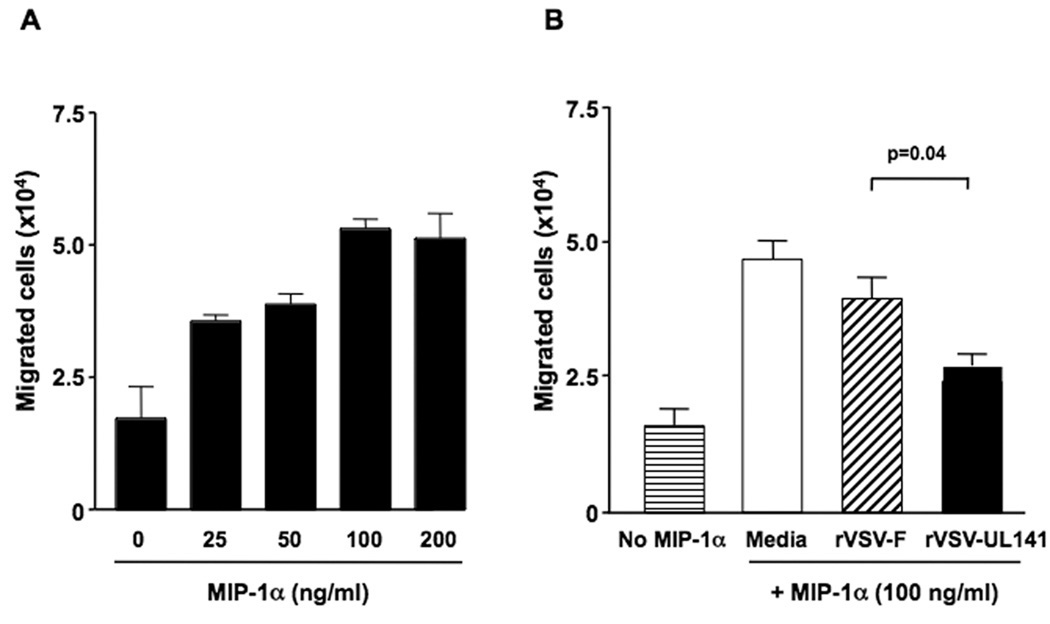
Panel A, dose response of rat NK cell migration in response to rat MIP-1α. The migration assays were performed using 24-well transwell plates. The migration of rat NK cells from the upper chamber to the lower chamber in response to serially diluted rat MIP-1α (0~200ng/ml) was monitored. Panel B, inhibition of NK cell migration in response to MIP-1α by conditioned media from rVSV infected rat HCC cells. The migration assays were performed using 24-well transwell plates. The migration of rat NK cells from the upper chamber to the lower chamber in response to 100ng/ml of MIP-1α was monitored in the presence of ultrafiltered and UV-inactivated supernatants from 105 HCC cells infected with rVSV-UL141 or rVSV-F. Data presented are the mean values of four independent experiments and the results were analyzed statistically by two-sided student t test.
Enhanced Replication of rVSV-UL141 in Orthotopic, Multifocal HCC Tumors in Syngeneic and Immune-competent Rats
To assess the in vivo effect of vector-mediated intratumoral UL141 production on oncolysis and viral replication within tumors, multi-focal HCC lesions were produced via injection of McA-RH7777 cells through the portal vein of male Buffalo rats as previously described17. 21 days post-implantation, the presence of multi-focal HCC lesions ranging from 1–10 mm in diameter was confirmed by laparotomy, and these animals were treated with either PBS, or 1×107 pfu of rVSV-UL141 or rVSV-F via hepatic artery infusion. Animals from each group were sacrificed on day 3 after treatment and tumor samples were collected and fixed for histological and immunohistochemical staining, as well as snap-frozen for intratumoral viral titer quantification. Immunohistochemical staining using a monoclonal antibody against VSV-G, revealed significantly more abundant expression of VSV-G protein within the tumors of rVSV-UL141 treated animals, compared to that observed in the rVSV-F group (Fig. 3A). To quantify the virus titers within the lesions, lysates prepared from snap-frozen tumor samples from each animal were subjected to TCID50 analysis. While rVSV-F infusion resulted in titers less than 104 TCID50/mg of tumor tissue, rVSV-UL141 replicated to yield titers of one-log higher at almost 105 TCID50/mg of tumor tissue (Fig. 3B, p=0.04).
Figure 3. rVSV-UL141 versus rVSV-F replication in HCC tumors in the livers of immune-competent Buffalo rats.
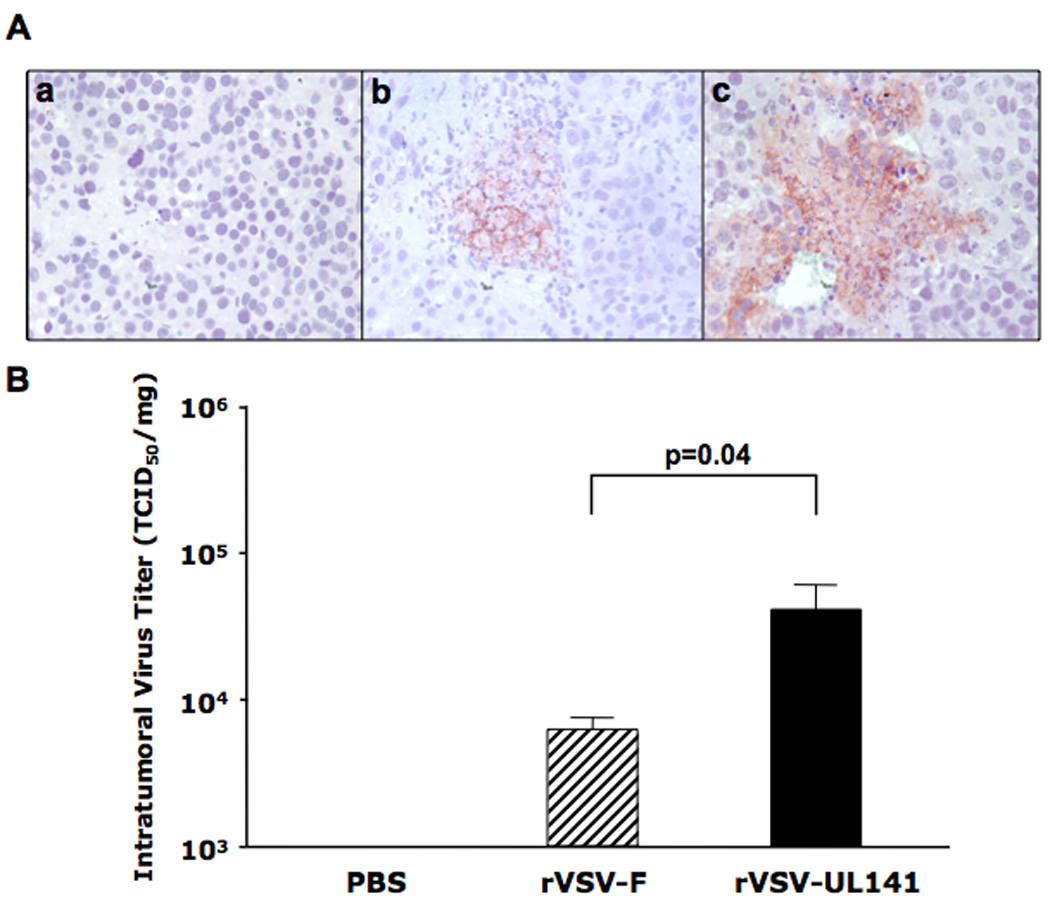
Multi-focal HCC-bearing Buffalo rats were treated with PBS (n=3), rVSV-F (n=4) or rVSV-UL141 (n=4) at 1×107 pfu/rat injected through the hepatic artery. Tumor samples were obtained from the treated rats at day 3 after virus infusion. 5 µm tumor sections were stained with a monoclonal anti-VSV-G antibody and counterstained with Hematoxylin (Panel A). Representative sections from rats treated with PBS, rVSV-F and rVSV-UL141 are shown in frames a, b and c, respectively (magnification=40×). In panel B, intratumoral virus titers were determined by TCID50 assays performed using tumor extracts on BHK-21 cells. Viral titers are expressed as TCID50/mg tissue (mean ± sstandard deviation), and the results were analyzed statistically by two-sided student t test.
Improved Tumor Response in Multi-focal HCC Lesions in the Livers of Rats Treated with rVSV-UL141
To determine the impact of enhanced intratumoral replication of the rVSV-UL141 vector on tumor viability, tumor-containing liver sections from the above experiment were examined by H/E staining and analyzed morphometrically for quantification of necrotic area. Using ImagePro analysis software, necrotic areas were measured and represented as a percentage of the entire tumor area. Tumors within the rVSV-UL141 treatment group were more than 50% necrotic, which represents a significant increase over the rVSV-F treatment group, which exhibited approximately 25% necrosis (Fig. 4B, p=0.01). In the PBS control group less than 15% necrosis was observed, which can be attributed to spontaneous necrosis that is a natural phenomenon in this tumor type in vivo.
Figure 4. Enhanced tumor response in rats treated with rVSV-UL141 versus those treated with rVSV-F.
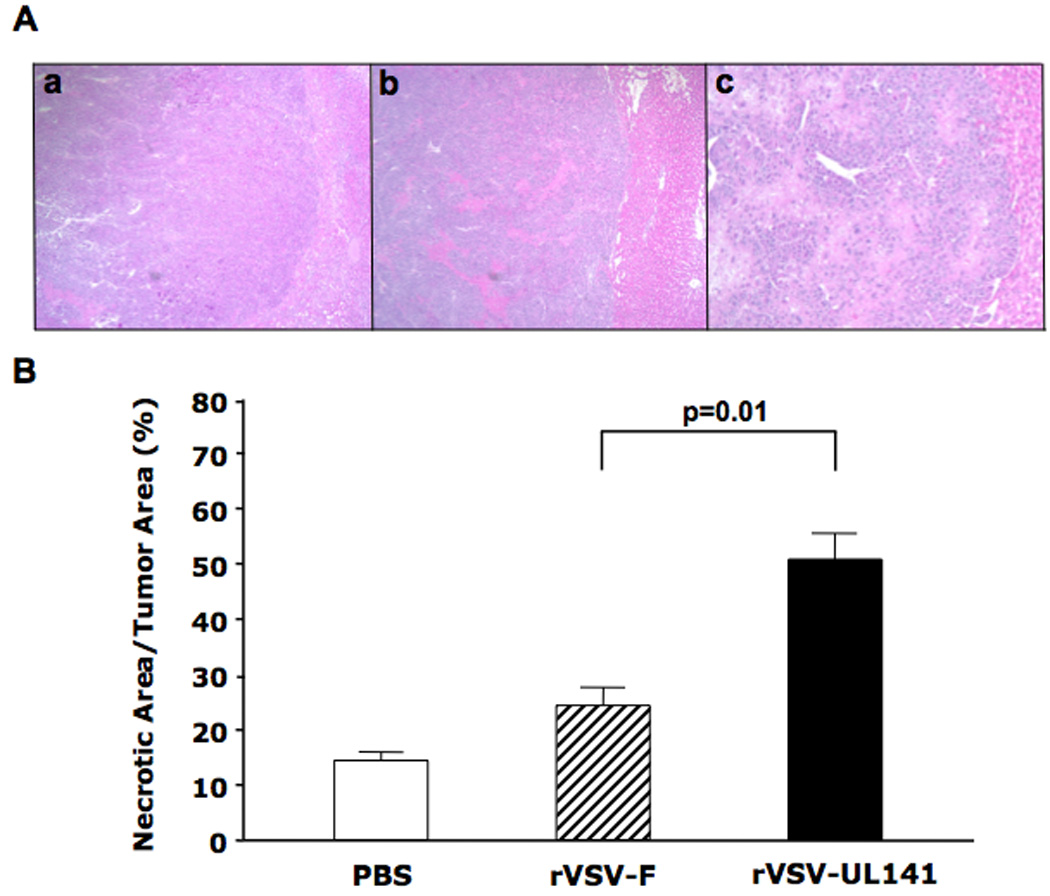
Multi-focal HCC-bearing Buffalo rats were injected via the hepatic artery with PBS (n=3), rVSV-F (n=4) or rVSV-UL141 (n=4) at 1×107 pfu/rat and sacrificed 3 days post-treatment. Panel A, 5 µm tumor sections were stained with H&E. Representative sections from rats treated with PBS, rVSV-F and rVSV-UL141 are shown in frames a, b and c, respectively (magnification=40×). Panel B, Percentage of necrotic areas in the tumors were measured morphometrically using ImagePro software. Data are shown as mean ± standard deviation, and the results were analyzed statistically by two-sided student t test.
Because this treatment strategy employs a transgene designed to inhibit a key component of the anti-viral inflammatory response in the host, safety was an obvious concern. To identify signs of hepatic toxicity, histopathological sections showing the border region between tumor and liver tissues and neighboring liver parenchyma were examined. It was determined that the surrounding liver histology was completely normal, with no evidence of abnormal pathology (results not shown).
Decreased Accumulation of NK and NKT Cells in Tumors Treated with rVSV-UL141 Compared with rVSV-F
Multi-focal HCC tumor-bearing Buffalo rats were treated with either PBS or 1×107 pfu of rVSV-UL141 or rVSV-F via the hepatic artery. On day 3 after treatment, animals were sacrificed, and tumor-containing liver sections were prepared for immunohistochemical staining of various immune cell types (Fig. 5A). Sections were stained for NK cells with anti-NKR-P1A (Frames a–c), neutrophils by anti-myeloperoxidase (Frames d–f), pan-T cells by anti-OX-52 (Frames g–i), and macrophages by anti-ED-1 (Frames j–l). Semi-quantification of marker-positive cells using ImagePro software revealed that there was substantial intra-tumoral accumulation of NK cells after rVSV-F infusion compared to PBS (Fig. 5Ba, p=0.04); however, this effect was substantially reduced in response to rVSV-UL141 treatment (Fig. 5Ba, p=0.002). In addition, there were significantly fewer pan-T marker-positive cells (Fig. 5Bc, p=0.04) in tumors treated with rVSV-UL141 compared to those treated with rVSV-F. To determine whether these were NKT cells or T-lymphocytes, indirect immunofluorescent staining was performed. Consecutive tumor sections from PBS, rVSV-UL141 or rVSV-F treated animals were stained with R-PE-conjugated mouse anti-rat CD3 antibody and FITC-conjugated mouse anti-rat NKR-P1A antibody (Fig. 6). Merged images indicate that the pan-T-positive cells present in the tumors after rVSV-F treatment are in fact NKT cells rather than T-lymphocytes. As expected, there were no statistically significant differences in neutrophil (p=0.6) or macrophage accumulation (p=0.08) within tumors of rats treated with the two rVSV vectors. Collectively, these results indicate that the inhibitory function of UL141 on inflammatory cells is specific for NK and NKT cells.
Figure 5. Immunohistochemical staining and semi-quantification of immune cells in tumors.
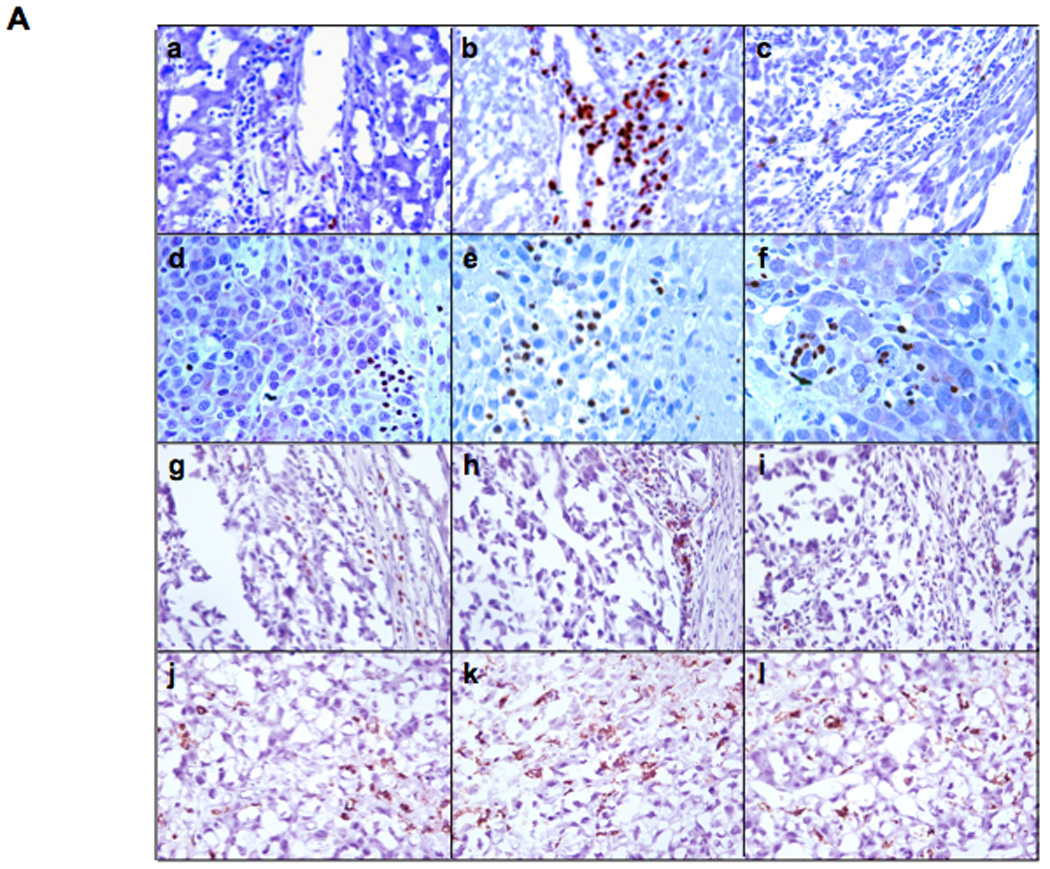
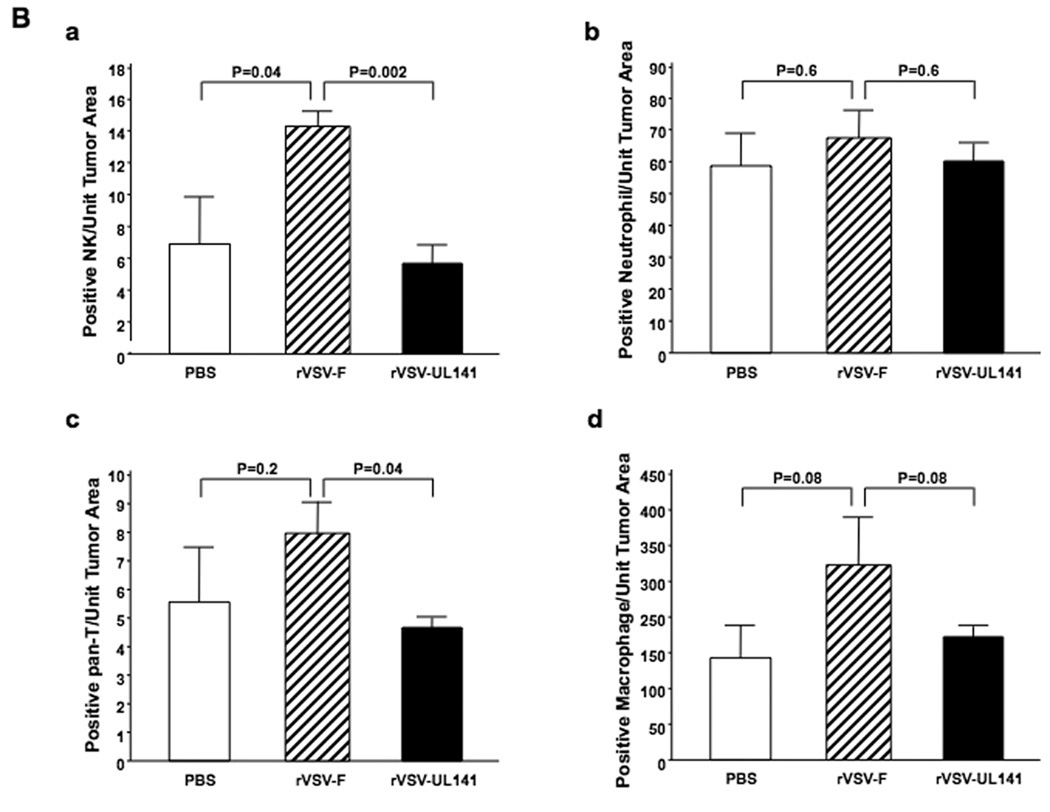
Panel A, representative immunohistochemical sections from tumors and surrounding tissues. Tumor-bearing rats were infused with PBS (frames a, d, g, j), rVSV-F (frames b, e, h, k) or rVSV-UL141 (frames c, f, i, l) at 1×107 pfu/ml/rat. Samples were obtained from rats at day 3 after virus infusion into the hepatic artery. 5 µm sections were stained with mouse monoclonal anti-NKR-P1A (frames a, b, c), polyclonal anti-myeloperoxidase (frames d, e, f), monoclonal anti-OX-52 (frames g, h, i) and monoclonal anti-ED-1 (frames j, k, l), (magnification=40×). Panel B, semi-quantification of immune cells in the lesions after virus treatment: NK cells (graph a), neutrophils (graph b), pan-T cells (graph c) and macrophages (graph d) by ImagePro software. Immune cell index was calculated as ratio of positive cell to unit tumor area (10,000 pixel as one unit tumor area), and the results were analyzed statistically by two-sided student t-test.
Figure 6. Immunofluorescent staining of T and NK cells in tumors.
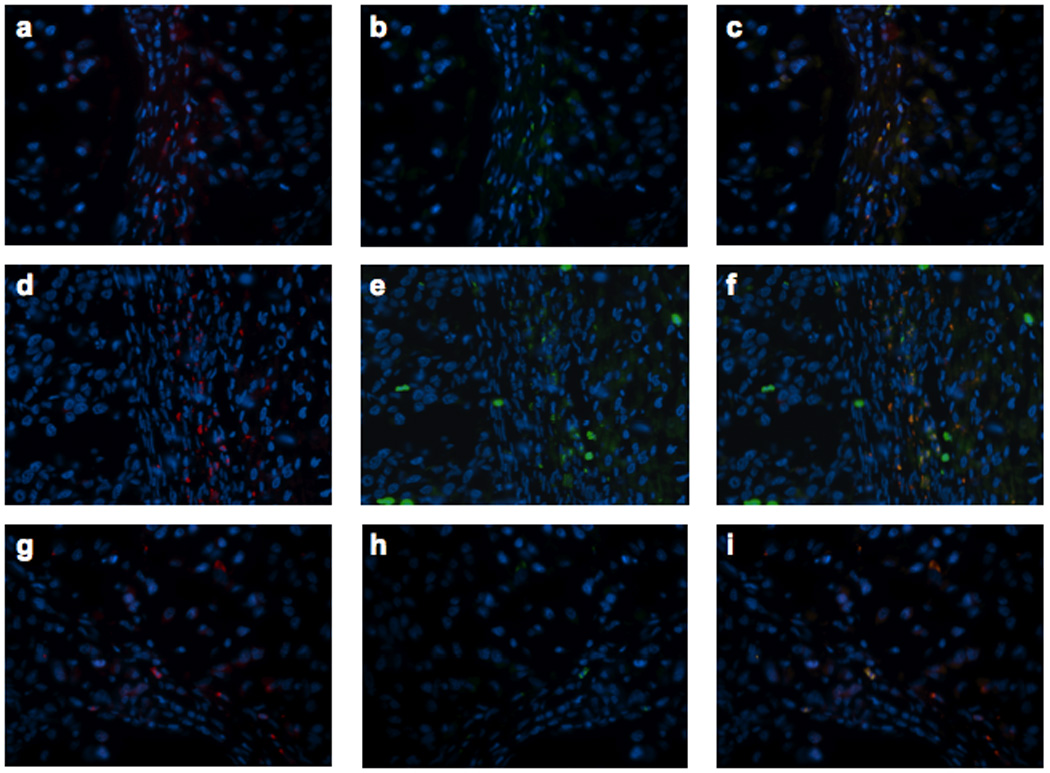
Tumor bearing Buffalo rats were infused via the hepatic artery with PBS (frames a to c), rVSV-F (frames d to f) or rVSV-UL141 (frames g to i) at 1×107 pfu/ml/rat. Samples were obtained at day 3 after virus infusion. 5 µm frozen sections were fixed with cold acetone and blocked with 4% goat serum, followed by staining with R-PE-conjugated mouse anti-rat CD3 monoclonal antibody (frames a, d and g) and FITC-conjugated mouse anti-rat NKR-P1A (frames b, e and h). Merged pictures are shown in frames c, f and i, respectively. Original magnification ×40.
Substantial Prolongation of Survival of Multi-focal HCC-Bearing Rats Treated with rVSV-UL141 versus rVSV-F
To assess the oncolytic potential of a rVSV vector expressing a transgene that inhibits NK and NKT cell function, rats bearing orthotopic multi-focal HCC tumors were randomly assigned to treatment groups to receive either PBS (n=8), 1×107 pfu of rVSV-UL141 (n=14), or an equal dose of the control rVSV-F vector (n=10). All animals received a single infusion of the respective treatment via the hepatic artery, and animals were monitored daily for survival (Fig. 7). In addition, all animals were carefully screened for clinical signs of treatment-associated toxicity to address safety concerns of the modified virus. While all animals in the PBS or rVSV-F treatment groups expired by day 21 or 29, respectively, rVSV-UL141 treatment resulted in a highly significant prolongation of survival (P<0.001), with 3 out of 14 animals (21.4%) achieving long-term survival of 150 days. Furthermore, the long-term surviving rats in the rVSV-UL141 treatment group were sacrificed on day 150 and evaluated for residual malignancy. Macroscopically, there was no visible tumor within the liver or elsewhere, and there was no histological evidence of residual tumor cells or hepatitis. These results indicate that even large multi-focal lesions (up to 10mm in diameter at the time of treatment) had undergone complete remission in these animals, which translated into long-term and tumor-free survival.
Figure 7.
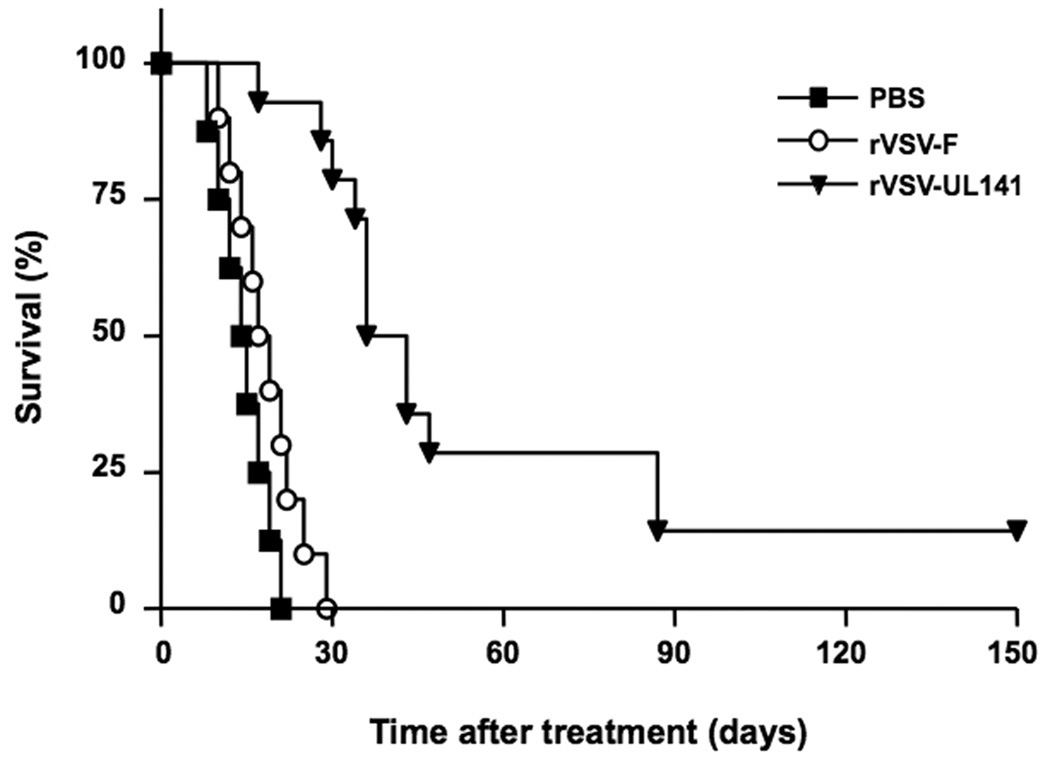
Kaplan-Meier survival curve of multi-focal HCC-bearing Buffalo rats after hepatic arterial infusion of PBS (n=8), rVSV-F (n=10) or rVSV-UL141 (n=14) at 1×107 pfu/ml/rat. Survival was monitored daily and the results were analyzed statistically by log rank test (p<0.001 for rVSV-UL141 versus rVSV-F).
DISCUSSION
Various strategies have been explored to enhance the oncolytic potency of recombinant VSV vectors, with the aim of improving the therapeutic potential of the virus. In the present study, we have presented a novel rVSV vector that encodes a heterologous viral protein, which specifically inhibits the function of NK cells. We developed this vector as an effective yet safe oncolytic agent for treatment of multi-focal HCC in an immune-competent rat model. The data presented here support our hypothesis that by inhibition of a specific aspect of the host inflammatory response, we can greatly enhance the oncolytic potency over the rVSV-F construct, without compromising the safety profile. This strategy translates into highly significant prolongation of survival with 20% of treated animals enjoying complete tumor regression and long-term survival, while all rVSV-F treated animals succumbed to the disease by day 30 post-treatment. Furthermore, these results were achieved after a single administration of the vector.
It is important to note that rVSV-F, and not wtVSV, was used as a control vector in these studies. It is our strategy when testing new recombinant vectors to compare them with our currently best available vector. In this way, we can directly assess whether or not the new vector represents an improvement over our prior work. Because we had previously reported a significant improvement in efficacy of the fusogenic rVSV-F vector over the wildtype version18, 29, it seemed intuitive to use this vector as a control for comparison of the rVSV-UL141 vector.
We have recently shown that vector-mediated suppression of anti-viral inflammatory responses is a novel and effective strategy for enhancing the potency of VSV for treatment of multi-focal HCC in rats. We hypothesized that the logarithmic decline in intra-tumoral virus replication observed at an early phase of infection was mediated by anti-viral inflammation, which was evident within the lesions after 1–2 days. This was confirmed by systemic suppression of NK cells using a specific antibody, which resulted in significantly enhanced and prolonged intra-tumoral viral titers23. We further demonstrated that suppression of chemotaxis of anti-viral inflammatory cells could be achieved by genetic modification of the rVSV genome to express a heterologous viral chemokine binding protein (vCKBP). In that study, we tested a rVSV vector encoding the secreted form of glycoprotein G from equine herpes virus-1 (gGEHV-1), which has demonstrated vCKBP activity and binds a broad range of chemokines with high affinity.25 The gGEHV-1 protein blocks chemokine activity by preventing interactions with specific receptors and glycosaminoglycans, which are required for the correct presentation and function of chemokines. We showed that rVSV-gG treatment resulted in an inhibition of intra-tumoral NK and NKT cells, which in turn led to elevated VSV-mediated oncolysis, tumor response and substantial survival prolongation. While the rVSV-gG vector proved to be a promising therapeutic agent for the treatment of multi-focal HCC, we faced concerns about the prospect of expressing a broad-range chemokine inhibitor, especially in light of the fact that our aim was to specifically inhibit NK cells. Therefore, we sought an alternate strategy in which we could specifically block the anti-viral function of NK cells, while leaving all other aspects of the inflammatory response intact.
UL141 is a viral gene product of human cytomegalovirus (HCMV). HCMV is a human herpesvirus, which causes persistent, lifelong infection, during which the host’s innate and adaptive immune responses work together to keep the virus in check30–32. In particular, NK cells are crucial in the control of cytomegalovirus infections; however, a key aspect of the capacity of HCMV to persist in the host is its ability to evade NK cell recognition33–35. In fact, at least six different HCMV genes encoding NK cell inhibitory function have been identified. One of these genes, UL141, modulates NK function by blocking cell surface expression of CD15526, which is a ligand for NK cell-activating receptors CD226 and CD9636, 37. It was shown that expression of a transfected, secreted form of UL141 resulted in efficient protection of cells against killing by a wide range of human NK cell populations tested26.
To test the potential of UL141 for the purpose of VSV-mediated evasion of the inhibitory effects of NK cells, we created a recombinant vector, rVSV-UL141, in which the secreted form of UL141 was expressed as an additional transcription unit. This vector was then subjected to a series of in vitro analyses to fully characterize its features. To test the growth kinetics of the recombinant vector, we performed multi-cycle growth curves to rule out the possibility that expression of the transgene could inadvertently attenuate viral replication. At all time points tested, rVSV-UL141 demonstrated equivalent viral yields as compared to the control rVSV-F vector. Furthermore, the cell viability (MTT) assays revealed similar levels of cell killing in response to both vectors in the rat HCC cell line, McA-RH7777. Although there is no available antibody to UL141, and we were therefore unable to confirm or quantify the production of UL141 protein produced by the vector, we confirmed the function of UL141 in the recombinant vector, by performing in vitro migration assays. While infection with the control vector, rVSV-F, induced the migration of NK cells, this effect was significantly impeded in the presence of media conditioned by rVSV-UL141 infection. This indicates that expression of UL141 resulted in functional inhibition of NK cells, as predicted.
To define the in vivo implications of the genetically modified rVSV vector expressing UL141, in terms of its ability to evade the inhibitory function of NK cells and allow enhanced viral replication and cell killing, we tested the vector in our multi-focal HCC rat model. On day 3 after treatment by hepatic artery infusion, we confirmed that positive staining for VSV-G protein was indeed augmented within tumors of rats treated with rVSV-UL141 as compared to the control rVSV-F vector, and intratumoral virus titers increased by one-log. Furthermore, rVSV-UL141 treatment led to significantly enhanced tumor necrosis. Together, these results indicate that the modified vector results in enhanced replication and improved tumor response, as compared with the rVSV-F vector. Although rVSV-UL141 was not quite as effective in prolonging survival as our previously reported rVSV-gG vector, we reason that a slight compromise in efficacy is justified by a more controlled regulation of anti-viral inflammation, which would minimize safety concerns. Furthermore, in addition to the potential clinical implications of this work, by creating a vector which specifically blocks the effects of NK and NKT cells, as opposed to the broad range chemokine binding function of the gG vector, we were able to elucidate the role of these cell types in oncolytic viral therapy.
To examine the mechanism whereby expression of UL141 resulted in substantially elevated intratumoral rVSV titers, we performed immunohistochemical staining of various immune cell types in tumor sections from animals treated with PBS, rVSV-UL141, or rVSV-F. As anticipated, natural killer cells seemed to be the major immune cell type recruited in response to rVSV-F infection. In contrast, rVSV-UL141 treatment resulted in a drastic reduction in NK cell accumulation to levels roughly equivalent to those observed in PBS treated tumors. In addition, we noted a decrease in pan-T positive cells within tumors treated with rVSV-UL141 compared with rVSV-F. Subsequent immunofluorescence staining revealed that the pan-T positive cells co-localized to NK marker-positive cells, indicating that these were in fact NKT cells. Finally, VSV-mediated expression of UL141 appeared to have no major impact on neutrophil or macrophage infiltration in HCC tumors.
Natural killer cells are a major component of the innate cellular immune response, and are crucial in the early defense against invading pathogens38, 39. NK cells are found primarily in the blood and spleen, as well as in non-lymphoid tissues such as liver, lung, and peritoneal cavity40, 41. These cells represent a distinct subset of the cytotoxic lymphocyte population, and act as a first line of defense against invading pathogens and viruses prior to the launch of the adaptive immune response39, 42, 43. Upon activation during viral infection, NK cells mediate the direct lysis of target cells by releasing cytotoxic granules containing lytic enzymes, or by binding to apoptosis-inducing receptors on the target cell33, 44. An in vitro study using herpes simplex virus and vaccinia virus in human colon adenocarcinoma cells (Colo-205) revealed that NK cells preferentially lyse virus infected cells at an early stage of infection, thereby preventing viral dissemination to neighboring cells43. These data support our hypothesis that the rapid recruitment of NK cells to VSV infected tumor tissue impedes the viral spread and, in turn, the oncolytic potential of VSV therapy. This is also consistent with our observation that vector-mediated expression of UL141HCMV, a heterologous viral protein with known NK inhibitory function, resulted in enhanced VSV replication in the tumors.
By means of immunofluorescent analysis, a second type of inflammatory cells inhibited by rVSV-UL141 was identified as NKT cells. NKT cells represent a subset of NK cells, found within the T cell population44. Although they represent only a small percentage of T cells found in the thymus and spleen, they constitute a significant proportion of those in the liver, accounting for approximately 25% of resident intrahepatic lymphocytes45. Although the mechanism and function of NKT cells in the inflammatory response is not completely understood, there is speculation that they may play a complementary role to NK cells in mediating early anti-viral responses44. Although an inhibitory function of UL141HCMV on NKT cells has not previously been reported, our results can be explained by the expression of CD226 on the surface of NKT cells. CD226 is an NK cell-activating receptor whose ligand, CD155, is directly blocked by UL141. Therefore, an inhibitory affect on NKT cells, in addition to classical NK cells could be predicted. Furthermore, because viruses continuously evolve in such a way to promote their own survival within the infected host, it might be postulated that the viral genome of human cytomegalovirus has selected for mechanisms that evade the anti-viral assault by NK and NKT cells in order to persist in immune-competent hosts.
While the potent anti-NK and –NKT properties of UL141 made it a powerful candidate for exploitation within the context of improved oncolytic VSV therapy for HCC, the safety implications of such a strategy must be carefully considered. One could argue that, because endogenous expression of UL141 by HCMV contributes to the ability of the virus to cause lifelong infections in humans, rVSV expression of UL141 could result in a similar outcome. This possibility would be detrimental to the development of such a vector for future clinical application. To address this concern, we previously demonstrated via comprehensive toxicity studies in immune-competent tumor-bearing rats, that our rVSV-gG vector introduced no additional toxicities compared to the rVSV-F vector, despite the fact that gGEHV-1 is known to be a broad-spectrum chemokine binding protein, and effectively inhibited the chemotaxis of NK and NKT, and potentially other undefined cell types, to virus-infected lesions. Because the function of UL141HCMV is specific for NK and NKT cells, we predicted that rVSV-UL141 should be similar to rVSV-gG in its safety profile. Furthermore, throughout the long-term survival experiments conducted in the present study, the animals were carefully monitored for clinical signs of toxicity. rVSV-UL141 treatment resulted in no significant weight loss, signs of dehydration, piloerection, limb paralysis, or lethality. This absence of toxicity, despite evasion of key players in the anti-viral inflammatory response, can be reconciled by the exquisite sensitivity of VSV to the type I interferon response in normal cells,44 which is un-altered in the modified vector.
A final consideration of the current study involves the complexity of NK function in immune-competent hosts. While we have discussed in detail the anti-viral function of NK cells, it is important to also consider the role of NK cells in anti-tumor immunity. Interestingly, others have shown that NK cells can actually augment the tumoricidal effects of oncolytic herpes simplex virus, acting synergistically with the robust adoptive anti-tumor immune response launched in response to viral antigens expressed by tumor cells21, 46, 47. Even more striking, it has recently been reported that NK cells are required for the efficacy of VSV therapy in a melanoma model48. Here it was proposed that the oncolytic potential of VSV is enhanced by anti-tumor immune responses. These contradictory results to our current study can be resolved by an understanding of the dual-function of NK cells. On the one hand, it is well established that NK cells are potent inhibitors of viral replication, and it is this function that is inhibited by UL141. On the other hand, NK cells play a role in anti-tumor immunity, a feature that is enhanced by the presence of viral antigens within tumor cells. While both strategies are scientifically sound, each approach contradicts the other. Although it would be quite interesting to compare both approaches side by side within the same tumor model, these studies are beyond the scope of the current investigation. Furthermore, to examine whether the impressive results obtained by rVSV-UL141 are limited to the treatment of HCC, or if they can also be applied to other cancers, it will be interesting to test this vector in other cancer models. These experiments are among our future plans.
In summary, we have presented a novel recombinant VSV vector, which exploits a specific NK and NKT cell inhibitor from human cytomegalovirus. We have provided conclusive evidence that this modified vector demonstrates superior replication and oncolysis, resulting in significant prolongation of survival in a multi-focal HCC tumor model in rats. Moreover, this vector maintains the same safety profile as the wildtype vector. Thus, rVSV-UL141 has the potential for development into an effective and safe therapeutic agent for the treatment of HCC and possibly other types of cancer in the future. Furthermore, since anti-viral inflammatory responses are universal obstacles limiting the potential of therapeutic viruses, the technology developed here is likely to be applicable in substantially enhancing the potency and efficacy of a wide spectrum of other oncolytic virus agents in cancer treatment as well.
ACKNOWLEDGEMENTS
We thank Dr. Tian-gui Huang for helpful discussions, Dr. Swan Thung for consultation on histological and immunohistochemical analyses of tissue samples, and Mr. Boxun Xie, Ms. Yafang Wang and Ms. Sonal Harbaran for excellent technical assistance.
Grant support: Supported in part by National Institutes of Health Grant CA100830 (S.L.C Woo), the German Research Aid (Max-Eder Research Program) (O.Ebert), and the Federal Ministry of Education and Research Grant 01GU0505 (O. Ebert).
REFERENCES
- 1.Parkin DM, Sthernsward J, Muir CS. Estimates of the worldwide frequency of twelve major cancers. Bull World Health Organ. 1984;62(2):163–182. [PMC free article] [PubMed] [Google Scholar]
- 2.Murray CJaL AD. Evidence-based health policy - lessons from the Global Burden of Disease Study. Science. 1996;274(5288):740–743. doi: 10.1126/science.274.5288.740. [DOI] [PubMed] [Google Scholar]
- 3.Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer. 2001;94:153–156. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 4.El-Serag HBaM AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340(10):745–750. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- 5.Dyer Z, Peltekian K, van Zanten SV. The changing epidemiology of hepatocellular carcinoma in Canada. Aliment Pharmacol Ther. 2005;22(1):17–22. doi: 10.1111/j.1365-2036.2005.02504.x. [DOI] [PubMed] [Google Scholar]
- 6.Yeung YP, Lo CM, Liu CL, et al. Natural history of untreated nonsurgical hepatocellular carcinoma. Am J Gastroenterol. 2005;100(9):1995–2004. doi: 10.1111/j.1572-0241.2005.00229.x. [DOI] [PubMed] [Google Scholar]
- 7.Cunningham S, Choti MA, Bellavance EC, Pawlik TM. Palliation of hepatic tumors. Surg Oncol. 2007;16(4):277–291. doi: 10.1016/j.suronc.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Rougier P, Mitry E, Barbare JC, Taieb J. Hepatocellular carcinoma (HCC): an update. Semin Oncol. 2007;34:S12–S20. doi: 10.1053/j.seminoncol.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: biological principles, risk management, and future directions. Nat Med. 2001;7:781–787. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- 10.Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- 11.Lorence RM, Katubig BB, Reichard KW, et al. Complete regression of human fibrosarcoma xenografts after local Newcastle disease virus therapy. Cancer Research. 1994;54:6017–6021. [PubMed] [Google Scholar]
- 12.Peng KW, Ahmann GJ, Pham L, et al. Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood. 2001;98:2002–2007. doi: 10.1182/blood.v98.7.2002. [DOI] [PubMed] [Google Scholar]
- 13.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6(7):821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 14.Letchworth GJ, Rodriguez LL, Del C, Barrera J. Vesicular Stomatitis. Vet J. 1999;157:239–260. doi: 10.1053/tvjl.1998.0303. [DOI] [PubMed] [Google Scholar]
- 15.Rose JK, Whitt MA. Rhabdoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. ed. 4th. Philidelphia: Lippincott Williams & Wilkins; 2001. pp. 1221–1242. [Google Scholar]
- 16.Ebert O, Shinozaki K, Huang TG, Savontaus MJ, Garcia-Sastre A, Woo SLC. Oncolytic vesicular stomatitis virus for treatment of orthotopic hepatocellular carcinoma in immune-competent rats. Cancer Research. 2003;63(13):611–613. [PubMed] [Google Scholar]
- 17.Shinozaki K, Ebert O, Kournioti C, et al. Oncolysis of multifocal hepatocellular carcinoma in the rat liver by hepatic artery infusion of vesicular stomatitis virus. Mol Ther. 2004;9:368–376. doi: 10.1016/j.ymthe.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Ebert O, Shinozakis K, Kournioti C, et al. Syncytia induction enhances the oncolytic potential of vesicular stomatitis virus in virotherapy for cancer. Cancer Research. 2004;64:3265–3270. doi: 10.1158/0008-5472.can-03-3753. [DOI] [PubMed] [Google Scholar]
- 19.Shinozaki K, Ebert O, Suriawinata A, Thung S, Woo S. Prophylactic alpha interferon treatment increases the therapeutic index of oncolytic vesicular stomatitis virus virotherapy for advanced hepatocellular carcinoma. J Virol. 2005a;79(21):13705–13713. doi: 10.1128/JVI.79.21.13705-13713.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guidotti LGaC FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annual Review of Immunology. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 21.Wakimoto H, Johnson PR, Knipe DM, et al. Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Therapy. 2003;10:983–990. doi: 10.1038/sj.gt.3302038. [DOI] [PubMed] [Google Scholar]
- 22.Wakimoto H, Fulci G, Tuminiski E, et al. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide's enhancement of viral oncolysis. Gene Therapy. 2004;11(2):214–223. doi: 10.1038/sj.gt.3302143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altomonte J, Wu L, Chen L, Meseck M, Ebert O, Garcia-Sastre A, Fallon J, Woo SLC. Exponential enhancement of oncolytic vesicular stomatitis virus potency by vector-mediated suppression of inflammatory responses in vivo. Mol Ther. 2008;16(1):146–153. doi: 10.1038/sj.mt.6300343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alcami A. Viral mimicry of cytokines, chemokines, and their receptors. Nature Immunology. 2003;3:36–50. doi: 10.1038/nri980. [DOI] [PubMed] [Google Scholar]
- 25.Bryant NA, Davis-Poynter N, Vanderplasschen Al, et al. Glycoprotein G isoforms from some alphaherpesviruses function as broad-spectrum chemokine binding proteins. Embo J. 2003;22:833–846. doi: 10.1093/emboj/cdg092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomasec P, Wang E, Davison A, Vojtesek B, Armstrong M, Griffin C, McSharry B, Morris R, Llewellyn-Lacey S, Rickards C, Nomoto A, Sinzger C, Wilkinson G. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nature Immunology. 2005;6(2):181–188. doi: 10.1038/ni1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawson ND, Stillman EA, Whitt MA, et al. Recombinant vesicular stomatitis viruses from DNA. Proc. Natl. Acad. Sci. 1995;92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whelan SP, Ball LA, Barr JN, et al. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. 1995;92:8388–8392. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shinozaki K, Ebert O, Woo SL. Eradication of advanced hepatocellular carcinoma in rats via repeated hepatic arterial infusions of recombinant VSV. Hepatology. 2005;41:196–203. doi: 10.1002/hep.20536. [DOI] [PubMed] [Google Scholar]
- 30.Guma M, Angulo A, Lopez-Botet M. NK cell receptors involved in the response to human cytomegalovirus infection. Curr Top Microbiol Immunol. 2006;298:207–223. doi: 10.1007/3-540-27743-9_11. [DOI] [PubMed] [Google Scholar]
- 31.Lin A, Xu H, Yan W. Modulation of HLA expression in human cytomegalovirus immune evasion. Cell Mol Immunol. 2007;4(2):91–98. [PubMed] [Google Scholar]
- 32.Lopez-Botet M, Llano M, Ortega M. Human cytomegalovirus and natural killer-mediated surveillance of HLA class I expression: a paradigm of host-pathogen adaptation. Immunol Rev. 2001;181:193–202. doi: 10.1034/j.1600-065x.2001.1810116.x. [DOI] [PubMed] [Google Scholar]
- 33.Orange JS, Fassett MS, Koopman LA, et al. Viral evasion of natural killer cells. Nature Immunology. 2002;3:1006–1012. doi: 10.1038/ni1102-1006. [DOI] [PubMed] [Google Scholar]
- 34.Rajagopalan S, Long EO. Viral evasion of NK-cell activation. Trends in Immunol. 2005;26(8):403–405. doi: 10.1016/j.it.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Wilkinson G, Tomasec P, Stanton RJ, Armstrong M, Prod'homme V, Aicheler R, McSharry BP, Rickards CR, Cochrane D, Llewellyn-Lacey S, Wang EC, Griffin CA, Davison AJ. Modulation of natural killer cells by human cytomegalovirus. J Clin Virol. 2007 doi: 10.1016/j.jcv.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, Cantoni C, Grassi J, Marcenano S, Reymond N, Vitale M, Moretta L, Lopez M, Moretta A. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med. 2003;198:557–567. doi: 10.1084/jem.20030788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuchs A, Cella M, Giurisato E, et al. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155) J Immunol. 2004;172:3994–3998. doi: 10.4049/jimmunol.172.7.3994. [DOI] [PubMed] [Google Scholar]
- 38.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annual Review of Immunology. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 39.Welsh RM. Regulation of virus infections by natural killer cells. Nat Immun Cell Growth Reg. 1986;5:169–199. [PubMed] [Google Scholar]
- 40.Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cerwenka A, Lanier LL. Natural killer cells, viruses and cancer. Nat Rev Immunol. 2001;1(1):41–49. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]
- 42.Brutkiewicz RR, Welsh RM. Major histocompatibility complex class I antigens and the control of viral infections by natural killer cells. J Virol. 1995;69:3967–3971. doi: 10.1128/jvi.69.7.3967-3971.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baraz L, Khazanov E, Condiotti R, Kotler M, Nagler A. Natural killer (NK) cells prevent virus production in cell culture. Bone Marrow Transplantation. 1999;24:179–189. doi: 10.1038/sj.bmt.1701825. [DOI] [PubMed] [Google Scholar]
- 44.Biron CA, Brossay L. NK cells and NKT cells in innate defense against viral infections. Current Opinion in Immunology. 2001;13:458–464. doi: 10.1016/s0952-7915(00)00241-7. [DOI] [PubMed] [Google Scholar]
- 45.Eberl G, Lees R, Smiley ST, Taniguchi M, Grusby MJ, MacDonald HR. Tissue-specific segregation of CD1d-dependent and CD1d-independent NK T-cells. J Immunol. 1999;162:6410–6419. [PubMed] [Google Scholar]
- 46.Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. 2001;98:6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas D, Fraser NW. HSV-1 therapy of primary tumors reduces the number of metastases in an immune-competent model of metastatic breast cancer. Mol Ther. 2003;8:543–551. doi: 10.1016/s1525-0016(03)00236-3. [DOI] [PubMed] [Google Scholar]
- 48.Diaz R, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, Valdes M, Barber G, Vile RG. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Research. 2007;67(7):2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]