

Abstract
Excessive activation of Na+/H+ exchanger isoform 1 (NHE-1) plays a role in cerebral ischemic injury. The current study investigated whether NHE-1 protein in ischemic brains is regulated by extracellular signal-regulated kinase (ERK)/90-kDa ribosomal S6 kinase (p90RSK) signaling pathways. A transient focal ischemia in mice was induced by a 60 min-occlusion of the middle cerebral artery followed by reperfusion for 3, 10, or 60 min (Rp). Expression of phosphorylated ERK 1/2 was significantly elevated in the ipsilateral hemispheres at 3 – 10 min Rp and reduced by 60 min Rp. An increase in phosphorylation of p90RSK, a known NHE-1 kinase, was also detected at 3 – 10 min Rp, which was accompanied with a transient elevation of NHE-1 phosphorylation (p-NHE-1). Stimulation of p90RSK in ischemic neurons was downstream of ERK activation because inhibition of MEK1 (MAP kinase/ERK kinase) with its inhibitor U0126 blocked phosphorylation of p90RSK. Moreover, direct inhibition of p90RSK by its selective inhibitor FMK not only reduced p-NHE-1 expression but also ischemic infarct volume. Taken together, our study revealed that reperfusion triggers a transient stimulation of the ERK/p90RSK pathway. p90RSK activation contributes to cerebral ischemic damage in part via activation of NHE-1 protein.
Keywords: brain damage, transient cerebral ischemia, reperfusion, U0126, FMK
INTRODUCTION
Excessive activation of Na+/H+ exchanger (NHE) plays a detrimental role in ischemia and reperfusion injury of myocardium, brain, and kidney (Humphreys et al. 1999; Luo et al. 2005; Masereel et al. 2003). Inhibition of NHE isoform 1 (NHE-1) activity is protective in cardiac ischemia (Engelhardt et al. 2002; Maekawa et al. 2006; Wang et al. 2003). Pharmacologic inhibition or genetic knockdown of NHE-1 is neuroprotective in the mouse model of transient focal cerebral ischemia (Luo et al. 2005; Wang et al. 2008). These findings suggest that NHE-1 activity is elevated following ischemia and reperfusion. Although activation of extracellular signal-related kinase (ERK)/90-kDa ribosomal S6 kinase (p90RSK) pathways have been shown to phosphorylate NHE-1 protein and increase its activity in cultured myocytes and cortical neurons in response to hypoxia and acidosis (Cuello et al. 2007; Luo et al. 2007), it remains unknown whether this regulatory mechanism takes place in regulating NHE-1 activity in ischemic brain tissues.
NHE-1 activity is increased markedly in response to sustained intracellular acidosis (Haworth et al. 2003; Yao et al. 2001). Such a regulation has been proposed to occur through the interaction of H+ with an allosteric modifier site (“pHi sensor”) within the transport domain of NHE-1 (Wang et al. 1995; Wakabayashi et al. 2000b; Wakabayashi et al. 2000a). The NHE-1 activity is also stimulated in response to various extracellular stimuli including mechanical stress, growth factors, changes in cell volume, cell proliferation, and cell migration (Meima et al. 2009; Shrode et al. 1997; Denker et al. 2000). These stimuli can regulate NHE-1 activity by direct protein phosphorylation. The cytosolic domain of the NHE-1 protein (501–815 amino acid residues) is involved in regulation of the activity of the exchanger (Slepkov et al. 2007). In particular, the distal C-terminal region of NHE-1 protein (700–815 amino acid residues) contains a number of serine and threonine residues which interact with protein kinases, including MAPK/ERK (Haworth et al. 2003) and p90RSK (Slepkov et al. 2007; Cuello et al. 2007).
p90RSK is the most important NHE-1 kinase and contains two functional kinase domains: one N-terminal kinase domain which phosphorylates substrates, and one C-terminal kinase domain which activates RSK via autophosphorylation (Vik and Ryder 1997). ERK1/2 activation can phosphorylate Thr573 of the C-terminal kinase domain of p90RSK, triggering phosphorylation of Ser380 and Thr359/Ser363 within the linker region of RSK and constitutively activating the kinase (Anjum and Blenis 2008; Maekawa et al. 2006). It has been established in rat ventricular myocytes that p90RSK is involved in regulating activity of NHE-1 protein (Snabaitis et al. 2006). p90RSK phosphorylates Ser770/771 or Ser703 within the regulatory domain of NHE-1 protein and thereby increases the exchanger activity (Malo et al. 2007; Takahashi et al. 1999). In the current study, we obtained the first line of evidence that transient phosphorylation of p90RSK (Ser380 and Thr359/Ser363) and NHE-1 (Ser703) occurred in ischemic brains during early reperfusion following a transient focal cerebral ischemia. Activation of p90RSK plays a role in cerebral ischemic damage at least in part via stimulation of NHE-1 activity.
EXPERMENTAL PROCEDURES
Transient focal ischemic animal model
Adult male mice (Black Swiss) weighing 25–30 g were used in this study. Focal cerebral ischemia was induced by occlusion of the left middle cerebral artery (MCA) as described previously (Wang et al. 2008). Mice were anesthetized with 3% isofluorane vaporized in N2O and O2 (3:2) for induction and 1.5% isofluorane for maintenance. After preparation of cerebral arteries, a rubber silicon-coated monofilament suture (6–0) was introduced into the internal carotid artery lumen and gently advanced approximately 9 to 9.5 mm to block MCA blood flow. The suture was withdrawn 60 min after MCAO. The body temperature was maintained at 36.5 ± 0.5°C during the surgery. The incision was closed and the mice recovered under a heating lamp to ensure that the core temperature was also maintained at 36.5 ± 0.5°C during recovery. After recovery, animals were returned to their cages with free access to food and water. A total of 40 mice were operated in this study. Sham control animals underwent the same surgical procedures except the insertion of the suture.
All animal procedures used in this study were conducted in strict compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of Wisconsin Center for Health Sciences Research Animal Care Committee.
Drug administration
To study the role of ERK1/2 in phosphorylation of p90RSK, MEK1 inhibitor U0126 was administered at a dose of 0.25 mg/kg body weight by intravenous injection (through the jugular vein) at 30 min prior to the onset of reperfusion. Vehicle control animals received equal volume of 0.1 M PBS containing 1% DMSO. A specific p90RSK inhibitor fluoromethyl ketone (FMK) was administered 30 min prior to reperfusion (intraperitoneally) at a dose of 40 mg/kg body weight. Vehicle control animals received DMSO.
Brain homogenate preparation
Mice were anesthetized with 3% isfluorane vaporized in N2O and O2 (3:2) and decapitated. The contralateral and ipsilateral hemispheres were dissected in ice-cold brain isolation buffer (pH 7.4, in mmol/L: 225 mannitol, 75 sucrose, 1 EGTA). Brain tissues were cut into small pieces in an anti-phosphatase buffer plus protease inhibitors (Wang et al. 2008). Brain tissues were gently homogenized with a tissue pestle grinder (Kontes, Vineland, N J, USA) for 10 strokes in five volumes of the anti-phosphatase buffer. The homogenized samples were centrifuged at 12,000 × g at 4 °C for 10 min and supernatants collected. The protein content was determined by the bicinchoninic acid method.
Gel electrophoresis and immunoblotting
Protein samples containing 80 μg of protein were denatured in sodium dodecyl sulfate (SDS) and mercaptoethanol and heated at 100°C for 8 min before gel electrophoresis. The samples were then electrophoretically separated on 8–10% SDS gels and the resolved proteins were electrophorectically transferred to polyvinylidene difluoride membrane (Millipore Corp., Bedford, MA). The blots were incubated in 7.5% non-fat dry milk prepared in Tris-buffered saline (TBST) for 1 h. Rabbit polycolonal anti-ERK1/2 (1:1000) and anti-phosphorylated ERK1/2 (1: 2000) antibodies were used for detection of total ERK1/2 and p-ERK1/2, respectively. Rabbit polyclonal antibodies against RSK (1:1000) or p-p90RSK (1:1000) at Thr359/Ser363 amino acid residue were used to detect total RSK and p-p90RSK, respectively. Polyclonal antibody against NHE-1 (G115, 1:1000) was used for detection of NHE-1 protein. The blots were rinsed with TBST and incubated with goat anti-rabbit (1:2000) horseradish peroxidase-conjugated secondary IgG for 1 h. Bound antibody was visualized using the enhanced chemiluminescence assay (ThermoScientific, Rockford, IL). Densitometric measurement of each protein band was performed using Image J software (NIH, Bethesda, MD).
Analysis of NHE-1 phosphorylation by immunoprecipitation
The phosphorylation level of NHE-1 was determined using the Pierce® Classic IP Kit (Thermo scientific, Rockford, IL, Chicago). The brain protein samples (0.4mg protein) were incubated with 20 μl of mouse monoclonal antibody against phospho-Ser 14-3-3 BM for 16 h at 4°C overnight. Immunocomplexes were mixed with 20 μl protein A/G beads (50% slurry, ThermoScientific, Rockford, IL) in a Pierce spin column and incubated for 2 h. The immunocomplexes were washed three times with PBS buffer. The immunocomplexes were dissociated from beads with the Laemmli sample buffer and heated for 8 min at 95°C. The resolved proteins were separated on 8% SDS-PAGE and probed with the rabbit polyclonal NHE-1 antibody as described above.
Immunofluorescence staining
At the end of the experiments, mice were deeply anesthetized and transcardially perfused with saline (containing 10 U/ml heparin), followed with ice-cold4 % paraformaldehyde in 0.1 M PBS (pH 7.4). Brains were post-fixed in the same fixative for 12 h at 4°C and cryoprotected with 30% sucrose in 0.1 M PBS for 24–36h. Coronal sections (30 μm) were cut on a freezing microtome cryostat microtome (Leica SM2000R, Heidelberger Str, Deutschland). Sections at the level of 0.26 mm posterior to bregma were selected and processed for immunofluorescence staining. The sections were washed in PBS-Triton X-100 (0.1%) for 10 min, and incubated with a blocking solution (5% bovine serum albumin, 0.1% Triton X-100, TBS) for 1.5 h. The sections were incubated with rabbit monoclonal antibody against p-p90RSK (Ser 380, 1:50 & Thr 359/Ser 363, 1:25) and p-BAD (Ser112, 1:50) in 3% blocking solution overnight at 4°C. After washing in TBS-Triton X-100 (0.1%) for 45 min, the sections were incubated with Alexa FluorTM 488 in goat anti-rabbit secondary antibody (1:150) in 1% blocking solution for 1.5 h. The sections were further incubated with TopRo-3 (1:1000) for 15 min. Sections were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Fluorescent images for p-p90RSK staining were captured under 40X lens using a Leica DMIRE2 inverted confocal laser-scanning microscope (Leica Software, Mannheim, Germany). Samples were excited at 488 nm (argon/krypton), and 633 nm and the emission fluorescence was recorded at 512–548 nm and 650–750 nm, respectively. The p-p90RSK-positive staining cells were counted from 3–4 evenly distributed areas (375 × 375 μm2) in striatum and cortex of the contralateral and ipsilateral hemispheres using Metamorph Software (Downingtown, Philadelphia). The total number of cells in the particular area was determined by the TopRo-3 staining. For negative controls, brain sections were incubated similarly but in the absence of the primary antibody. Identical digital imaging acquisition parameters were used throughout the study.
Fluoro-Jade staining
Brain sections (30 μm) on slides were dried on a slide warmer at 50°C for 30 min. Sections were stained with 0.06% KMnO4 for 15 min. After a brief rinse in ddH2O, the sections were stained with 0.001% Fluro-Jade C (FJ-C) in 1% acetic acid for 25 min on a shaker. Sections were rinsed in ddH2O, followed by air-drying on the slide warmer at 50°C for 8 min. Slides were then rinsed in CitriSolv and mounted with DPX mounting medium. FJ-C staining images were taken under 40X lens using the Leica DMIRE2 inverted confocal laser-scanning microscope. In a blind manner, numbers of FJ-C positive cells were counted in the brain regions of interest in each slice using the Metamorph image analysis software.
Infarct volume estimation
After 72 h of reperfusion, mice were anesthetized and decapitated. Brain slices (2 mm) were stained for 20 min at 37°C with 2 % 2, 3, 5,-triphenyltetrazolium chloride monohydrate (TTC) and scanned using a scanner as described in our previous study (Wang et al. 2008).
In parallel, Nissl staining was also performed for estimating infarct volume. Brain sections (30 μm) were stained with 0.1 % cresyl violet for 1.5 min after rehydration through 100% and 95% alcohol to distilled H2O. Bright-field images were taken under 2.5X lens using a Zeiss epifluorescence microscope (Stereo Investigator 9 software, Williston, VT). Volume of the ischemic lesion was computed by numeric integration of data from four sections at different levels (0.62 mm, 0.02 mm, −1.06 mm,−1.94 mm posterior from bregma) of each brain (Satriotomo et al. 2006). The infarct areas were summed across four sections, and multiplied by the section thickness (30 μm) giving the infarct volume. To account for edema and differential shrinkage resulting from tissue processing (Swanson et al. 1990), the ischemic area for each slice was calculated by subtracting the non-infarct area in the ipsilateral hemisphere from the total area of the contralateral hemisphere.
Statistical analysis
Statistical significance was determined by student’s t-test or an ANOVA (Bonferroni post-hoc test) in the case of multiple comparisons. A p-value smaller than 0.05 was considered statistically significant. N values represent the number of animals for each group.
RESULTS
Elevation of p-ERK1/2 during early reperfusion after focal cerebral ischemia
To test whether the ERK/p90RSK signaling pathway plays a role in regulation of NHE-1 activity in ischemic brain tissues, we first determined time-dependent changes of p-ERK1/2 (44/42 kDa proteins) and total ERK1/2 (44/42 kDa proteins) in contralateral and ipsilateral hemispheres. Immunoblotting analysis showed a similar expression level of p-ERK1/2 in sham controls as well as in the contralateral hemispheres of the ischemic brains at 3, 10 or 60 min Rp (Figure 1A). However, the levels of p-ERK1/2 were selectively increased in the ispilateral hemispheres at 3 min Rp and remained elevated at 10 min Rp. p-ERK1/2 returned to the baseline level by 60 min Rp (Figure 1A, B). Total ERK1/2 protein levels were not significantly altered. The quantitative analysis shows an approximate 2.5-fold increase in p-ERK1/2 in the ipsilateral hemispheres at 3 min Rp (p < 0.05), which remained at the similar level at 10 min Rp (p < 0.05, Figure 1B).
Figure 1. Activation of ERK1/2 in ischemic brain tissues.
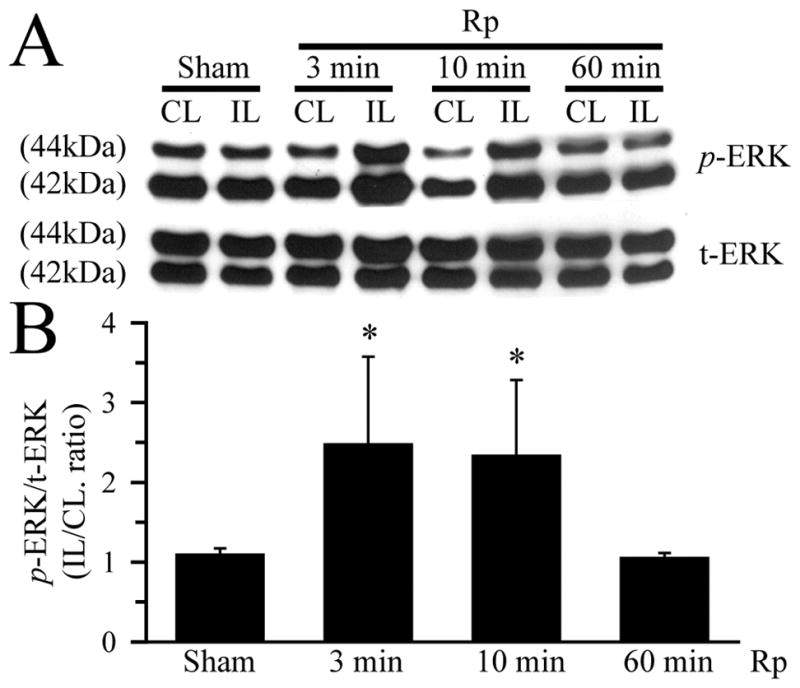
A. Changes of 44/42 kDa ERK proteins (t-ERK1/2) and phosphorylated ERK proteins (p-ERK1/2). Contralateral (CL) and ipsilateral (IL) hemisphere samples were obtained from Sham control or animals at 3 min, 10 min, and 60 min reperfusion (Rp) following 60 min MCAO. The same blots were probed with t-ERK antibody and anti-p-ERK1/2 antibody, respectively. B. Summary data of densitometric analysis of immunoblots. Data are means ± SD (n = 5). * p < 0.05 vs. Sham.
p90RSK was stimulated during early reperfusion after focal cerebral ischemia
Activation of p90RSK in ischemic brains was determined by measuring the phosphorylation status of p90RSK. There was a low level of p-p90RSK (Thr 359/Ser363) detected in the Sham control and contralateral hemisphere brain tissues (Figure 2A). p-p90RSK expression was rapidly increased in the ipsilateral hemispheres at 3 min Rp (approx 3.2-fold of the control level, p < 0.05). By 10 min Rp, p-p90RSK protein was reduced to approximately 1.4-fold of the control and further returned to the baseline level at 60 min Rp (Figure 2A, B). There were no significant changes in the t-RSK protein expression in the contralateral and ipsilateral hemispheres, compared to the Sham controls.
Figure 2. Activation of p90RSK protein in ischemic brain tissues.
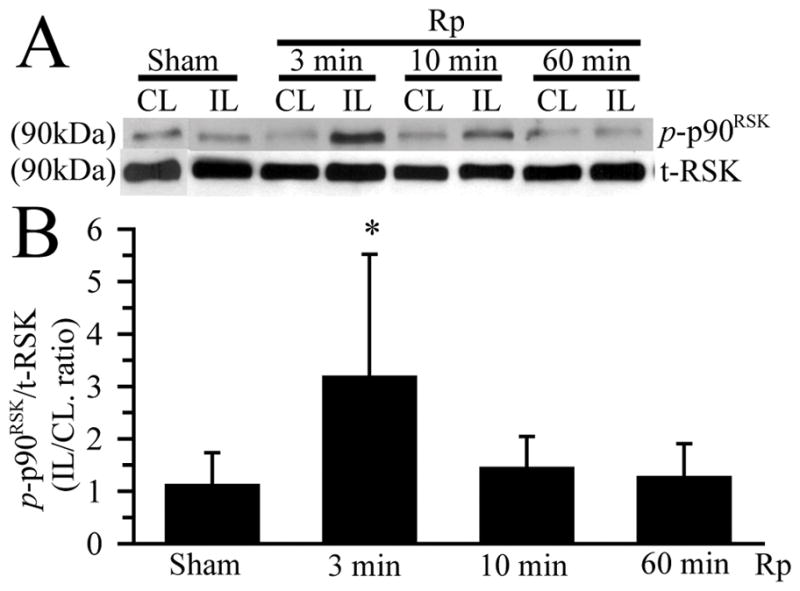
A. Changes of p-p90RSK and non-phospho-p90RSK (t-RSK) expression. CL and IL brain hemisphere samples were prepared as described in Figure 1 legend. The blots were probed with anti-p-p90RSK (Thr359/Ser363) antibody and anti-p90RSK (t-RSK) antibody, respectively. B. Summary data of densitometric analysis. Data are means ± SD (n = 3). * p < 0.05 vs. Sham.
Phosphorylation of NHE-1 protein during early reperfusion
We hypothesized that activation of p90RSK during early reperfusion may trigger phosphorylation of NHE-1 protein in ischemic brain tissues. First, we examined phosphorylation of Ser770/Ser771/703 within the regulatory domain of NHE-1 protein, which binds to the phospho-Ser 14-3-3 and can be immunoprecipitated by the phospho-Ser 14-3-3 BM antibody. This method has been established in cultured myocytes and neurons (Snabaitis et al. 2006; Luo et al. 2007). As illustrated in Figure 3A, a trace level of p-NHE-1 was detected in the immunoprecipitate samples of the Sham control. p-NHE-1 protein level in the ipsilateral hemispheres was dramatically elevated at 3 min Rp (approx 2.4-fold of the control, p< 0.05, Figure 3B). Interestingly, p-NHE-1 remained elevated at 10 min Rp (p < 0.05) and returned to the basal level by 60 min Rp (Figure 3B).
Figure 3. p90RSK-mediated phosphorylation of NHE-1 protein in ischemic brain tissues.

A. Changes of phosphorylated NHE-1 (p-NHE-1) in ischemic brain tissues. p-NHE-1 level was determined by immunoprecipitation with an antibody recognizing the p14-3-3 BM which binds to p-NHE-1 mediated specifically by p90RSK activation (Snabaitis et al. 2006; Luo et al. 2007). The blots were subsequently probed with anti-NHE-1 antibody. B. Summary data of densitometric analysis. Data are means ± SD (n = 3). * p < 0.05 vs. Sham.
Detection of p-p90RSK protein expression in ischemic neurons
We further investigated whether expression of p-p90RSK (Ser380) was altered in neurons of ischemic cortex and striatum by immunofluorescence staining analysis. As shown in Figure 4B, a, c (arrowhead), at 3 min Rp, contralateral cortex and striatum exhibited a diffuse pattern of background immunoreactive signals of p-p90RSK. Omitting the primary antibody abolished all the fluorescence signals (Figure 4B, c, inset). Interestingly, 3 min Rp triggered a sharp increase in p-p90RSK expression in ipsilateral cortical and striatal neurons (Figure 4B, b, d, arrow). Some cortical neurons exhibited nuclear p-p90RSK translocation (Figure 4B, b, open arrowhead). Summary data in Figure 4C demonstrate that the number of neurons with the increased p-p90RSK expression was significantly elevated in both the ipsilateral cortex and striatum at 3 min Rp (p < 0.01). This elevation of p-p90RSK persisted in some of the striatal and cortical neurons at 10 min Rp (data not shown). Most importantly, inhibition of ERK1/2 with MEK1 inhibitor U0126 (30 min prior to reperfusion) reduced the p-p90RSK expression in the ipsilateral striatum and cortex (Figure 4B, e, f, C). These findings further suggest that activation of p90RSK in ischemic brains results from MEK1/ERK stimulation.
Figure 4. Activation of p90RSK (Ser380) in cortical and striatal neurons during early reperfusion.

Expression of p-p90RSK (Ser380) was detected in neurons in cortex and striatum at 3 min Rp. A. The TTC stained brain slice illustrates ROI in cortex (yellow box) and in striatum (green box). B. Green: p-p90RSK (Ser380). Blue: Topro3 nuclear staining. a, c: CL; b, d: IL. Inset image in c: a control study by omitting the primary antibody. e, f: IL hemispheres of U0126-treated brains (U0126 administered at 0.25 mg/kg body weight, iv; 30 min prior to induction of Rp). Arrowhead: low level of p-p90RSK staining. Arrow: increased immunoreactive signals of p-p90RSK. Open arrowhead: nuclear localization of p-p90RSK. Scale bar: 10 μm. C. Summary data of neurons with the positive immunoreactivity for p-p90RSK. Data are means ± SD (n = 5). * p < 0.05 vs. CL.
In addition, we also evaluated expression of p90RSK at phosphorylation site The359/Ser363. As shown in Figure 5A, B, many cortical and striatal neurons showed an increased p-p90RSK (Thr359/Ser363) expression at 3 min Rp (arrow), compared to contralateral striatal neurons (inset in B). Moreover, most of the neurons at 10 min Rp exhibited nuclear translocation of p-p90RSK (Thr359/Ser363) (Figure 5C, D, open arrowhead). Taken together, these findings clearly show that p90RSK in cortical and striatal neurons was stimulated during the initial reperfusion period.
Figure 5. Sustained activation of p90RSK (Thr359/Ser363) in cortical and striatal neurons at 10 min reperfusion.
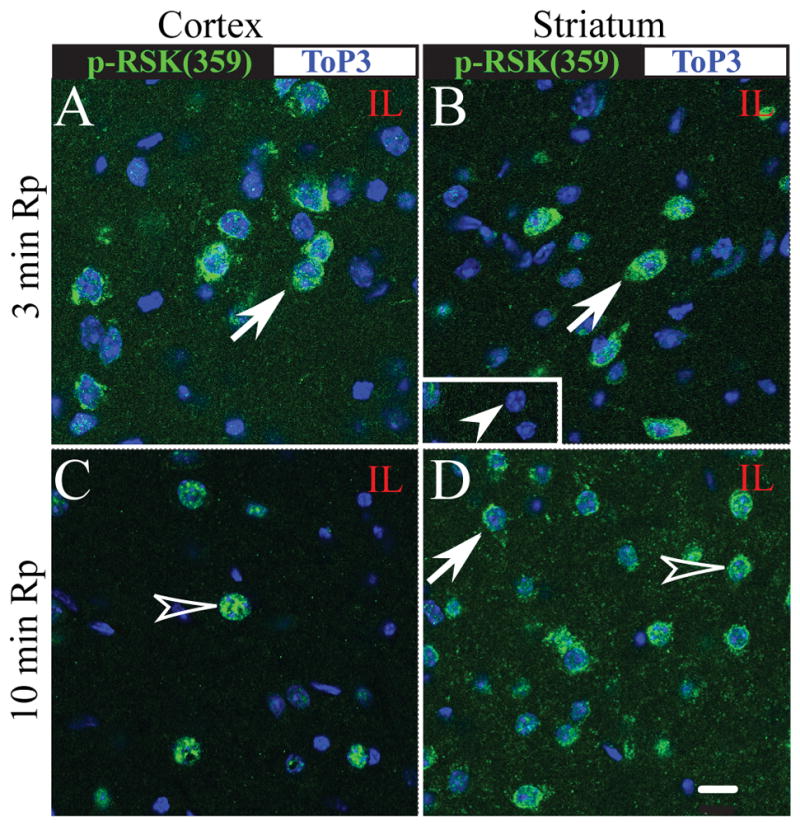
p-p90RSK (Ser359/363) expression (green) and Topro3 nuclear staining (blue) were shown in the ipsilateral cortex and striatum at 3 min Rp (A, B) or 10 min Rp (C, D). Arrow: increased immunoreactive signals of p-p90RSK. Open arrowhead: nuclear localization of p-p90RSK. Arrowhead in inset: an absence of p-p90RSK staining in the contralateral striatum. Identical digital imaging acquisition parameters were used in all images. Scale bar: 10 μm.
Direct inhibition of p90RSK with its selective inhibitor FMK reduces infarct volume
We then investigated whether selective blocking p90RSK function with the potent p90RSK inhibitor FMK is neuroprotective. First, FMK had no effects on rCBF. Induction of MCAO caused rCBF dropped to less than 6% of the control in both the vehicle-control group and FMK-treatment group (p < 0.05, Supplemental Figure 1). Administration of FMK (40 mg/kg, ip) 30 min prior to reperfusion did not affect rCBF reduction (determined at 45 min and 60 min). FMK decreased infarct volume and neuronal degeneration as evidenced by TTC staining and cresyl violet staining, respectively (Figure 6A, B). The infarct volume was 36.2 ± 7.2 % in the DMSO-vehicle treated group. In contrast, the FMK-treated group exhibited a significantly reduced infarction volume (14.0 ± 4.2%, n = 3 – 4, p < 0.05, Figure 6C). A decreased expression of p-NHE-1 protein in the ipsilateral hemispheres was also confirmed in the FMK-treated brains (Figure 6D).
Figure 6. Direct inhibition of p90RSK by its potent inhibitor FMK is neuroprotective.
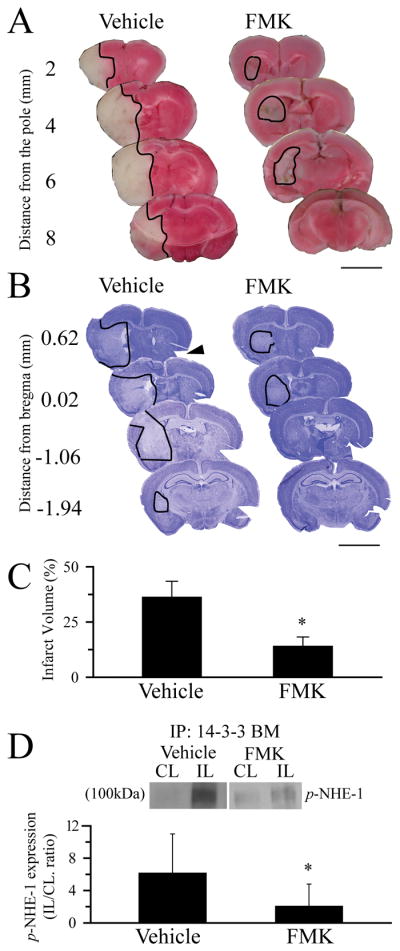
A. Representative TTC staining images of coronal sections at 72 h Rp. FMK (40 mg/kg wt, i.p) was administered at 30 min prior to induction of reperfusion. DMSO was used as a vehicle control. Scale Bar: 5 mm. B. Cresyl violet staining. Arrow: a cut as a marker for CL. Scale Bar: 5 mm. Line trace: brain lesion. C. Summary data of infarction volume calculated in the cresyl violet stained brain sections. Data are means ± SD (n = 3 – 4). * p < 0.05 vs. CL. D. p-NHE-1 level in the control and the FMK-treated brains. Data are means ± SD (n = 3 – 4). * p < 0.05 vs. CL.
Neurodegeneration was further evaluated with FJ-C staining. As illustrated in Figure 7A, B, FJ-positive cells were evaluated in the contralateral and ipsilateral cortex and striatum at 72 h Rp. In the vehicle-control brains, neurons in the contralateral hemispheres exhibited a low level of FJ background staining signal in a diffuse pattern (Figure 7B, a, e, arrowhead). In contrast, the ipsilateral cortex and striatum (ischemic core) showed many cells with strong, condensed FJ fluorescence staining (Figure 7B, b, f, arrow), which reflects degenerated neurons at 72 h Rp. In the FMK-treated brains, the FJ staining in the ipsilateral cortex and striatum was weak and diffuse, similar to the contralaterial hemisphere staining (Figure 7B, d, h). Not many cells showed the strong, condensed FJ staining in the FMK-treated brains. FJ-positive cells with the condensed staining morphology were counted and the summary data were shown in Figure 7C. At 72 h Rp, the ipsilateral cortex in the ischemic core area had 234.7 ± 73.7 cells/mm2, while the ipsilateral striatum showed 290.4 ± 162.6 cells/mm2 (n = 4). In contrast, the FMK-treated brains revealed a significantly reduced number of FJ-positive cells in both ipsilateral cortex (8.3 ± 11.2 cells/mm2, p < 0.05) and striatum (61.9 ± 54.1 cells/mm2, p < 0.05). These findings further confirm that inhibition of p90RSK with FMK indeed reduced cortical and striatal neuronal degeneration following ischemia/reperfusion injury.
Figure 7. Less neurodegeneration in brains treated with p90RSK inhibitor FMK.

A. Cresyl violet staining image of the vehicle-control brain at 72 h Rp. CL: contralateral hemisphere. IL: ipsilateral hemisphere. Dashed line: the ischemic core area. Square box: regions where the FJ-C staining was evaluated. Scale bar: 2 mm. B. Representative images of FJ-C staining at 72 h Rp after 1 h MCAO. a, b, e, f: vehicle-control. c, d, g, h: FMK-treated. Arrowhead: non-degenerating cells with diffuse FJ staining. Arrow: degenerated cells with the condensed FJ staining. Scale bar: 75 μm. C. Summary data. FJ-C positive cells with the condensed cellular morphology were counted in the ipsilateral core area (cortex, striatum). Data are means ± SD (n = 4). * p < 0.05 vs. vehicle-control.
p-BAD expression was not affected by inhibition of p90RSK with FMK
RSK-mediated phosphorylation of BAD at Ser-112 is reported to involve in survival of hippocampal neurons (Watson and Fan 2005). To further understand the neuroprotective effects afforded by FMK, we investigated changes of p-BAD expression (Ser 112) in the vehicle control and FMK-treated brains. Figure 8A (a, c) illustrates a basal level of p-BAD expression in the contralateral cortex and striatum in the vehicle control brains at 72 h Rp (arrow). p-BAD expression was sharply reduced in the ischemic core of the vehicle control (Figure 8A, b, d, B, p = 0.057). In the FMK-treated brains, the p-BAD expression pattern in the ischemic core was also reduced (Figure 8A, e, f, B). Reduction of p-BAD in the ischemic core areas was less in the FMK-treated brains, compared to the control (p = 0.08). Interestingly, there was an up-regulation of p-BAD expression in the ipsilateral cortical peri-infarct areas in the vehicle control and FMK-treated brains (Figure 8A, g, h). Summary data were shown in Figure 8B. Taken together, these findings imply that administration of FMK prior to reperfusion had no significant effects on p-BAD (Ser112) expression in ischemic brains.
Figure 8. p-BAD expression in ischemic brains.

A. Representative images of p-BAD expression. The specific brain regions were chosen as illustrated in Figure 7. CL: contralateral hemisphere. IL: ipsilateral hemisphere. a – d, g: vehicle-control. e, f, h: FMK-treated. Inset in b: a control study by omitting the primary antibody. Arrow: a basal level of immunoreactive p-BAD staining. Arrowhead: reduced p-BAD immunostaining. Scale bar: 75 μm. B. Summary data. Data are means ± SD (n = 5). * p < 0.05 vs. CL. # p < 0.05 vs. the ischemic core area.
DISCUSSION
ERK/p90RSK signaling is responsible for NHE-1 phosphorylation in ischemic brains
ERK/p90RSK signaling pathway is responsible for NHE-1 activation in many cell types (Luo et al. 2007; Wang et al. 1997; Maekawa et al. 2006). The use of the specific p90RSK inhibitor FMK or cardiac-specific overexpression of dominant negative RSK further establishes that p90RSK is the principal regulator of NHE-1 phosphorylation in cardiac cells (Cuello et al. 2007; Maekawa et al. 2006). However, it remains unknown whether this ERK/p90RSK signaling pathway governs NHE-1 regulation in ischemic brain tissues.
In the current study, for the first time, we detected a transient increase in p-NHE-1 (Ser703) expression in the ischemic brain tissues during early reperfusion (3–10 min) following a transient ischemia. This was accompanied with a concurrent elevation of p-ERK1/2 and p-p90RSK expression. Immunofluorescence staining analysis revealed a robust increase of p-p90RSK (Ser380) expression in the ischemic striatal neurons and cortical neurons at 3–10 min reperfusion. Inhibition of the upstream MEK1 by U0126 abolished activation of p90RSK in the ischemic striatal neurons and cortical neurons. Most importantly, elevation of p-NHE-1 expression was reduced by a direct inhibition of p90RSK with its inhibitor FMK. Taken together, our study clearly demonstrates that early reperfusion can trigger ERK/p90RSK activation and NHE-1 phosphorylation in ischemic neurons.
ERK1/2 can rapidly phosphorylate p90RSK at the Thr359/Ser363 in cardiac myocytes (Haworth et al. 2006; Cuello et al. 2007) and in cultured ischemic neurons (Luo et al. 2007). Autophosphosphorylation of p90RSK at Ser380 by the C-terminal RSK1 kinase domain can be triggered upon docking and activation of ERK (Vik and Ryder 1997). This autophosphorylation event contributes to the formation of a fully active RSK enzyme (Vik and Ryder 1997). In the current study, we detected phosphorylation of p90RSK at Thr359/Ser363 as well as at Ser380. Therefore, these data suggest that ischemia/reperfusion-mediated activation of p90RSK in brains results from ERK-mediated phosphorylation and autophosphorylation.
Because of the wide-range of potential molecular substrates of ERK/p90RSK present in cells, the precise subcellular location in which p90RSK activation occurs determines its cellular function. Transient membrane localization of p90RSK occurs prior to its translocation into the nucleus (Richards et al. 2001). In our current study, the transient elevation of immunoreactive signals of p-p90RSK (Ser 380) was localized at the plasma membrane/cytosol, with a few neurons exhibiting a nuclear RSK localization. This suggests that early reperfusion following ischemia may selectively trigger the RSK-NHE-1 activation without affecting RSK-induced gene transcription signaling in nuclei. This would allow neurons to quickly correct ischemic acidosis by activating NHE-1-meditaed H+ extrusion. However, over-stimulation of NHE-1 activity exacerbates reperfusion-mediated brain damage via disruption of Na+/Ca2+ ionic homeostasis. This view is further supported by our recent findings that administration of NHE-1 inhibitor HOE 642 only during reperfusion reduces neuronal death (Ferrazzano et al. 2010).
However, nuclear translocation of RSK may regulate transcription by phosphorylating a number of transcription factors, including CREB and nuclear factor-κB, and immediate-early gene products such as FOS, and JUN (Anjum and Blenis 2008). Thus, RSK may play an important role in transcriptional regulation, cell growth, and cell survival in ischemic brains. It is conceivable to speculate that sustained activation of RSK could affect ischemic brain damage as well as repair. Future study is needed to dissect these processes by blocking RSK function at different time periods following ischemia.
In the current study, activation of ERK1/2 and p90RSK was rapid but short lived. This is consistent with the reports about the robust transient activation of ERK at 3 min reperfusion following a transient focal cerebral ischemia (Alessandrini et al. 1999), or at 4 h reperfusion after spinal cord ischemia (Shackelford and Yeh 2003). The short life of ERK1/2 activation may result from a concurrent stimulation of MAPK phosphatases during reperfusion. ERK activity is regulated by its phosphorylation state, which is the result of the balanced action of both ERK kinases and ERK-directed protein phosphatases (Silverstein et al. 2002; Kim et al. 2003). Early reperfusion following a transient ischemia induces an increase in PP2A expression as early as at 0–10 min reperfusion, which sustained through 24 h reperfusion (Hu et al. 2009). Therefore, a concurrent activation of phosphatase function would dephosphorylate ERK and p90RSK to prevent a sustained activation.
Inhibition of p90RSK is neuroprotective in ischemic brains
Many studies show that activation of ERK1/2 and p90RSK plays a role in cell death following ischemia or oxidative stress. A transient activation of p90RSK (Thr359/Ser363) occurs at 20 min reperfusion of ischemic myocardium and inhibition of p90RSK activity by cardiac-specific dominant negative RSK expression reduces apoptosis and improves myocardium function (Maekawa et al. 2006). Phosphorylation of cardiac troponin I following a transient activation of p90RSK depresses the acto-myosin interaction in response to oxidative stress (Itoh et al. 2005). Elevation of ERK1/2 and p90RSK also leads to GSH-depletion associated apoptosis in retinal pigmented epithelial cells (Glotin et al. 2006). In the current study, we observed a robust neuroprotective effect of inhibiting p90RSK by its potent p90RSK (RSK 1, 2) inhibitor FMK [IC50 of 15 nM, (Cohen et al. 2005)]. We found that administration of FMK prior to reperfusion reduced the infarct volume by 60%. The FMK-treated brains exhibited ~ 80% less number of degenerative neurons, compared to the vehicle controls. These findings clearly illustrate that inhibition of p90RSK (RSK 1, 2) during early reperfusion is neuroprotective.
Several reports have demonstrated that inhibition of ERK1/2 with the MEK inhibitors SL327, PD98058, or U0126 reduces ischemic neuronal death (Alessandrini et al. 1999; Namura et al. 2001; Wang et al. 2008). The underlying mechanisms are not completely understood. Activation of ERK plays a role in Ca2+-dependent glutamate release via phosphorylation of a synaptic vesicle protein synapsin I (Jovanovic et al. 2001). Moreover, p90RSK (RSK2) binds and phosphorylates several postsynaptic density fraction 95/discs large/ZO-1 (PDZ) domain proteins, which increases AMPA-R synaptic transmission (Thomas et al. 2005). Phosphorylation of phospholipase D via p90RSK enhances fusion of secretory vesicles with the plasma membrane in the exocytosis of neurotransmitters (Zeniou-Meyer et al. 2008). Therefore, it is possible that the neuroprotective effects offered by p90RSK inhibition in the current study may also result from attenuating glutamate release in ischemic brains.
Role of p90RSK in regulation of p-BAD in ischemic brains
The MEK/ERK signaling pathway affects many cellular targets. The strength and duration of the activation of the different components of the cascade would dictate the final biological outcome following stimulation of the MEK/ERK pathway. For example, p90RSK links MEK/ERK survival signals to the apoptotic cell-death machinery through RSK-dependent phosphorylation and inactivation of the mitochondrial pro-apoptotic protein BAD, thus, promoting cell survival (Anjum and Blenis 2008). Phosphorylation of BAD leads to the association with the 14-3-3 family of proteins, which prevents BAD from interacting with the anti-apoptotic proteins Bcl-2 and Bcl-XL on the outer membrane of mitochondria. BAD function is modulated by phosphorylatation at four sites, Ser 112, Ser 136, Ser 155, and Ser 170(Shichinohe et al. 2004). BAD phosphorylation at Ser 112 involves in ERK/p90RSK pathway, whereas phosphorylation at Ser 136 involves PI3K/Akt pathway. One important issue here is whether inhibition of p90RSK with FMK would reduce BAD phosphorylation and therefore facilitate ischemic damage via enhancing apoptotic cell death in ischemic brains. In the current study, we examined changes of p-BAD (Ser112) in the vehicle control and FMK-treated brains. Up-regulation of p-BAD (Ser112) expression in the peri-infarct area was not significantly affected by FMK. These data imply that BAD phosphorylation state is regulated by phosphatase-mediated dephosphorylation and kinase-induced phosphorylation. The loss of p-BAD (Ser112) expression in the ischemic core suggests that ischemia-induced dephosphorylation of BAD via phosphatase function may also be involved. This view is supported by the report that inhibition of calcineurin with FK506 prolongs p-BAD expression during reperfusion and reduces ischemic apoptosis (Li et al. 2006). Regarding to the lack of significant effects of FMK on p-BAD expression, we can not rule out the possibility that other kinases (PKC, PKA, PI3K/Akt), which are known to act on more than one of the phosphorylation sites on BAD (Shichinohe et al. 2004), may compensate the inhibition of RSK function in the FMK-treated brains.
Summary
Our current study demonstrates a transient increase in phosphorylation of p90RSK, a known NHE-1 kinase, as well as NHE-1 protein. Stimulation of p90RSK in ischemic neurons was downstream of ERK activation. Moreover, direct inhibition of p90RSK by its selective inhibitor FMK not only reduced NHE-1 phosphorylation but also ischemic infarct volume. Taken together, our study revealed that reperfusion triggers a transient stimulation of the p90RSK pathway which contributes to cerebral ischemic damage in part via phosphorylation of NHE-1 protein.
Supplementary Material
Acknowledgments
This work was supported in part by an NIH grant RO1NS48216 and AHA EIA 0540154 (D.S.), and NIH grant P30 HD03352 (Waisman Center). The authors would like to thank Douglas Kintner for his assistance in preparation of figures.
Reference List
- Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci U S A. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9:747–758. doi: 10.1038/nrm2509. [DOI] [PubMed] [Google Scholar]
- Chen H, Luo J, Kintner DB, Shull GE, Sun D. Na+-dependent chloride transporter (NKCC1)-null mice exhibit less gray and white matter damage after focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:54–66. doi: 10.1038/sj.jcbfm.9600006. [DOI] [PubMed] [Google Scholar]
- Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello F, Snabaitis AK, Cohen MS, Taunton J, Avkiran M. Evidence for direct regulation of myocardial Na+/H+ exchanger isoform 1 phosphorylation and activity by 90-kDa ribosomal S6 kinase (RSK): effects of the novel and specific RSK inhibitor fmk on responses to alpha1-adrenergic stimulation. Mol Pharmacol. 2007;71:799–806. doi: 10.1124/mol.106.029900. [DOI] [PubMed] [Google Scholar]
- Denker SP, Huang DC, Orlowski J, Furthmayr H, Barber DL. Direct binding of the Na--H exchanger NHE1 to ERM proteins regulates the cortical cytoskeleton and cell shape independently of H+ translocation. Mol Cell. 2000;6:1425–1436. doi: 10.1016/s1097-2765(00)00139-8. [DOI] [PubMed] [Google Scholar]
- Engelhardt S, Hein L, Keller U, Klambt K, Lohse MJ. Inhibition of Na(+)-H(+) exchange prevents hypertrophy, fibrosis, and heart failure in beta(1)-adrenergic receptor transgenic mice. Circ Res. 2002;90:814–819. doi: 10.1161/01.res.0000014966.97486.c0. [DOI] [PubMed] [Google Scholar]
- Ferrazzano P, Shi Y, Manhas N, et al. Inhibiting the sodium/hydrogen exchanger reduces reperfusion injury: a small animal MRI Study. Frontiers of Bioscience. 2010 doi: 10.2741/e222. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotin AL, Calipel A, Brossas JY, Faussat AM, Treton J, Mascarelli F. Sustained versus transient ERK1/2 signaling underlies the anti- and proapoptotic effects of oxidative stress in human RPE cells. Invest Ophthalmol Vis Sci. 2006;47:4614–4623. doi: 10.1167/iovs.06-0297. [DOI] [PubMed] [Google Scholar]
- Haworth RS, Dashnyam S, Avkiran M. Ras triggers acidosis-induced activation of the extracellular-signal-regulated kinase pathway in cardiac myocytes. Biochem J. 2006;399:493–501. doi: 10.1042/BJ20051628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth RS, McCann C, Snabaitis AK, Roberts NA, Avkiran M. Stimulation of the plasma membrane Na+/H+ exchanger NHE1 by sustained intracellular acidosis. Evidence for a novel mechanism mediated by the ERK pathway. J Biol Chem. 2003;278:31676–31684. doi: 10.1074/jbc.M304400200. [DOI] [PubMed] [Google Scholar]
- Hu X, Wu X, Xu J, Zhou J, Han X, Guo J. Src kinase up-regulates the ERK cascade through inactivation of protein phosphatase 2A following cerebral ischemia. BMC Neurosci. 2009;10:74. doi: 10.1186/1471-2202-10-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys RA, Haist JV, Chakrabarti S, Feng Q, Arnold JM, Karmazyn M. Orally administered NHE1 inhibitor cariporide reduces acute responses to coronary occlusion and reperfusion. Am J Physiol. 1999;276:H749–H757. doi: 10.1152/ajpheart.1999.276.2.H749. [DOI] [PubMed] [Google Scholar]
- Itoh S, Ding B, Bains CP, et al. Role of p90 ribosomal S6 kinase (p90RSK) in reactive oxygen species and protein kinase C beta (PKC-beta)-mediated cardiac troponin I phosphorylation. J Biol Chem. 2005;280:24135–24142. doi: 10.1074/jbc.M413015200. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Jr, Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci. 2001;21:7944–7953. doi: 10.1523/JNEUROSCI.21-20-07944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Song MC, Kwak IH, Park TJ, Lim IK. Constitutive induction of p-Erk1/2 accompanied by reduced activities of protein phosphatases 1 and 2A and MKP3 due to reactive oxygen species during cellular senescence. J Biol Chem. 2003;278:37497–37510. doi: 10.1074/jbc.M211739200. [DOI] [PubMed] [Google Scholar]
- Li JY, Furuichi Y, Matsuoka N, Mutoh S, Yanagihara T. Tacrolimus (FK506) attenuates biphasic cytochrome c release and Bad phosphorylation following transient cerebral ischemia in mice. Neuroscience. 2006;142:789–797. doi: 10.1016/j.neuroscience.2006.06.064. [DOI] [PubMed] [Google Scholar]
- Luo J, Chen H, Kintner DB, Shull GE, Sun D. Decreased neuronal death in Na+/H+ exchanger isoform 1-null mice after in vitro and in vivo ischemia. J Neurosci. 2005;25:11256–11268. doi: 10.1523/JNEUROSCI.3271-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Kintner DB, Shull GE, Sun D. ERK1/2-p90RSK-mediated phosphorylation of Na(+)/H(+) exchanger isoform 1. A role in ischemic neuronal death. J Biol Chem. 2007;282:28274–28284. doi: 10.1074/jbc.M702373200. [DOI] [PubMed] [Google Scholar]
- Maekawa N, Abe J, Shishido T, Itoh S, Ding B, Sharma VK, Sheu SS, Blaxall BC, Berk BC. Inhibiting p90 ribosomal S6 kinase prevents (Na+)-H+ exchanger-mediated cardiac ischemia-reperfusion injury. Circulation. 2006;113:2516–2523. doi: 10.1161/CIRCULATIONAHA.105.563486. [DOI] [PubMed] [Google Scholar]
- Malo ME, Li L, Fliegel L. Mitogen-activated Protein Kinase-dependent Activation of the Na+/H+ Exchanger Is Mediated through Phosphorylation of Amino Acids Ser770 and Ser771. J Biol Chem. 2007;282:6292–6299. doi: 10.1074/jbc.M611073200. [DOI] [PubMed] [Google Scholar]
- Masereel B, Pochet L, Laeckmann D. An overview of inhibitors of Na(+)/H(+) exchanger. Eur J Med Chem. 2003;38:547–554. doi: 10.1016/s0223-5234(03)00100-4. [DOI] [PubMed] [Google Scholar]
- Meima ME, Webb BA, Witkowska HE, Barber DL. The sodium-hydrogen exchanger NHE1 is an Akt substrate necessary for actin filament reorganization by growth factors. J Biol Chem. 2009;284:26666–26675. doi: 10.1074/jbc.M109.019448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci U S A. 2001;98:11569–11574. doi: 10.1073/pnas.181213498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards SA, Dreisbach VC, Murphy LO, Blenis J. Characterization of regulatory events associated with membrane targeting of p90 ribosomal S6 kinase 1. Mol Cell Biol. 2001;21:7470–7480. doi: 10.1128/MCB.21.21.7470-7480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satriotomo I, Bowen KK, Vemuganti R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J Neurochem. 2006;98:1353–1368. doi: 10.1111/j.1471-4159.2006.04051.x. [DOI] [PubMed] [Google Scholar]
- Shackelford DA, Yeh RY. Activation of extracellular signal-regulated kinases (ERK) during reperfusion of ischemic spinal cord. Brain Res Mol Brain Res. 2003;115:173–186. doi: 10.1016/s0169-328x(03)00206-7. [DOI] [PubMed] [Google Scholar]
- Shichinohe H, Kuroda S, Yasuda H, Ishikawa T, Iwai M, Horiuchi M, Iwasaki Y. Neuroprotective effects of the free radical scavenger Edaravone (MCI-186) in mice permanent focal brain ischemia. Brain Res. 2004;1029:200–206. doi: 10.1016/j.brainres.2004.09.055. [DOI] [PubMed] [Google Scholar]
- Shrode LD, Klein JD, Douglas PB, O’Neill WC, Putnam RW. Shrinkage-induced activation of Na+/H+ exchange: role of cell density and myosin light chain phosphorylation. Am J Physiol. 1997;272:C1968–C1979. doi: 10.1152/ajpcell.1997.272.6.C1968. [DOI] [PubMed] [Google Scholar]
- Silverstein AM, Barrow CA, Davis AJ, Mumby MC. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc Natl Acad Sci U S A. 2002;99:4221–4226. doi: 10.1073/pnas.072071699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepkov ER, Rainey JK, Sykes BD, Fliegel L. Structural and functional analysis of the Na+/H+ exchanger. Biochem J. 2007;401:623–633. doi: 10.1042/BJ20061062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snabaitis AK, D’Mello R, Dashnyam S, Avkiran M. A novel role for protein phosphatase 2A in receptor-mediated regulation of the cardiac sarcolemmal Na+/H+ exchanger NHE1. J Biol Chem. 2006;281:20252–20262. doi: 10.1074/jbc.M600268200. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Takahashi E, Abe J, Gallis B, Aebersold R, Spring DJ, Krebs EG, Berk BC. p90(RSK) is a serum-stimulated Na+/H+ exchanger isoform-1 kinase. Regulatory phosphorylation of serine 703 of Na+/H+ exchanger isoform-1. J Biol Chem. 1999;274:20206–20214. doi: 10.1074/jbc.274.29.20206. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Rumbaugh GR, Harrar DB, Huganir RL. Ribosomal S6 kinase 2 interacts with and phosphorylates PDZ domain-containing proteins and regulates AMPA receptor transmission. Proc Natl Acad Sci U S A. 2005;102:15006–15011. doi: 10.1073/pnas.0507476102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vik TA, Ryder JW. Identification of serine 380 as the major site of autophosphorylation of Xenopus pp90rsk. Biochem Biophys Res Commun. 1997;235:398–402. doi: 10.1006/bbrc.1997.6794. [DOI] [PubMed] [Google Scholar]
- Wakabayashi S, Pang T, Su X, Shigekawa M. A novel topology model of the human Na(+)/H(+) exchanger isoform 1. J Biol Chem. 2000a;275:7942–7949. doi: 10.1074/jbc.275.11.7942. [DOI] [PubMed] [Google Scholar]
- Wakabayashi S, Pang T, Su X, Shigekawa M. Second mutations rescue point mutant of the Na(+)/H(+) exchanger NHE1 showing defective surface expression. FEBS Lett. 2000b;487:257–261. doi: 10.1016/s0014-5793(00)02348-6. [DOI] [PubMed] [Google Scholar]
- Wang D, Balkovetz DF, Warnock DG. Mutational analysis of transmembrane histidines in the amiloride-sensitive Na+/H+ exchanger. Am J Physiol. 1995;269:C392–C402. doi: 10.1152/ajpcell.1995.269.2.C392. [DOI] [PubMed] [Google Scholar]
- Wang H, Silva NL, Lucchesi PA, Haworth R, Wang K, Michalak M, Pelech S, Fliegel L. Phosphorylation and regulation of the Na+/H+ exchanger through mitogen-activated protein kinase. Biochemistry. 1997;36:9151–9158. doi: 10.1021/bi970802f. [DOI] [PubMed] [Google Scholar]
- Wang Y, Luo J, Chen X, Chen H, Cramer SW, Sun D. Gene inactivation of Na+/H+ exchanger isoform 1 attenuates apoptosis and mitochondrial damage following transient focal cerebral ischemia. Eur J Neurosci. 2008;28:51–61. doi: 10.1111/j.1460-9568.2008.06304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Meyer JW, Ashraf M, Shull GE. Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ Res. 2003;93:776–782. doi: 10.1161/01.RES.0000094746.24774.DC. [DOI] [PubMed] [Google Scholar]
- Watson K, Fan GH. Macrophage inflammatory protein 2 inhibits beta-amyloid peptide (1–42)-mediated hippocampal neuronal apoptosis through activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase signaling pathways. Mol Pharmacol. 2005;67:757–765. doi: 10.1124/mol.104.004812. [DOI] [PubMed] [Google Scholar]
- Yao H, Gu XQ, Douglas RM, Haddad GG. Role of Na+/H+ exchanger during O2 deprivation in mouse CA1 neurons. Am J Physiol Cell Physiol. 2001;281:C1205–C1210. doi: 10.1152/ajpcell.2001.281.4.C1205. [DOI] [PubMed] [Google Scholar]
- Zeniou-Meyer M, Liu Y, Begle A, Olanich ME, Hanauer A, Becherer U, Rettig J, Bader MF, Vitale N. The Coffin-Lowry syndrome-associated protein RSK2 is implicated in calcium-regulated exocytosis through the regulation of PLD1. Proc Natl Acad Sci U S A. 2008;105:8434–8439. doi: 10.1073/pnas.0710676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.