

Abstract

A synthetic pathway leading to (+)-fusicoauritone (1) is highlighted by the use of a Julia condensation for preparation of an eleven-membered dolabelladienone precursor for subsequent Nazarov cyclization to yield the 5-8-5 tricyclic diterpene skeleton.
Keywords: Fusicoccanes, Julia condensation, Medium-ring compounds, Nazarov cyclization, Total synthesis
The fusicoccanes are representative of a family of terpenes which feature a fused 5-8-5 carbocyclic skeleton.[1] Substances exhibiting this structural motif have been isolated from a variety of sources including wax secretions of scale insects, fungi, liverworts and more recently, from higher plants. Fusicoccanes,[2] cotylenins[3] and fusicoplagins[4] are diterpenoid examples, whereas the ophiobolins[5] and ceroplastols[6] are sesterterpenes. Fusicoccins and cotylenins exhibit significant phytohormonal activities associated with the activation of plasma membrane H+-ATPase, and fusicoccin-binding proteins are considered to be key elements in intracellular signal transduction pathways. [7] The putative biogenesis of this tricyclic framework is described by a π-cation cyclization to form an eleven-membered ring, and further transannular events from a [9.3.0]cyclotetradecane precursor lead to fusicoccanes as exemplified by fusicoauritone (1) (Figure 1).[8] The initial carbocyclization of geranylgeranyl pyrophosphate yields the dolabellanes, a widely distributed class of marine natural products, via hydration or elimination from cation 2.[9] In fact, 3,7-dolabelladienes are key biogenic precursors to fusicoccanes, and neodolabellanes as well as the dolastanes (clavularanes), a class of 5-7-6 tricyclic marine terpenes.[10] Interestingly, marine sources produce relatively few examples of secondary metabolites bearing the 5-8-5 skeleton.[11]
Figure 1.

Merging concepts of biosynthesis with a retrosynthetic design toward 1.
Recognizing the central importance of the dolabellane nucleus in the biosynthesis of several families of diterpenes, we have examined strategies for its preparation,[12] and our efforts have described stereocontrolled transannular reactions to dolastanes and relevant rearrangement products.[13] Recently, synthesis studies toward dolabellane and dolastane diterpenes have been reviewed.[14] A convergent strategy toward the fusicoccanes has been described in a body of work by Kato, Takeshita and co-workers culminating in the total synthesis of (−)-cotylenol.[15] Kishi[16] and Boeckman[17] have independently developed syntheses of ophiobolin C and (±)-ceroplastol I, respectively, and distinctive routes for the synthesis of epoxydictymene have also been reported.[18] Unabated interest in the synthesis of the dicyclopenta[a,d]cyclooctane ring system continues.[19] Herein, we communicate the successful application of our retrosynthetic hypothesis (Figure 1), which features eleven-membered ring formation via an intramolecular alkylation from 3 for utilization of the Nazarov reaction[20] of a dolabelladienone 4 to provide a 5-8-5 tricyclic skeleton as the basis for an effective total synthesis of fusicoauritone (1).
Preparation of the appropriately functionalized cyclopentane of 1 required elaboration of three contiguous stereogenic sites (C-10, C-11 and C-14). This task was undertaken (Scheme 1) beginning with optically active cyclopentenyl alcohol 5[21] via the Johnson ortho-ester Claisen rearrangement. The sigmatropic process occurred with high stereoselectivity,[22] and hydride reduction with subsequent protection gave 7 (MEM = CH2OCH2CH2OCH3).
Scheme 1.

[a] (EtO)3CCH3, H3CCH2COOH (cat.), 145 °C, 79%, then LiAlH4, Et2O, 0 °C, 95%; [b] MEM–Cl, iPr2NEt, DMAP, CH2Cl2, 92%; [c] BH3•THF, 0 °C, then aq. H2O2, NaOH, 75%; [d] (COCl)2, DMSO, CH2Cl2 at −78 °C, then Et3N at −50 °C, 99%; [e] (Z)-1-propenyllithium, Et2O, −50 °C, 65%; [f] (EtO)3CCH3, H3CCH2COOH (cat.), 145 °C; then LiBH4, MeOH, 70%; [g] Na°, HMPA, tBuOH, 97%; [h] TsCl, Et3N, DMAP, CH2Cl2, 89%; [i] NaI, H3CCOC2H5, reflux, then NaO2SC6H4CH3, DMF, 70 °C, 93%.
Hydroboration of the exocyclic olefin 7 led to a primary alcohol that proved to be a single C-10 diastereomer and low temperature Swern oxidation[23] gave an aldehyde that was directly utilized without chromatographic purification to avoid epimerization. Our efforts to elongate the alkyl chain and introduce the remote stereocenter at C-7 were rewarded by further studies of the Claisen rearrangement. Towards this end, (Z)-propenyllithium was prepared according to the Whitesides procedure,[24] and nucleophilic addition with crude aldehyde in ether at −50 °C afforded approximately a 6:1 mixture of (Z)-allylic alcohols (82% yield). Upon chromatographic separation, the major product 8 was obtained in 65% yield.[25]
Treatment of 8 with triethylorthoacetate in the presence of catalytic propanoic acid at 145 °C produced a facile Claisen rearrangement, and direct hydride reduction of the resulting ethyl ester gave alcohol 9 as a single diastereomer (70% for 2 steps).[26] Subsequent reduction of the (E)-alkene in 9 proved to be a challenging problem. A variety of hydrogenation catalysts led to products of double bond migration. Rhodium on alumina efficiently transformed 9 into the corresponding endocyclic (C10–C14) tetrasubstituted olefin. Other techniques, such as the use of diimide were totally ineffective. Fortunately, a dissolving metal reduction using sodium in a concentrated solution of HMPA and tert-butyl alcohol[27] cleanly yielded the saturated alcohol 10 (97%) for straightforward conversion to the desired sulfone 12.
To effect carbocyclization of the eleven-membered [9.3.0] tetradecane system, we utilized modifications of the Julia condensation.[12] Thus, primary alcohol 13 was prepared via deprotection of 12 [HBF4; aq. MeOH (85%)], and oxidation followed by Wittig olefination gave the E-unsaturated ester 15. Conversion to the aldehydic sulfone 16 was followed by rapid addition into a vigorously stirred solution of sodium tert-amylate (0.15 M in benzene at 35 °C). The gold-colored solution was stirred for an additional one to two minutes and quenched with glacial acetic acid leading to the β-hydroxysulfones 17 (dr 5:1) in yields ranging from 73% to 82% with the recovery of additional amounts of enal 16 (12%). The major cyclization product proved to be the trans-β-hydroxysulfone[28] although this stereochemistry was inconsequential for our studies. Transformation of 17 to an appropriate divinylketone substrate required an initial Swern oxidation[23] yielding the corresponding α-sulfonylketones (5:1 ratio). However, the dehydro-elimination of the α-sulfonyl substituent was not feasible. On the other hand, oxidation of the resulting enolate of this system with Davis oxaziridine[29] directly produced α-diketone 18 (85%). Although enolic tautomerization is not evident in the 1H NMR data of 18, treatment with BF3 etherate catalyzed formation of the putative divinylketone intermediate for facile Nazarov cyclization yielding α-hydroxy cyclopentenone 19.[30] Characterization of 19 with extensive use of NMR data, using nOe difference experiments, confirmed the all syn stereochemistry of HA, HB and HC as well as the relationship of the bridgehead (C11) methyl and the newly created α-methylketone.[31]
To effect ring contraction of the dolabelladiene precursor via the Nazarov cyclization with control of enone regiochemistry as presented in 1, we chose to examine the less substituted divinylketone substrate 23.[32] As illustrated in Scheme 3, phenylselenation of α-sulfonyl ketone 20 (major product from 17, Scheme 2) gave 21 for immediate oxidation yielding exclusively the E-α,β-unsaturated sulfone 22.[33] This electron-deficient system exhibited diminished reactivity under Lewis-acid catalyzed Nazarov conditions. A three-step sequence effected replacement of the sulfonyl group with hydrogen. Carbonyl reduction gave solely the β-alcohol,[34] and desulfonylation with sodium naphthalide at −78 °C in THF selectively occurred with retention of double bond geometry. Mild allylic oxidation provided the Z-enone of 23 as indicated by vicinal coupling (JAB = 10.7 Hz) in the proton NMR spectrum. Finally, treatment of 23 under protic or Lewis acid conditions resulted in a smooth Nazarov reaction to 24 (and its C-6 epimer).[35] Upon standing for several days, chloroform solutions of 24 yielded colorless crystals which were unambiguously identified by X-ray crystallography as the hydroperoxide 25,[36] and subsequent reduction with sodium hydrogen sulfite gave 1. Autoxidation of 24 was postulated to occur via enolization and capture of the conjugated enol by dissolved oxygen. This slow, serendipitous reaction to 25 was not pursued as a viable synthetic conversion. Completion of the total synthesis of fusicoauritone was more efficiently rendered by direct oxidation of a mixture of 24 and its C-6 epimer with tert-butylhypochlorite in aqueous acetone providing a 40% yield of synthetic 1 which proved to be identical in all respects with optical rotation data and spectroscopic characterizations of the natural substance.[37]
Scheme 3.

[a] NaHMDS, THF, −78 °C, PhSeCl, 0 °C, then aq. H2O2, 98%; [b] DIBAL, −78 °C, 95%; [c] sodium naphthalide, THF, −78 °C, 1 min, then MnO2, PhH, 70%; [d] TsOH (cat.), ClCH2CH2Cl, 92%; [e] tBuOCl, aq. acetone, 40%; [f] air oxidation; [g] CH2Cl2; aq. NaHSO3 (95%).
Scheme 2.

[a] (COCl)2 DMSO, −78 °C, then Et3N; [b] (carbethoxyethylidene) triphenylphosphorane, CH2Cl2, 22 °C, 96%; [c] DIBAL, CH2Cl2, −78 °C, then PCC, CH2Cl2, 86%; [d] Add 15 to benzene, sodium t-amylate, 35 °C, 5 min, then quench HOAc, 73%; [e] (COCl2), DMSO, −78 °C, then Et3N, followed by K+−OtBu, THF, 2-[(p-chlorophenyl)sulfonyl]-3-(p-chlorophenyl)oxaziridine, −78 °C, 85%; [f] BF3•Et2O, ClCH2CH2Cl, reflux, 77%.
Supplementary Material
Footnotes
We thank the National Institutes of Health [National Institute of General Medical Sciences (GM42897)] for support of this research. We also thank Dr. Josef Zapp (Saarland University) for providing us with NMR and MS spectra of authentic 1.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Petasis NA, Patane MA. Tetrahedron. 1992;48:5757. [Google Scholar]
- 2. Barrow KD, Barton DHR, Chain EB, Ohnsorge UFW, Thomas R. Chem. Commun. 1968:1198. Ballio A, Casinovi CG, D’Alessio V, Grandolini G, Randazzo G, Rossi C. Experimentia. 1974;30:844. doi: 10.1007/BF01938313. c) For recent examples: Kim S, Shin D-S, Lee T, Oh K-B. J. Nat. Prod. 2004;67:448. doi: 10.1021/np030384h.
- 3.For a leading reference: Sassa T, Ooi T, Nukina M, Kato N. Biosci. Biotech. Biochem. 1998;62:1815. doi: 10.1271/bbb.62.1815.
- 4.Hashimoto T, Tori M, Taira Z, Asakawa Y. Tetrahedron Lett. 1985;26:6473. [Google Scholar]
- 5.Nozoe S, Morisaki M, Tsuda K, Iitaka Y, Takahashi N, Tamura S, Ishibashi K, Shirasaka M. J. Am. Chem. Soc. 1965;87:4968. doi: 10.1021/ja00949a061. [DOI] [PubMed] [Google Scholar]
- 6.Iitaka Y, Watanabe I, Harrison IT, Harrison S. J. Am. Chem. Soc. 1968;90:1092. [Google Scholar]
- 7.a) DeBoer B. Trends Plant Sci. 1997;2:60. [Google Scholar]; b) Asahi K, Honma Y, Hazeki K, Sassa T, Kubohara Y, Sakurai A, Takahashi N. Biochem. Biophys. Res. Commun. 1997;238:758. doi: 10.1006/bbrc.1997.7385. [DOI] [PubMed] [Google Scholar]
- 8.a) Liu H-J, Wu C-L, Becker H, Zapp J. Phytochem. 2000;53:845. doi: 10.1016/s0031-9422(99)00609-3. [DOI] [PubMed] [Google Scholar]; b) Zapp J, Burkhardt G, Becker H. Phytochem. 1994;37:787. doi: 10.1016/s0031-9422(00)89573-4. [DOI] [PubMed] [Google Scholar]; c) Huneck S, Baxter G, Cameron AF, Connolly JD, Rycroft DS. Tetrahedron Lett. 1983;24:3787. [Google Scholar]
- 9. Banjeri A, Jones RB, Mellows G, Phillips L, Sim K-Y. J. Chem. Soc. Perkin Trans I. 1976:2221. doi: 10.1039/p19760002221. b) For the production of these metabolites in liverwort: Asakawa Y, Lin X, Tori M, Kondo K. Phytochem. 1990;29:259. Williams DR, Heidebrecht RW., Jr J. Am. Chem. Soc. 2003;125:1843. doi: 10.1021/ja0279803. and references therein.
- 10.Rodriguez AD, González E, Ramirez C. Tetrahedron. 1998;54:11683. [Google Scholar]
- 11.a) Wahlberg I, Eklund A-M, Nishida T, Enzell CR, Berg J-E. Tetrahedron Lett. 1983;24:843. [Google Scholar]; b) Enoki N, Furusaki A, Suehiro K, Ishida R, Matsumoto T. Tetrahedron Lett. 1983;24:4341. [Google Scholar]
- 12.Williams DR, Coleman PJ, Nevill CR, Robinson LA. Tetrahedron Lett. 1993;34:7895. [Google Scholar]
- 13.a) Williams DR, Coleman PJ, Henry SS. J. Am. Chem. Soc. 1993;115:11654. [Google Scholar]; b) Williams DR, Coleman PJ. Tetrahedron Lett. 1995;36:39. [Google Scholar]
- 14.Hiersemann M, Helmboldt H. Top. Curr. Chem. 2005;243:73. [Google Scholar]
- 15.For leading references: Kato N, Okamoto H, Takeshita H. Tetrahedron. 1996;52:3921. Kato N, Nakanishi K, Takeshita H. Bull. Chem. Soc. Jpn. 1986;59:1109.
- 16.Rowley M, Tsukamoto M, Kishi Y. J. Am. Chem. Soc. 1989;111:2735. [Google Scholar]
- 17. Boeckman RK, Jr, Arvanitis A, Voss ME. J. Am. Chem. Soc. 1989;111:2737. b) See also: Paquette LA, Wang T-Z, Vo NH. J. Am. Chem. Soc. 1993;115:1676.
- 18.a) Jamison TF, Shambayati S, Crowe WE, Schreiber SL. J. Am. Chem. Soc. 1994;116:5505. [Google Scholar]; b) Paquette LA, Sun L-Q, Friedrich D, Savage PB. Tetrahedron Lett. 1997;38:195. [Google Scholar]
- 19.a) Mehta G, Krishnamurthy N. Chem. Commun. 1986:1319. [Google Scholar]; b) Dauben WG, Warshawsky AM. J. Org. Chem. 1990;55:3075. [Google Scholar]; c) Snider BB, Yang K. J. Org. Chem. 1992;57:3615. [Google Scholar]; d) Rigby JH, McGuire T, Senanayake C, Khemani K. J. Chem. Soc. Perkin Trans. I. 1994:3449. [Google Scholar]; e) Chase CE, Bender JA, West FG. Synlett. 1996:1173. [Google Scholar]; f) Wender PA, Nuss JM, Smith DB, Suárez-Sobrino A, Vågberg J, Decosta D, Bordner J. J. Org. Chem. 1997;62:4908. [Google Scholar]; g) Sieburth SM, McGee KF, Jr, Al-Tel TH. J. Am. Chem. Soc. 1998;120:587. [Google Scholar]; h) Blake AJ, Highton AJ, Majid TN, Simpkins NS. Org. Lett. 1999;1:1787. [Google Scholar]; i) Bader SJ, Snapper ML. J. Am. Chem. Soc. 2005;127:1201. doi: 10.1021/ja044542i. [DOI] [PubMed] [Google Scholar]
- 20.Tius MA. Eur. J. Org. Chem. 2005:2193. [Google Scholar]
- 21.Using slight modifications of a known procedure, (S)-limonene oxide was converted into 5 in 50% overall yield. White JD, Ruppert JF, Avery MA, Torii S, Nokami J. J. Am. Chem. Soc. 1981;103:1813.
- 22. Mehta G, Krishnamurthy N. Tetrahedron Lett. 1987;28:5945. b) For a related [2,3]-sigmatropic rearrangement from 5:Wright J, Drtina GJ, Roberts RA, Paquette LA. J. Am. Chem. Soc. 1988;110:5806.
- 23.Mancuso AJ, Huang S-L, Swern DJ. J. Org. Chem. 1978;43:2480. [Google Scholar]
- 24.Linstrumelle G, Krieger JK, Whitesides GM. In: Organic Synthesis. Masamune S, editor. Vol. 55. New York: J. Wiley and Sons; 1976. pp. 103–113. [Google Scholar]
- 25.The diastereomeric allylic alcohols were separately subjected to the orthoester Claisen conditions leading to the assignment of stereochemistry as described for 8. Inversion of the undesired alcohol epimer to provide additional quantities of 8 was not feasible.
- 26.The stereochemistry of the newly formed chiral center (C7) was confirmed via degradation. The γ,δ-unsaturated ester obtained from Claisen rearrangement of 8 was reduced (LiBH4), followed by ozonolysis with a NaBH4 quench to produce (R)-2-methylbutane-1,4-diol, +22.6 (c 0.6, CHCl3).Lautens M, Stammers TA. Synthesis. 2002;14:1993.
- 27.Whitesides GM, Ehmann WJ. J. Org. Chem. 1970;35:3565. [Google Scholar]
- 28.The trans-β-hydroxysulfone is distinguished by a large vicinal coupling constant (J = 10 Hz) in the proton NMR spectrum of the major product indicating a diaxial disposition of methine hydrogens. Each sulfone diastereomer independently underwent desulfonylation with 6% Na-Hg in methanol to yield identical samples of allylic alcohol.
- 29.For a leading reference: Williams DR, Robinson LA, Amato GS, Osterhout MH. J. Org. Chem. 1992;57:3740.
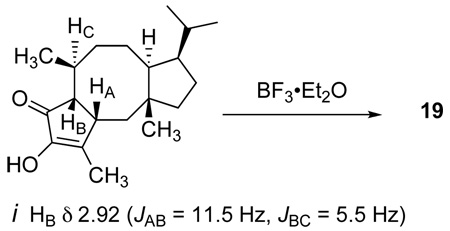
-
30.Initial Nazarov cyclization attempts also gave varying amounts of isomeric ketone i, with tentative stereochemical assignments as shown. This cis-fused arrangement is found in naturally occurring ophiobolins. Treatment of i with BF3•Et2O led to tautomerization to ketone 19.
- 31.NMR studies of 19 demonstrated that irradiation of HB (δ 3.00) gave a 6% nOe enhancement of HA (δ 2.61) and no nOe was observed for the bridgehead C11 methyl substituent. Irradiation of HA gave an 8% enhancement of HB (JAB = 6.0 Hz) and induced a negative 3% nOe of the axial HC (δ 2.85).
- 32.A reviewer requested some insight into alternative strategies which were explored toward enone 24. In fact, we also investigated the use of a Pausen–Khand reaction for cyclization of the eight-membered ring as well as the desired cyclopentenone in a single operation. The required alkyne was readily prepared, but the cyclization was not successful in our hands.
- 33.The E-double bond geometry in 22 was established by irradiation of aromatic hydrogens inducing an nOe with the β-HB.
- 34.Assignment of stereochemistry for the hydride reduction of 22 was made following treatment with 6% Na-Hg in EtOH which produced the C-4 diastereomeric allylic alcohol as compared with the desulfonylation product from 17.
- 35.Cyclopentenone 24 undergoes facile C-6 epimerization and flash silica gel chromatography leads to an incomplete separation of these diastereomers affording fractions of pure 24 for complete characterization.
- 36.Crystal data for 25: colorless block, 0.3×0.3×0.2mm, C20H32O3, M = 320.47, monoclinic, 12.571(3) Å, b = 8.897(2) Å, c = 16.858(4) Å, β = 109.545(10)°, V = 1776.8(8) Å3, T = 108(2) K, space group P21/n, Z = 4, ρcalcd = 1.198 Mg m−3, μ = 0.0781mm−1, MoKα (λ = 0.71073). A total of 3045 reflections were measured. The final residuals was R = 0.0565 with GOF = 1.341 and largest residual peak 0.21 e Å−3.
- 37.Synthetic and natural 1 were identical in all respects. Optical rotation for natural fusicoauritone was recorded as +14.0 (c 0.16, CHCl3). Characterization of synthetic 1 is as follows: Rf 0.28 (30% EtOAc/hexanes); +13.3 (c 0.21, CHCl3); IR (neat) 3445, 2970, 2935, 2880, 1702, 1468, 1390, 1050, 1020, 975, 955 cm−1; 1H NMR (400 MHz, CDCl3) δ 2.71 (d, J = 12.8 Hz, 1H), 2.46 (d, J = 18.4 Hz, 1H), 2.27 (d, J = 18.4 Hz, 1H), 2.22 (d, J = 13.2 Hz, 1H), 2.19-2.10 (m, 3H), 2.05 (s, 1H), 1.73 (s, 3H), 1.71-1.55 (m, 6H), 1.30-1.25 (m, 3H), 1.11 (d, J = 7.2 Hz, 3H), 0.90 (d, J = 6.4 Hz, 3H), 0.84 (d, J = 6.8 Hz, 3H), 0.79 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 207.8, 173.6, 139.6, 83.1, 48.5, 47.2, 46.9, 44.3, 44.0, 40.3, 33.4, 30.2, 28.0, 24.5, 24.4, 22.9, 20.0, 19.5, 17.6, 9.5; MS (CI, NH3) m/z (rel. intensity) 304 (M+), 179 (100), 137 (80), 95 (72); HRMS (CI, NH3) calcd for C20H33O2 [M++1]: 305.2475, found: 305.2471.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.