

Abstract
Cigarette smoke contains a high concentration of free radicals and induces oxidative stress in the lung and other tissues. Several transcription factors are known to be activated by oxidative stress, including nuclear factor-κB (NF-κB), activator protein-1 (AP-1), and hypoxia-inducible factor (HIF). Studies were therefore undertaken to examine if cigarette smoke could activate these transcription factors, as well as other transcription factors that may be important in lung carcinogenesis. Female A/J mice were exposed to cigarette smoke for 2, 5, 10, 15, 20, 42, or 56 days (6 hr/day, 5 days/wk). Cigarette smoke did not increase NF-κB activation at any of these times, but NF-κB DNA binding activity was lower after 15 days and 56 days of smoke exposure. The DNA binding activity of AP-1 was lower after 10 days and 56 days but was not changed after 42 days of smoke exposure. The DNA binding activity of HIF was quantitatively increased after 42 days of smoke exposure but decreased after 56 days. Whether the activation of other transcription factors in the lung could be altered after exposure to cigarette smoke was subsequently examined. The DNA binding activities of FoxF2, myc-CF1, RORE, and p53 were examined after 10 days of smoke exposure. The DNA binding activities of FoxF2 and p53 were quantitatively increased, but those of myc-CF1 and RORE were unaffected. These studies show that cigarette smoke exposure leads to quantitative increases in DNA binding activities of FoxF2 and p53, while the activations of NF-κB, AP-1, and HIF are largely unaffected or reduced.
Introduction
Cigarette smoking is the predominant risk factor in the development of lung cancer, which is the main cause of cancer deaths in men and women in the USA and the world. Estimates indicate that the mortality from tobacco use will rise globally roughly 3-fold during the next 20 years. Tobacco smoke is a complex chemical mixture containing over 4,000 different compounds, and more than 50 of which are known carcinogens, co-carcinogens and/or mutagens (Hoffmann and Hoffmann 1997). The presence of high levels of pro-oxidants like free radicals in smoke is well documented, and cigarette smoke is estimated to contain 1016 free radicals/cigarette, which are detectable in both mainstream and sidestream cigarette smoke (Pryor et al. 1983).
It has been generally difficult to induce respiratory cancers in animals by inhalation exposure to cigarette smoke (Coggins 2007, 2002). Witschi and coworkers (2002) were the first to establish a reproducible lung tumorigenicity model for inhaled cigarette smoke using the A/J strain of mice. Exposure to environmental tobacco smoke, composed of 89% diluted sidestream smoke and 11% mainstream smoke, for 5 months followed by a recovery period of 4 months in filtered air increased the incidence and multiplicity of lung tumors in this sensitive strain of mice and showed a relationship between the exposure dose of smoke and tumorigenicity (Witschi et al. 2004). Currently, this model is commonly used for smoke-induced lung tumor studies and was found to be reproducible in different labs (Witschi 2007; Curtin et al. 2004; Stinn et al. 2005). Traditionally, cigarette smoke constituents, such as polycyclic aromatic hydrocarbons (PAH) and 4-(methylnitrosamino) - 1-(3-pyridyl)-1-butanone (NNK), are believed to be the main carcinogens in smoke. However, studies in A/J mouse model suggest that constituents other than these classical tobacco carcinogens may also play a role in smoke-induced lung tumorigenesis (Witschi 2007). Exposure to gas phase of smoke alone, which is largely devoid of the particulate carcinogens, increased lung tumors in A/J mice to a similar extent as whole smoke, showing that PAH- and NNK-mediated lung tumorigenesis in the A/J strain of mouse did not truly represent tobacco smoke tumorigenesis (Witschi et al. 1997). Free radicals and other pro-oxidants of tobacco smoke, which induce oxidative stress, therefore might be expected to play a role in lung tumorigenesis.
Increased oxidative stress in cigarette smokers is well documented in the literature. Inhalation of smoke exposes the lung tissue to high levels of free radicals that produce oxidative stress. In addition, various endogenous oxidative processes are activated, which promote chronic pulmonary inflammation characterized by increased levels of activated leukocytes in circulation and tissues (Johnson et al. 1990). The presence of elevated levels of various oxidized DNA, protein and lipid products, like 8-hydroxy deoxyguanosine, inactivated α1-antiprotease and F2-isoprostanes, and depletion of plasma antioxidants, e.g., vitamin C, in active and passive smokers as well as smoke-exposed animals (Howard et al. 1998; Chow 1993) substantiates induction of increased oxidant burden by inhaled smoke.
Increased oxidative stress may also influence carcinogenesis by altering gene expression. One family of transcription factors known to be activated by active oxygen is the nuclear factor-κB (NF-κB) family. Reactive oxygen species (ROS), including hydrogen peroxide, are potent activators of NF-κB (Gabbita et al. 2000). Consistent with these data, antioxidants are effective inhibitors of NF-κB (Pahl and Baeuerle 1994). These effects by hydrogen peroxide and antioxidants, and the broad range of stimuli which activate NF-κB, suggest that NF-κB may be an important active oxygen-induced transcription factor. Further, AP-1 activity can be regulated by the redox status of a cell (Meyer et al. 1993; Abate et al. 1990), suggesting that AP-1 activity could be modulated by H2O2 levels. Hypoxia-inducible factor (HIF)-1 is a transcription factor that is activated by hypoxic conditions; it can also be activated by hydrogen peroxide (Lopez-Lazaro 2006; Paul et al. 2004). HIF-1 increases the expression of genes related to angiogenesis and cell survival (Lopez-Lazaro 2006; Paul et al. 2004).
In this study, it was postulated that oxidative stress induced by exposure to cigarette smoke activates transcription factors related to oxidative stress, including NF-κB, AP-1, and HIF-1, thereby triggering a series of downstream events contributing to lung tumor development. In addition, the DNA binding activity of several other transcription factors in the lung was examined, including the forkhead transcription factor FoxF2 (also known as FREAC-2 or LUN), which has high expression in the lung (Aitola et al. 2000; Hellqvist et al. 1996); myc common factor 1 (myc-CF1), which binds the c-myc promoter (Riggs et al. 1991); retinoic acid receptor-related orphan receptor (RORE), which acts as a negative regulator of lung inflammation (Stapleton et al. 2005); and p53, which is frequently inactivated in lung cancer (Robles et al. 2002).
Material and Methods
Materials
Biodyne B blotting membranes were purchased from Pall Life Sciences, East Hills, NY. Transignal Protein/DNA arrays were purchased from Panomics, Redwood City, CA. All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Smoke Exposure
Female A/J mice, 8-9 weeks of age weighing 18-20 g, were purchased from The Jackson Laboratory (Bar Harbor, Maine) and were fed the AIN-93G purified diet (Harlan Teklad, Madison, WI) and water ad libitum. All procedures were approved by the Institutional Animal Care and Use Committee at the University of Kentucky, Lexington, KY. After a one-week acclimation period, mice (6 per group) were either maintained in HEPA-filtered ambient air (sham) or exposed to cigarette-smoke in whole-body chambers (6 hr per day, 5 days per week) for 2 days, 5 days (one week), 10 days (2 weeks), 15 days (3 weeks), 20 days (4 weeks), 30 days (6 weeks), or 40 days (8 weeks). Side stream smoke was generated from the University of Kentucky reference cigarette (2R4F), with a suspended smoke particulate concentration in the chamber atmosphere of approximately 40-45 mg/m3. Although the particulate concentration used in our study falls at the lower end of the concentrations used in other ETS studies (Witschi et al. 2002; Curtin et al. 2004; Stinn et al. 2005), a rise in the incidence and multiplicity of lung tumors was consistently noted in A/J mice exposed to pure SSCS using the Witschi protocol (Gupta et al. 2005; Martin et al. 2010). It should be noted that because of the use of a whole-body exposure system, animals probably also received some exposure to smoke particulates through ingestion of tar particles during grooming and through dermal absorption. Further, it is pertinent to note that chemical composition of mainstream and sidestream are qualitatively similar but levels of some constituents may differ (Adams et al. 1987). Exposure of mice to smoke was ascertained by measuring levels of urinary cotinine by ELISA and CYP1A1 protein levels in lung microsomes by Western blot. For the 2 day - 20 day exposure periods, mice were euthanized within one hr of completing the final smoke exposure. For the 6 and 8 week exposure periods, mice were euthanized within one hr and 20 hr after completing the final smoke exposure. At the time of euthanasia, lungs from sham and cigarette-smoke exposed mice were quickly excised, and frozen immediately at -80°C for analyses.
Electrophoretic mobility shift assays (EMSAs)
Nuclear extracts were made with a Panomics nuclear extraction kit using a Panomics protocol. Protein quantitation was performed by the BCA method using a Pierce BCA protein assay kit. For EMSAs, 5 μg of each sample were incubated in a binding buffer containing 50 mM KCl, 10 mM Hepes, pH 7.9, 6.5 mM DTT, and 10% glycerol along with 0.5 ug poly (dI-dC) for 10 min on ice and for 20 min at room temperature with 1 ng or approximately 20,000 cpm of radioactively labeled probe. The NF-κB (5′-AGT TGA GGG GAC TTT CCC AGG C-3′), AP-1 (5′-CGC TTG ATG ACT CAG CCG GAA-3′), HIF-1 (5′-AGCTTGCCCTACGTGCTGTCTCAGA-3′), Freac2 (5′-CAAACGTAAACAATCC-3′), Myc-CF1 (5′-GGCCAGAGAAAATGGAGAGAAAATGGCCGAG-3′), RORE (5′-AGTGACCCTTTTAACCAGGTCAGTGA-3′), and P53 (5′-TACAGAACATGTCTAAGCATGCTGGGG-3′) probes were made from the respective consensus oligonucleotides (Promega for NF-κB, Santa Cruz for the AP-1 and Panomics for HIF-1, Freac2, Myc-CF1, RORE and P53). The gels were run using the protocol of Tharappel et al. (2002). The shifted bands were quantified using densitometry.
Western Blotting
The nuclear extracts were denatured by boiling with 2× gel loading buffer (173 mL/L glycerol, 1.25 mol/L ß-mercaptoethanol, 52 g/L SDS, 220 mmol/L Tris pH 6.8, 1 mg bromophenol blue). Ten μg of protein per sample were loaded to a western gel and electrophoresed at 175 volts for 55 min. The proteins were transferred to a PVDF membrane (Biorad, Hercules, CA). The membranes were blocked with 5% blocking solution (10 mmol/L Tris pH 7.5, 150 mmol/L NaCl, 500 μL/LTween-20, 50 g/L nonfat dry milk) for 1 hr at room temperature. The membranes were then incubated with antibodies diluted in 5% blocking solution for one hr. The membranes were washed 3× in a washing buffer (10 mmol/L Tris pH 7.5, 150 mmol/L NaCl, 500 μL/L Tween-20) before the primary and secondary antibody incubations. The membranes were probed with anti-actin antibodies to normalize the sample quantities. For the detection of membrane bound antibodies, Pierce SuperSignal West Pico chemiluminescent reagents were used (Pierce, Rockford, IL).
Statistical Analysis
Results were analyzed by Student's t-test or by analysis of variance. Results were considered significant at p < 0.05. All results are expressed as means ± standard error of the mean (SEM).
Results
The purpose of this study was to examine whether cigarette smoke could activate transcription factors in the lung related to oxidative stress, including NF-κB, AP-1, and HIF, as well as several additional factors associated with tumorigenesis. In our initial study, mice were exposed to cigarette smoke for 2-20 days, euthanized within one hr after the end of the final smoke exposure, and the DNA binding activity of NF-κB in the lung was quantified. Cigarette smoke did not increase NF-κB activation at any of these times (Figure 1); the DNA binding activity of NF-κB was significantly lower only after 15 days (3 weeks).
Figure 1. Effect of smoke exposure on the DNA binding activity of NF-κB.

Mice were exposed to cigarette smoke 6 hr/day, 5 days/wk, for 2, 5, 10, 15, and 20 days. The DNA binding activity of NF-κB was determined by EMSA. Values were obtained by densitometry. Results are expressed as percent of control ± SEM.
*Significant effect of smoke treatment (p < 0.05).
The activation of two other transcription factors that can be activated by oxidative stress, AP-1 and HIF, as well as NF-κB, was then examined after 6 and 8 weeks of smoke exposure. Mice were euthanized either the same day (SD), within one hr after smoke exposure ended, or the day after (DA) (20 hr later). After 6 weeks, neither cigarette smoke nor time of euthanasia significantly affected NF-κB binding activity (Figure 2). However, after 8 weeks of treatment, cigarette smoke decreased the NF-κB binding activity whereas the timing of euthanasia exerted no effect (Figure 3). In the 6 week smoke treatment groups, AP-1 binding activity showed no marked change between control and smoke groups (Figure 4), whereas in mice exposed to smoke for 8 weeks AP-1 binding activity was reduced in smoke-treated animals (Figure 5) when compared to control animals. The timing of euthanasia exerted no effect at either 6 or 8 weeks. The HIF-1 DNA binding activity also showed a time-specific change. After 6 weeks, this transcription factor showed a quantitative increase in its binding activity regardless of the time of euthanasia (Figure 6). After 8 weeks of treatment, the DNA binding activity of HIF was significantly decreased by smoke exposure. Euthanizing the animals the following day rather than immediately after ending smoke exposure also significantly decreased the DNA binding activity of HIF.
Figure 2. Effect of 6 weeks' smoke exposure and timing of euthanasia on the DNA binding activity of NF-κB.
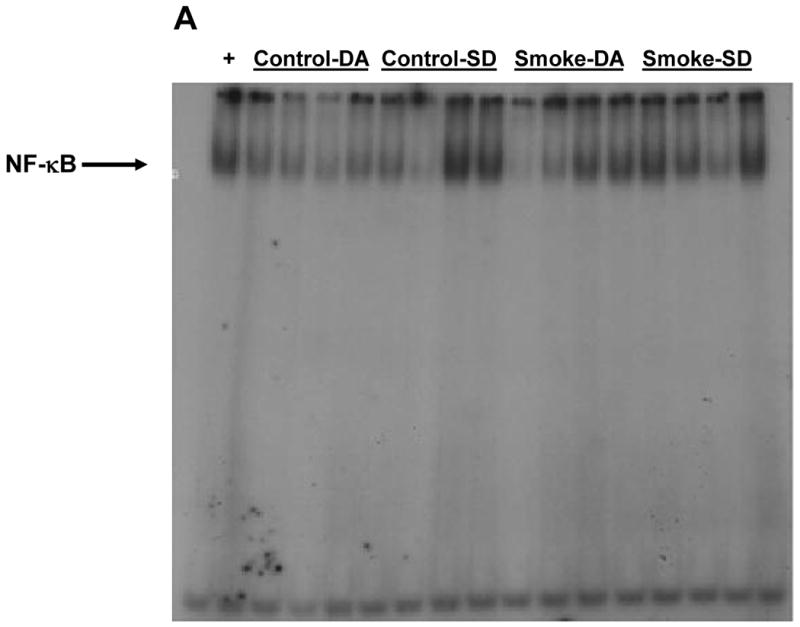

Mice were euthanized on the same day (SD) of the final day of smoke exposure or the day after (DA). A. EMSA. B. Quantitation of NF-κB bands from EMSA. Values are means ± SEM.
*Significant effect of smoke treatment (p< 0.05).
#Significant effect of timing of euthanasia (p < 0.05).
Figure 3. Effect of 8 weeks' smoke exposure and timing of euthanasia on the DNA binding activity of NF-κB.
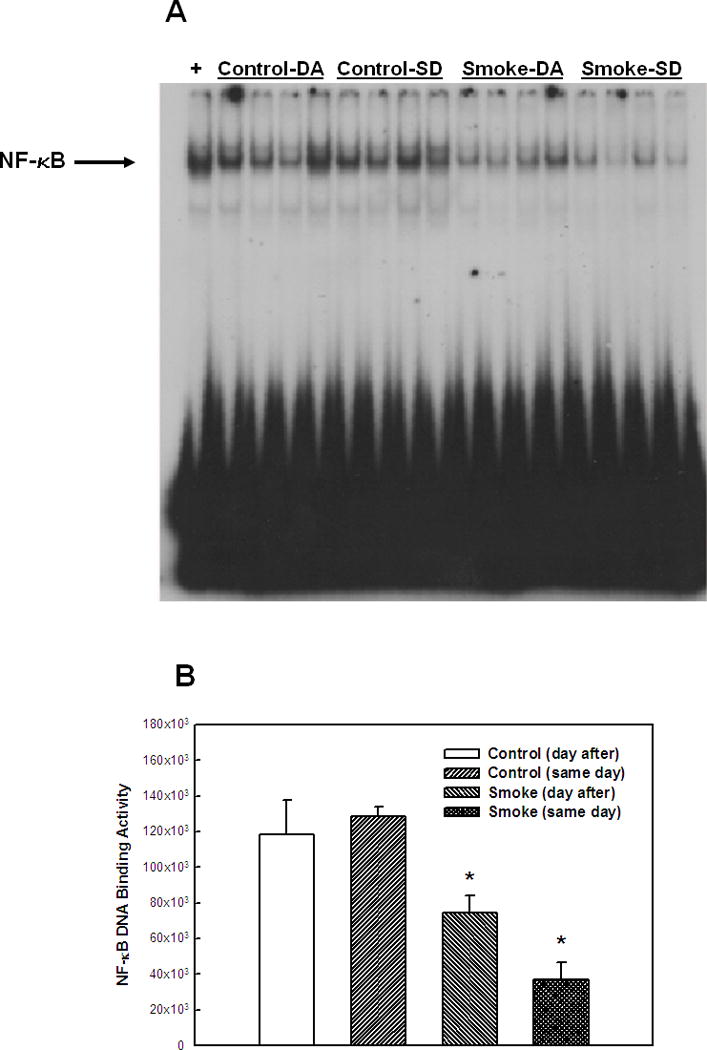
Mice were euthanized on the same day (SD) of the final day of smoke exposure or the day after (DA). A. EMSA. B. Quantitation of NF-κB bands from EMSA. Values are means ± SEM.
*Significant effect of smoke treatment (p < 0.05).
#Significant effect of timing of euthanasia (p < 0.05).
Figure 4. Effect of 6 weeks' smoke exposure and timing of euthanasia on the DNA binding activity of AP-1.
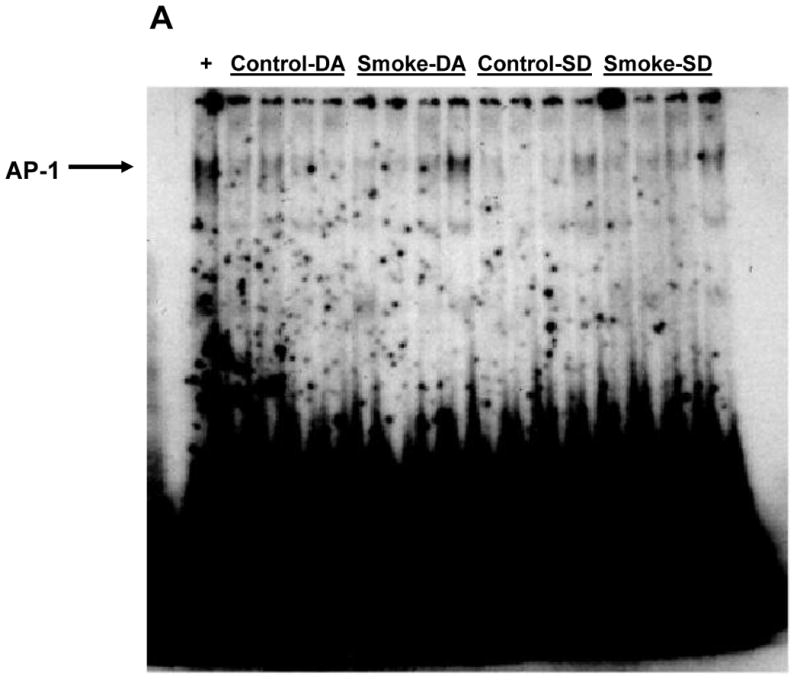

Mice were euthanized on the same day (SD) of the final day of smoke exposure or the day after (DA). A. EMSA. B. Quantitation of AP-1 bands from EMSA. Values are means ± SEM.
*Significant effect of smoke treatment (p < 0.05).
#Significant effect of timing of euthanasia (p < 0.05).
Figure 5. Effect of 8 weeks' smoke exposure and timing of euthanasia on the DNA binding activity of AP-1.
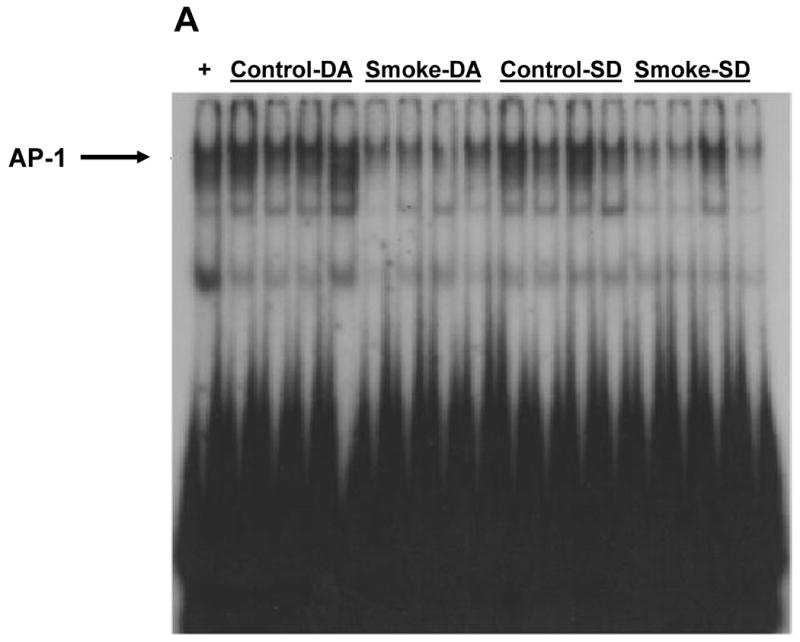

Mice were euthanized on the same day (SD) of the final day of smoke exposure or the day after (DA). A. EMSA. B. Quantitation of AP-1 bands from EMSA. Values are means ± SEM.
*Significant effect of smoke treatment (p < 0.05).
#Significant effect of timing of euthanasia (p < 0.05).
Figure 6. Effect of smoke exposure and timing of euthanasia on the DNA binding activity of HIF.

Mice were exposed to cigarette smoke for 6 weeks (A) or 8 weeks (B) and were euthanized on the same day of the final day of smoke exposure or the day after. The DNA binding activity of HIF was determined by EMSA. Values were obtained by densitometry. Values are means ± SEM.
*Significant effect of smoke treatment (p < 0.05)
#Significant effect of timing of euthanasia (p< 0.05).
Whether the activation of other transcription factors in the lung could be altered after exposure to cigarette smoke was subsequently examined. The DNA binding activities of FoxF2, myc-CF1, RORE, and p53, as well as that of AP-1, were examined after 10 days of smoke exposure. The data showed that 4 transcription factors had quantitatively elevated DNA binding activity after exposure to cigarette smoke: FoxF2 was increased 50%, whereas myc-CF1 rose 12%, RORE was elevated 10%, and p53 was increased 50%, with none of these 4 changes being significant (Figure 7). The DNA binding activity of AP-1 was significantly decreased.
Figure 7. Effect of 10 days' smoke exposure on the DNA binding activity of FoxF2, myc-CF1, RORE, p53, and AP-1.

Mice were exposed to cigarette smoke 6 hr/day, 5 days/wk, for 10 days. The DNA binding activity of the above transcription factors was determined by EMSA. Values were obtained by densitometry, and are expressed as means ± SEM.
*Significant effect of smoke treatment (p < 0.05).
Discussion
Studies were undertaken to examine whether transcription factors whose activation is related to oxidative stress were activated in the lung following exposure to cigarette smoke in mice. The three transcription factors examined--NF-κB, AP-1, and HIF--were not activated by smoke exposure, and for some time points their levels were in fact decreased. The minor changes in the data between time points that were observed are likely due to normal statistical variation. Four additional transcription factors were also analyzed; of these, FoxF2 and p53 were elevated about 50%, although neither increase was statistically significant.
Several other studies have examined if cigarette smoke can activate NF-κB or AP-1 in the lung in vivo (Yao et al. 2008; Thatcher et al. 2007; Valenca et al. 2006; Vlahos et al. 2006; Li et al. 2009; Marwick et al. 2004; Zhong et al., 2006; 2008). No other studies examining the effect of smoke on HIF, FoxF2, myc-CF1, or RORE have been published. Several studies reported that NF-κB was activated after smoke exposure in BALB/c or C57BL/6 mice, or in Ah receptor -/- mice on a C57BL/6 background (Yao et al. 2008; Thatcher et al. 2007; Valenca et al. 2006; Vlahos et al. 2006). NF-κB was activated to a lesser extent in A/J, AKR/J, and CD-1 mice (Yao et al. 2008), but was not activated in 129SvJ mice (Yao et al. 2008) or C57BL/6 mice (Li et al. 2009). In Sprague-Dawley rats, cigarette smoke increased the DNA binding activities of NF-κB (Marwick et al. 2004), but decreased NF-κB activation was observed after smoke exposure in spontaneously hypertensive rats (Zhong et al. 2008). Zhong et al. (2006) found that exposing pregnant rhesus monkeys and their offspring to cigarette smoke decreased the DNA binding activity of NF-κB in the offspring as well as the protein levels of the NF-κB activation pathway and of NF-κB-activated genes. In the present study, which was performed in A/J mice, the DNA binding activity of NF-κB was mainly unaffected but was reduced after 15 and 56 days of exposure. The reasons for the variable results in the present study and in other studies are not clear.
The results of studies examining AP-1 in the lung in vivo generally observed activation after smoke exposure. Cigarette smoke increased AP-1 DNA binding activity in mice and male Sprague-Dawley rats (Marwick et al. 2004; Li et al. 2009). Wang et al. (1999) examined the effect of cigarette smoke on the expression of AP-1 in ferrets and observed that the protein levels of both c-Jun and c-Fos were elevated. These results contrast with those in the present study. Laan et al. (2004), however, found that cigarette smoke condensate (CSC) inhibited the LPS-mediated rise in DNA binding activity of AP-1 in BEAS-2B bronchial epithelial cells.
The DNA binding activity of HIF was quantitatively increased after 6 weeks of smoke exposure but was significantly decreased after 8 weeks. The importance of these findings is not clear. No other studies have examined effects of cigarette smoking on HIF expression or DNA binding in the lung.
No other studies examined the effects of cigarette smoke on the DNA binding activity of p53 in the lung in vivo. The expression and phosphorylation of p53 in vivo was increased to a maximum of about 2-fold by cigarette smoke in rats (Kuo et al. 2005). In RAT-1 murine fibroblasts, CSC was shown to elevate the protein levels of p53 as well as the protein levels of p53-regulated genes (Palozza et al. 2004). In addition, exposing human lung A549 cells to the cigarette smoke component benzo[a]pyrene increased p53 DNA binding activity and protein levels (Biswal et al. 2003). In the present study, a 50% rise in the DNA binding activity of p53 was observed, which was not statistically significant. This possible increase may be in response to damage produced by cigarette smoke.
The 50% rise in FoxF2 DNA binding activity is intriguing. FoxF2 is expressed in the GI tract and in lung epithelium (Chu et al. 2001; Hellqvist et al. 1996; Miura et al. 1998). The lungs are not affected in FoxF2 knockout mice, however, with the primary defects being cleft palate and an abnormal tongue (Ormestad et al. 2004; Wang et al. 2003); the mouse strain using in this study (A/J) is also used as a model for cleft palate (Johnston and Bronsky 1991). FoxF2 binds to DNA sequences identical to those of regulatory regions of the E-cadherin and Talin genes (Chu et al. 2001), whose presence or absence may be important in lung tumorigenesis (Bremnes et al. 2002; Pitterle et al. 1998). E-cadherin expression is inhibited by exposure of A549 lung cells to the smoke component benzo[a]pyrene (Yoshino et al. 2007). However, FoxF2 expression is downregulated in the lung tissues from smokers as well as in human lung cancer tissue (Oyanagi et al. 2004).
In summary, data demonstrated that cigarette smoke exposure leads to quantitative increases in the DNA binding activities of FoxF2 and p53 (after 10 days exposure), while the activations of NF-κB (2-56 days exposure), AP-1 (10, 42, and 56 days of exposure), and HIF (42 and 56 days of exposure) are largely unaffected or decreased. Therefore, the activation of NF-κB, AP-1, or HIF does not appear to be important in the carcinogenic activity of cigarette smoke in mice. The mechanisms and consequences of FoxF2 and p53 DNA binding require further study.
Acknowledgments
These studies were supported by the Kentucky Lung Cancer Research Program. Jill Cholewa was supported by a training grant from the National Institutes of Health (DK07778).
References
- Abate C, Patel L, Rauscher F, Curran T. Redox regulation of fos and jun DNA binding activity in vitro. Science. 1990;248:189–194. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- Adams JD, O'Mara-Adams KJ, Hoffmann D. Toxic and carcinogenic agents in undiluted mainstream smoke and sidestream smoke of different types of cigarettes. Carcinogenesis. 1987;8:729–731. doi: 10.1093/carcin/8.5.729. [DOI] [PubMed] [Google Scholar]
- Aitola M, Carlsson P, Mahlapuu M, Enerback S, Pelto-Huikko M. Forkhead transcription factor FoxF2 is expressed in mesodermal tissues involved in epithelio-mesenchymal interactions. Dev Dyn. 2000;218:136–149. doi: 10.1002/(SICI)1097-0177(200005)218:1<136::AID-DVDY12>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Biswal S, Maxwell T, Rangasamy T, Kehrer JP. Modulation of benzo[a]pyrene-induced p53 DNA activity by acrolein. Carcinogenesis. 2003;24:1401–1406. doi: 10.1093/carcin/bgg061. [DOI] [PubMed] [Google Scholar]
- Bremnes RM, Veve R, Hirsch FR, Franklin WA. The E-cadherin cell-cell adhesion complex and lung cancer invasion, metastasis, and prognosis. Lung Cancer. 2002;36:115–124. doi: 10.1016/s0169-5002(01)00471-8. [DOI] [PubMed] [Google Scholar]
- Chow CK. Cigarette smoking and oxidative damage in the lung. Ann N Y Acad Sci. 1993;686:289–298. doi: 10.1111/j.1749-6632.1993.tb39189.x. [DOI] [PubMed] [Google Scholar]
- Chu D, Kakazu N, Gorrin-Rivas MJ, Lu HP, Kawata M, Abe T, Ueda K, Adachi Y. Cloning and characterization of LUN, a novel ring finger protein that is highly expressed in lung and specifically binds to a palindromic sequence. J Biol Chem. 2001;276:14004–14013. doi: 10.1074/jbc.M010262200. [DOI] [PubMed] [Google Scholar]
- Coggins CR. A minireview of chronic animal inhalation studies with mainstream cigarette smoke. Inhal Toxicol. 2002;14:991–1002. doi: 10.1080/08958370290084746. [DOI] [PubMed] [Google Scholar]
- Coggins CR. An updated review of inhalation studies with cigarette smoke in laboratory animals. Int J Toxicol. 2007;26:331–338. doi: 10.1080/10915810701490190. [DOI] [PubMed] [Google Scholar]
- Curtin GM, Higuchi MA, Ayres PH, Swauger JE, Mosberg AT. Lung tumorigenicity in A/J and rasH2 transgenic mice following mainstream tobacco smoke inhalation. Toxicol Sci. 2004;81:26–34. doi: 10.1093/toxsci/kfh175. [DOI] [PubMed] [Google Scholar]
- Gabbita SP, Robinson KA, Stewart CA, Floyd RA, Hensley K. Redox regulatory mechanisms of cellular signal transduction. Arch Biochem Biophys. 2000;376:1–13. doi: 10.1006/abbi.1999.1685. [DOI] [PubMed] [Google Scholar]
- Gupta RC, Stoner GD, Gairola CG. Inhibition of cigarette smoke-mediated lung tumorigenesis in A/J mice by dietary berries. Proc Am Assoc Cancer Res. 2005;46:3895. [Google Scholar]
- Hellqvist M, Mahlapuu M, Samuelsson L, Enerback S, Carlsson P. Differential activation of lung-specific genes by two forkhead proteins, FREAC-1 and FREAC-2. J Biol Chem. 1996;271:4482–4490. doi: 10.1074/jbc.271.8.4482. [DOI] [PubMed] [Google Scholar]
- Hoffmann D, Hoffmann I. The changing cigarette, 1950-1995. J Toxicol Environ Health. 1997;50:307–364. doi: 10.1080/009841097160393. [DOI] [PubMed] [Google Scholar]
- Howard DJ, Ota RB, Briggs LA, Hampton M, Pritsos CA. Environmental tobacco smoke in the workplace induces oxidative stress in employees, including increased production of 8-hydroxy-2′-deoxyguanosine. Cancer Epidemiol Biomarkers Prev. 1998;7:141–146. [PubMed] [Google Scholar]
- Johnson JD, Houchens DP, Kluwe WM, Craig DK, Fisher GL. Effects of mainstream and environmental tobacco smoke on the immune system in animals and humans: a review. Crit Rev Toxicol. 1990;20:369–395. doi: 10.3109/10408449009089870. [DOI] [PubMed] [Google Scholar]
- Johnston MC, Bronsky PT. Animal models for human craniofacial malformations. J Craniofac Genet Dev Biol. 1991;11:277–291. [PubMed] [Google Scholar]
- Kuo WH, Chen JH, Lin HH, Chen BC, Hsu JD, Wang CJ. Induction of apoptosis in the lung tissue from rats exposed to cigarette smoke involves p38/JNK MAPK pathway. Chem Biol Interact. 2005;155:31–42. doi: 10.1016/j.cbi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Laan M, Bozinovski S, Anderson GP. Cigarette smoke inhibits lipopolysaccharide-induced production of inflammatory cytokines by suppressing the activation of activator protein-1 in bronchial epithelial cells. J Immunol. 2004;173:4164–4170. doi: 10.4049/jimmunol.173.6.4164. [DOI] [PubMed] [Google Scholar]
- Li YT, He B, Wang YZ. Exposure to cigarette smoke upregulates AP-1 activity and induces TNF-alpha overexpression in mouse lungs. Inhal Toxicol. 2009;21:641–647. doi: 10.1080/08958370802322596. [DOI] [PubMed] [Google Scholar]
- Lopez-Lazaro M. HIF-1: hypoxia-inducible factor or dysoxia-inducible factor? FASEB J. 2006;20:828–832. doi: 10.1096/fj.05-5168hyp. [DOI] [PubMed] [Google Scholar]
- Martin JB, Li J, Tharappel JC, Gillespie HD, Cantor AH, Gairola CG, Glauert HP. Effect of dietary selenium on cigarette smoke-induced lung cancer in mice. FASEB J. 2010;24:728.3. [Google Scholar]
- Marwick JA, Kirkham PA, Stevenson CS, Danahay H, Giddings J, Butler K, Donaldson K, Macnee W, Rahman I. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol. 2004;31:633–642. doi: 10.1165/rcmb.2004-0006OC. [DOI] [PubMed] [Google Scholar]
- Meyer M, Schreck R, Baeuerle PA. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993;12:2005–2015. doi: 10.1002/j.1460-2075.1993.tb05850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura N, Kakinuma H, Sato M, Aiba N, Terada K, Sugiyama T. Mouse forkhead (winged helix) gene LUN encodes a transactivator that acts in the lung. Genomics. 1998;50:346–356. doi: 10.1006/geno.1998.5288. [DOI] [PubMed] [Google Scholar]
- Ormestad M, Astorga J, Carlsson P. Differences in the embryonic expression patterns of mouse Foxf1 and -2 match their distinct mutant phenotypes. Dev Dyn. 2004;229:328–333. doi: 10.1002/dvdy.10426. [DOI] [PubMed] [Google Scholar]
- Oyanagi H, Takenaka K, Ishikawa S, Kawano Y, Adachi Y, Ueda K, Wada H, Tanaka F. Expression of LUN gene that encodes a novel RING finger protein is correlated with development and progression of non-small cell lung cancer. Lung Cancer. 2004;46:21–28. doi: 10.1016/j.lungcan.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Pahl HL, Baeuerle PA. Oxygen and the control of gene expression. Bioessays. 1994;16:497–502. doi: 10.1002/bies.950160709. [DOI] [PubMed] [Google Scholar]
- Palozza P, Serini S, Di Nicuolo F, Boninsegna A, Torsello A, Maggiano N, Ranelletti FO, Wolf FI, Calviello G, Cittadini A. beta-Carotene exacerbates DNA oxidative damage and modifies p53-related pathways of cell proliferation and apoptosis in cultured cells exposed to tobacco smoke condensate. Carcinogenesis. 2004;25:1315–1325. doi: 10.1093/carcin/bgh142. [DOI] [PubMed] [Google Scholar]
- Paul SA, Simons JW, Mabjeesh NJ. HIF at the crossroads between ischemia and carcinogenesis. J Cell Physiol. 2004;200:20–30. doi: 10.1002/jcp.10479. [DOI] [PubMed] [Google Scholar]
- Pitterle DM, Jolicoeur EM, Bepler G. Hot spots for molecular genetic alterations in lung cancer. In Vivo. 1998;12:643–658. [PubMed] [Google Scholar]
- Pryor WA, Prier DG, Church DF. Electron-spin resonance study of mainstream and sidestream cigarette smoke: nature of the free radicals in gas-phase smoke and in cigarette tar. Environ Health Persp. 1983;47:345–355. doi: 10.1289/ehp.8347345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs KJ, Merrell KT, Wilson G, Calame K. Common factor 1 is a transcriptional activator which binds in the c-myc promoter, the skeletal alpha-actin promoter, and the immunoglobulin heavy-chain enhancer. Mol Cell Biol. 1991;11:1765–1769. doi: 10.1128/mcb.11.3.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles AI, Linke SP, Harris CC. The p53 network in lung carcinogenesis. Oncogene. 2002;21:6898–6907. doi: 10.1038/sj.onc.1205563. [DOI] [PubMed] [Google Scholar]
- Stapleton CM, Jaradat M, Dixon D, Kang HS, Kim SC, Liao G, Carey MA, Cristiano J, Moorman MP, Jetten AM. Enhanced susceptibility of staggerer (RORalphasg/sg) mice to lipopolysaccharide-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2005;289:L144–L152. doi: 10.1152/ajplung.00348.2004. [DOI] [PubMed] [Google Scholar]
- Stinn W, Teredesai A, Kuhl P, Knorr-Wittmann C, Kindt R, Coggins C, Haussmann HJ. Mechanisms involved in A/J mouse lung tumorigenesis induced by inhalation of an environmental tobacco smoke surrogate. Inhal Toxicol. 2005;17:263–276. doi: 10.1080/08958370590922544. [DOI] [PubMed] [Google Scholar]
- Tharappel JC, Lee EY, Robertson LW, Spear BT, Glauert HP. Regulation of cell proliferation, apoptosis, and transcription factor activities during the promotion of liver carcinogenesis by polychlorinated biphenyls. Toxicol Appl Pharmacol. 2002;179:172–184. doi: 10.1006/taap.2001.9360. [DOI] [PubMed] [Google Scholar]
- Thatcher TH, Maggirwar SB, Baglole CJ, Lakatos HF, Gasiewicz TA, Phipps RP, Sime PJ. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am J Pathol. 2007;170:855–864. doi: 10.2353/ajpath.2007.060391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenca SS, Castro P, Pimenta WA, Lanzetti M, Silva SV, Barja-Fidalgo C, Koatz VL, Porto LC. Light cigarette smoke-induced emphysema and NFkappaB activation in mouse lung. Int J Exp Pathol. 2006;87:373–381. doi: 10.1111/j.1365-2613.2006.00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos R, Bozinovski S, Jones JE, Powell J, Gras J, Lilja A, Hansen MJ, Gualano RC, Irving L, Anderson GP. Differential protease, innate immunity, and NF-kappaB induction profiles during lung inflammation induced by subchronic cigarette smoke exposure in mice. Am J Physiol Lung Cell Mol Physiol. 2006;290:L931–L945. doi: 10.1152/ajplung.00201.2005. [DOI] [PubMed] [Google Scholar]
- Wang T, Tamakoshi T, Uezato T, Shu F, Kanzaki-Kato N, Fu Y, Koseki H, Yoshida N, Sugiyama T, Miura N. Forkhead transcription factor Foxf2 (LUN)-deficient mice exhibit abnormal development of secondary palate. Dev Biol. 2003;259:83–94. doi: 10.1016/s0012-1606(03)00176-3. [DOI] [PubMed] [Google Scholar]
- Wang XD, Liu C, Bronson RT, Smith DE, Krinsky NI, Russell M. Retinoid signaling and activator protein-1 expression in ferrets given beta-carotene supplements and exposed to tobacco smoke. J Natl Cancer Inst. 1999;91:60–66. doi: 10.1093/jnci/91.1.60. [DOI] [PubMed] [Google Scholar]
- Witschi H. Tobacco smoke-induced lung cancer in animals--a challenge to toxicology (?) Int J Toxicol. 2007;26:339–344. doi: 10.1080/10915810701490380. [DOI] [PubMed] [Google Scholar]
- Witschi H, Espiritu I, Dance ST, Miller MS. A mouse lung tumor model of tobacco smoke carcinogenesis. Toxicol Sci. 2002;68:322–330. doi: 10.1093/toxsci/68.2.322. [DOI] [PubMed] [Google Scholar]
- Witschi H, Espiritu I, Maronpot RR, Pinkerton KE, Jones AD. The carcinogenic potential of the gas phase of environmental tobacco smoke. Carcinogenesis. 1997;18:2035–2042. doi: 10.1093/carcin/18.11.2035. [DOI] [PubMed] [Google Scholar]
- Witschi H, Espiritu I, Uyeminami D, Suffia M, Pinkerton KE. Lung tumor response in strain a mice exposed to tobacco smoke: some dose-effect relationships. Inhal Toxicol. 2004;16:27–32. doi: 10.1080/08958370490258372. [DOI] [PubMed] [Google Scholar]
- Yao H, Edirisinghe I, Rajendrasozhan S, Yang SR, Caito S, Adenuga D, Rahman I. Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1174–L1186. doi: 10.1152/ajplung.00439.2007. [DOI] [PubMed] [Google Scholar]
- Yoshino I, Kometani T, Shoji F, Osoegawa A, Ohba T, Kouso H, Takenaka T, Yohena T, Maehara Y. Induction of epithelial-mesenchymal transition-related genes by benzo[a]pyrene in lung cancer cells. Cancer. 2007;110:369–374. doi: 10.1002/cncr.22728. [DOI] [PubMed] [Google Scholar]
- Zhong CY, Zhou YM, Joad JP, Pinkerton KE. Environmental tobacco smoke suppresses nuclear factor-kappaB signaling to increase apoptosis in infant monkey lungs. Am J Respir Crit Care Med. 2006;174:428–436. doi: 10.1164/rccm.200503-509OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong CY, Zhou YM, Pinkerton KE. NF-kappaB inhibition is involved in tobacco smoke-induced apoptosis in the lungs of rats. Toxicol Appl Pharmacol. 2008;230:150–158. doi: 10.1016/j.taap.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]