

Abstract
An isoreticular metal-organic framework (IRMOF-3) containing 2-amino-1,4-benzenedicarboxylic acid (NH2–BDC) as a building block is shown to undergo chemical modification with a diverse series of anhydrides and isocyanates. The modification of IRMOF-3 by these reagents has been evidenced by using a variety of methods, including NMR and electrospray ionization mass spectrometry, and the structural integrity of the modified MOFs has been confirmed by thermogravimetric analysis, powder X-ray diffraction, and gas sorption analysis. The results show that a variety of functional groups can be introduced onto the MOF including amines, carboxylic acids, and chiral groups. Furthermore, it is shown that tert-butyl-based asymmetric anhydrides can be used to selectively deliver chemical payloads to the IRMOF. Finally, the results demonstrate that at least four different chemical modifications can be performed on IRMOF-3 and that the reaction conditions can be modulated to control the relative abundance of each group. The findings presented here demonstrate several important features of postsynthetic modification on IRMOF-3, including (1) facile introduction of a wide range of functional groups using simple reagents (e.g., anhydrides and isocyanates), (2) the introduction of multiple (as many as four different) substituents into the MOF lattice, and (3) control over reaction conditions to preserve the crystallinity and microporosity of the resultant MOFs. The findings clearly illustrate that postsynthetic modification represents a powerful means to access new MOF compounds with unprecedented chemical complexity, which may serve as the basis of multifunctional materials.
Introduction
Metal-organic frameworks (MOFs) are crystalline materials composed of metal ions or metal cluster nodes connected by organic ligands.1–17 A multitude of structures and topologies are viable through the judicial use of metals and ligands.11,18,19 Throughout the past several years, MOFs have been increasingly investigated for gas adsorption as a result of their thermal stability and inherent porosity.20–31 Moreover, MOFs have shown promise in a diverse set of applications including catalysis,32–36 sensors,37 separation,38 and drug delivery.39–41 Significant efforts have been spent on developing MOFs for novel applications through the use of functionalized ligands.42,43 However, the use of functional linkers is limited by the conventional solvothermal synthesis of MOFs. The incorporation of functional groups on the linking ligands can introduce steric, solubility, and metal-coordinating characteristics that can interfere with MOF formation. Our group44–48 and others49–58 have recently demonstrated the practical use of postsynthetic modification to produce functionalized systems by targeting the organic linking group of prefabricated MOF crystals. More specifically, our efforts have focused on the heterogeneous acylation of the pendant amino group of 2-amino-1,4-benzenedicarboxylate (NH2–BDC) inside a variety of MOFs.48 The generality of post-synthetic covalent modification provides an opportunity to develop MOFs with a variety of functionalities. Herein, we demonstrate a number of new postsynthetic approaches that allow for the incorporation of more complex functional entities into the MOF structure. Among the new findings described here are (1) the introduction of amine derivatives by use of N-Boc (Boc=tert-butyloxycarbonyl) aminoanhydrides, (2) the generation of free carboxylic groups using cyclic anhydrides, (3) the generation of chiral MOFs by modification with chiral anhydrides, (4) the selective transformation of a MOF with t-butyl-containing asymmetric anhydrides, and (5) functionalization of a single MOF lattice with multiple reagents. Most of the previous studies on MOF postsynthetic modification have shown the introduction of only a single chemical modification into the MOF lattice. Very few have demonstrated that more than one chemical reagent could be used to covalently modify a single MOF structure.45,58 In one example, two different anhydrides were used to successfully modify isoreticular metal-organic frame-work-3 (IRMOF-3, Chart 1). To further understand the ability of IRMOF-3 to be modified with multiple reagents and create multifunctional materials, postsynthetic modification was explored using as many as four reagents, including a mixture of anhydrides and isocyanates.
Chart 1.
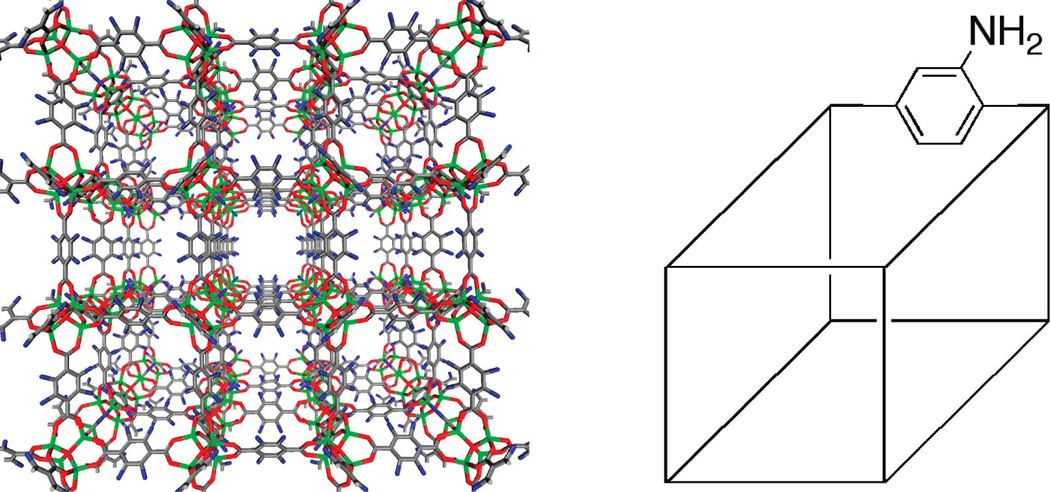
Structure of IRMOF-3 (Left) Showing the Disordered Amino Group (Blue) in All Four Possible Positions on the Organic Linkera
aThe pore diameter of IRMOF-3 is ~9.7Å (ref 59). Schematic representationof IRMOF-3 (right) that is used throughout this manuscript toillustrate postsynthetic modification reactions is also shown.
Experimental Methods
General
Starting materials and solvents were purchased and used without further purification from commercial suppliers (Sigma-Aldrich, Alfa Aesar, EMD, TCI, Cambridge Isotope Laboratories, Inc., and others). IRMOF-3 was synthesized and activated as described previously.48,59 Boc-β-alanine anhydride was synthesized as described previously60.
Synthesis. Butyric Pivalic Anhydride
Butyric acid (0.92 mL, 10 mmol) was added to a solution of THF (20 mL) containing triethylamine (1.39 mL, 10 mmol) and placed in an ice bath. Trimethylacetyl chloride (1.23 mL, 10 mmol) was added to the mixture and stirred for 30 min at 0 °C The reaction was allowed to reach room temperature and stirred for an additional 30 min. The white triethylamine chloride salt was filtered, and the solvent was removed under a vacuum producing an oil. Yield = 85%. 1H NMR (500 MHz, CDCl3): δ 2.4 (t, 2 H), 1.7 (q, 2H), 1.2 (s, 9H), 0.9 (t, 3H). ESI-MS: m/z 189.98 [M+NH4]+, 195.01 [M+Na]+.
Boc-Aminovaleric Acid
5-Aminovaleric acid (1.31 g, 11.2 mmol) was dissolved in 2:1 dioxane/H2O (30 mL). A total of 10 mL of a 1.0 M NaOH solution was added, and the mixture was placed in an ice bath. Di-tert-butyl dicarbonate (2.61 g, 12.3 mmol) was added to the mixture at 0 °C The solution was allowed to reach room temperature and was stirred for ~18 h. The solvent was removed under a rotary vacuum and redis-solved with 10 mL of EtOAc. A total of 10 mL of a 5% citric acid solution was added, and the organic layer was collected and then washed with brine. The organic layer was dried over MgSO4, filtered, and dried under a vacuum producing a colorless oil. Approximately 20 mL of pentane was added, precipitating a white solid. The mixture was filtered, producing a white solid. Yield = 27%. 1H NMR (500 MHz, CDCl3): δ 4.6 (s, br, 1H), 3.1 (d, 2H), 2.3 (t, 2H), 1.6 (q, 2H), 1.5 (q, 2H), 1.4 (s, 9H). ESI-MS: m/z 216.0 [M–H]−.
Boc-Aminovaleric Anhydride
Boc-aminovaleric acid (0.65 g, 3 mmol) was dissolved with 3 mL of anhydrous CH2Cl2 and placed in an ice bath. A solution of DCC (0.3 g, 1.4 mmol) in 2 mL of anhydrous CH2Cl2 was added. The mixture was stirred for 15 min at 0 °C The solution was allowed to warm to room temperature and was stirred for 2 h. The mixture was filtered and dried under a vacuum. The colorless oil was dissolved in EtOAc to precipitate any remaining DCC and was filtered. The solvent was removed by rotary evaporation producing an oil. Yield=49%. 1H NMR (500 MHz, CDCl3): δ 4.6 (s, br, 1H), 3.1 (d, 2H), 2.5 (t, 2H), 1.67 (q, 2H) 1.54 (q, 2H), 1.4 (s, 9H). ESI-MS: m/z 416.91 [M+H]+.
Modification of MOFs. IRMOF-3-AM βAla
Approximately 60 mg of IRMOF-3 (ca. 0.20 mmol equiv of –NH2) was placed in a vial with 2 equiv (0.4 mmol) of Boc-protected β-alanine anhydride dissolved in 2 mL of CHCl3. After the sample was allowed to stand for 2 days, the solution was decanted, and the crystals were washed with 3×6 mL of CHCl3 then soaked in 10 mL of pure CHCl3 (i.e., without anhydride) for three days, with fresh CHCl3 added every 24 h. After three days of soaking, the crystals were stored in the last CHCl3 solution until being analyzed.
IRMOF-3-AMAval
Approximately 60 mg of IRMOF-3 (ca. 0.20 mmol equiv of -NH2) was placed in a vial with 2 equiv (0.4 mmol) of Boc-aminovaleric anhydride dissolved in 4 mL of CHCl3. After the sample was allowed to stand for 2 days, the solution was decanted, and the crystals were washed with 3×6 mL of CHCl3 then soaked in 10 mL of pure CHCl3 for three days, with fresh CHCl3 added every 24 h. After three days of soaking, the crystals were stored in the last CHCl3 solution until being analyzed.
IRMOF-3-(S)-AM3Me
Approximately 60 mg of IRMOF-3 (ca. 0.20 mmol equiv of –NH2) was placed in a vial with 2 equiv (0.4 mmol) of (S)-(+)-2-methylbutyric anhydride dissolved in 2 mL of CHCl3. After the sample was allowed to stand for 5 days, the solution was decanted, and the crystals were washed with 3×6 mL of CHCl3 then soaked in 10 mL of pure CHCl3 for three days, with fresh CHCl3 added every 24 h. After three days of soaking, the crystals were stored in the last CHCl3 solution until being analyzed.
IRMOF-3-AMMal, IRMOF-3-AMSuc, IRMOF-3-(S)-AM-SucAcO
Approximately 60 mg of IRMOF-3 (ca. 0.20 mmol equiv of –NH2) was placed in a vial with 0.5 equiv (0.1 mmol) of the cyclic anhydrides (maleic, succinic, and (S)-(−)-2-acetoxy-succinic anhydride) dissolved in 5 mL of CHCl3. After the sample was allowed to stand for 1 day, the solution was decanted, and the crystals were washed with 3×6 mL of CHCl3 then soaked in 10 mL of pure CHCl3 for three days, with fresh CHCl3 added every 24 h. After three days of soaking, the crystals were stored in the last CHCl3 solution until being analyzed.
IRMOF-3-AM3
Approximately 60 mg of IRMOF-3 (ca. 0.20 mmol equiv of –NH2) was placed in a vial with 2 equiv (0.4 mmol) of butyric pivalic anhydride dissolved in 8 mL of CHCl3. After the sample was allowed to stand for 3 days, the solution was decanted, and the crystals were washed with 3sx 6 mL of CHCl3 then soaked in 10 mL of pure CHCl3 for three days, with fresh CHCl3 added every 24 h. After three days of soaking, the crystals were stored in the last CHCl3 solution until being analyzed.
IRMOF-3-AM9/URPh
Approximately 60 mg of IRMOF-3 (ca. 0.20 mmol equiv of –NH2) was placed in a vial with 2 equiv (0.4 mmol) of decanoic anhydride dissolved in 8 mL of CHCl3. After the sample was allowed to stand for 1 day, the solution was decanted, and the crystals were washed with 3×6 mL of CHCl3. The crystals were then treated with 8 equiv (1.6 mmol) of phenyl isocyanate dissolved in 2 mL of a 10% MeOH/CHCl3 solution. After the samples were allowed to stand for 1 day, the solution was decanted, and the crystals were washed with 3×6 mL of CHCl3. Samples not undergoing subsequent modification were then soaked in 10 mL of pure CHCl3 for three days, with fresh CHCl3 added every 24 h.
IRMOF-3-AM9/URPh/URAl
Allyl isocyanate (1.6 mmol) was added to approximately 60 mg of IRMOF-3-AM9/URPh in 2 mL of CHCl3. After the sample was allowed to stand for 1 day, the solution was decanted, and the crystals were washed with 3 × 6 mL of CHCl3. Samples not undergoing subsequent modification were then soaked in 10 mL of pure CHCl3 for three days, with fresh CHCl3 added every 24 h.
IRMOF-3-AM9/URPh/URAl/AMCrot
Crotonic anhydride (0.4 mmol) was added to approximately 60 mg of IRMOF-3-AM9/URPh/URAl in 4 mL of CHCl3. After the sample was allowed to stand for 3 days, the crystals were washed with 3 × 6 mL of CHCl3 then soaked in 10 mL of pure CHCl3 for three days, with fresh CHCl3 added every 24 h.
IRMOF-3-AM9/UR3
Approximately 60 mg of IRMOF-3 (ca. 0.20 mmol equiv of –NH2) was placed in a vial with 2 equiv (0.4 mmol) of decanoic anhydride dissolved in 8 mL of CHCl3. After the samples were allowed to stand for 1 day, the solution was decanted, and the crystals were washed with 3×6 mL of CHCl3. The crystals were then treated with 8 equiv (1.6 mmol) of propyl isocyanate dissolved in 2 mL of CHCl3 solution. After the samples were allowed to stand for 1 day, the solution was decanted, and the crystals were washed with 3×6 mL of CHCl3. Samples not undergoing subsequent modification were then soaked in 10 mL of pure CHCl3 for three days, with fresh CHCl3 added every 24 h.
IRMOF-3-AM9/UR3/URAl
Allyl isocyanate (1.6 mmol) was added to approximately 60 mg of IRMOF-3-AM9/UR3 in 2 mL of CHCl3. After the sample was allowed to stand for 1 day, the solution was decanted, and the crystals were washed with 3 × 6 mL of CHCl3. Samples not undergoing subsequent modification were then soaked in 10 mL of pure CHCl3 for three days, with fresh CHCl3 added every 24 h.
IRMOF-3-AM9/UR3/URAl/AMCrot-a
Crotonic anhydride (0.4 mmol) was added to ~60 mg of IRMOF-3-AM9/UR3/URAl in 4 mL of CHCl3. After the sample was allowed to stand for 3 days, the crystals were washed with 3× 6 mL of CHCl3 then soaked in 10 mL of pure CHCl3 for three days, with fresh CHCl3 added every 24 h.
IRMOF-3-AM9/UR3/URAl/AMCrot-b
Approximately 60 mg of IRMOF-3 (ca. 0.20 mmol equiv of –NH2) was placed in a vial with 2 equiv (0.4 mmol) of decanoic anhydride dissolved in 8 mL of CHCl3. After the samples were allowed to stand for 1 day, the solution was decanted, and the crystals were washed with 3×6 mL of CHCl3. The crystals were then treated with 8 equiv (1.6 mmol) of propyl isocyanate dissolved in 2 mL of a CHCl3 solution. After the samples were allowed to stand for 3 h, the solution was decanted, and the crystals were washed with 3×6 mL of CHCl3. The crystals were then treated with 8 equiv (1.6 mmol) of allyl isocyanate dissolved in 2 mL of CHCl3. After the samples were allowed to stand for 3 h, the solution was decanted, and the crystals were washed with 3×6 mL of CHCl3. Subsequently, the samples were treated with 2 equiv (0.4 mmol) of crotonic anhydride dissolved in 4mLofCHCl3. After 1 day, the crystals were washed with 3×6mL of CHCl3 then soaked in 10 mL of pure CHCl3 for three days, with fresh CHCl3 added every 24 h.
Anaylsis of MOFs. Digestion and Analysis by 1H NMR
1H NMR spectra were recorded on a JEOL ECA spectrometer (500 MHz). Approximately 5 mg of modified IRMOF-3 samples was dried under vacuum at room temperature or at 90 °C overnight and digested with sonication in 500 µL of DMSO-d6 and 100 µL of dilute DCl (23 µL of 35% DCl in D2O diluted with 1.0 mL of DMSO-d6).
Digestion and Analysis by Electrospray Ionization Mass Spectrometry
Electrospray ionization mass spectrometry (ESI-MS) was performed using a ThermoFinnigan LCQ-DECA mass spectrometer, and the data were analyzed using the Xcalibur software suite. Crystals of modified IRMOF-3 (~0.1–1 mg) were digested in 1 mL of MeOH (or H2O) with sonication.
Digestion and Analysis by Liquid Chromatography Ultraviolet Spectroscopy/Mass Spectrometry
Liquid chromatography ultraviolet spectroscopy/mass spectrometry (LC-UV/MS) was performed using a ThermoFinnigan LCQ-deca mass spectrometer with an electrospray ionization source and was operated under the negative ion mode. The data were analyzed using the Xcalibur software suite. Crystals of modified IRMOF-3 (~5 mg) were digested in 1.5 mL of 10% MeOH/H2O with sonication and filtered. A total of 10 µL of this solution was injected into an Agilent Eclipse XBD-C18 column (5 µm, 4.6 × 150 mm). The solvent system consisted of MeOH in H2O (5% at 0 min rising to 90% at 20 min then 5% after 26 min) with a with a flow rate of 1.0 mL/min. The chromatogram was monitored at λ = 254 nm over 30 min.
Thermal Gravimetric Analysis
Samples were analyzed under a stream of dinitrogen using a TA Instrument Q600 SDT running from room temperature to 600 °C with a scan rate of 5 °C/min. Approximately 10–20 mg of modified IRMOF-3 samples was used for thermogravimetric analysis (TGA) measurements.
Powder X-Ray Diffraction Analysis
Powder X-ray diffraction (PXRD) data were collected at ambient temperature on a Bruker Advance D8 diffractometer at 40 kV and 40 mA for Kα(λ= 1.5418 Å) with a scan speed of 0.3 sec/step, a step size of 0.02° in 2θ, and a 2θ range of 3–40°. Approximately 15mg of modified IRMOF-3 samples (typically soaked in CHCl3) were air-dried before PXRD analysis. The experimental backgrounds were corrected using the Jade 5.0 software package.
Brunauer–Emmett–Teller Surface Area Analysis
Brunauer–Emmett–Teller (BET) surface area (m2/g) measurements were collected at 77 K using dinitrogen on an ASAP 2020 using volumetric techniques. Approximately 40–60 mg of modified IRMOF-3 samples were evacuated on a vacuum line for 5–18 h. The sample was then transferred to a preweighed sample tube and degassed at 105 °C for approximately 24 h or until the outgas rate was <5 µmHg/min. The sample tube was reweighed to obtain a consistent mass for the degassed modified IRMOF-3.
Results and Discussion
Modification of MOFs with Functionalized Anhydrides
In previous studies, IRMOF-3 was shown to undergo modification with linear alkyl chain anhydrides44,46 and isocyanates47 to produce amide and urea functionalized materials. In this present study, seven diverse anhydrides were examined for their ability to modify IRMOF-3 and generate new functionalized MOF systems. The anhydrides studied included (Scheme 1) two Boc-protected (Boc = t-butyloxycarbonyl) aminoanhydrides (β-alanine anhydride and aminovaleric anhydride), a chiral alkyl anhydride (S)-(+)-2-methylbutyric anhydride), three cyclic anhydrides (maleic anhydride, succinic anhydride, and (S)-(−)-2-acetoxysuccinic anhydride), and an asymmetric anhydride (butyric pivalic anhydride). The modifications were performed under various reaction conditions, given the diverse properties of the reagents (e.g., solubility in CHCl3, reactivity), in order to achieve high conversions and maintain good crystal quality with each reagent. Initial characterization of modified IRMOF-3 samples was performed by digestion of the MOF (in acidic media), followed by ESI-MS and 1H NMR analysis (Figures S1–S12, S14, S15, Supporting Information). 1H NMR analysis was further used to determine the degree of conversion. Conversion (Table 1) was determined by using peak integration by comparing the relative areas of the singlet aromatic resonance (corresponding to the C-3 position) of the modified or unmodified dicarboxylate ligands. The bulk crystallinity of each modified IRMOF-3 was evaluated by PXRD—modified IRMOF-3 with high crystallinity has been shown to display powder patterns essentially identical to unmodified IRMOF-3 (Figure 1). Finally, the thermal stability of modified samples was evaluated by TGA, which was compared to that of IRMOF-3 (Td ~430 °C; Figure S16, Supporting Information).
Scheme 1.
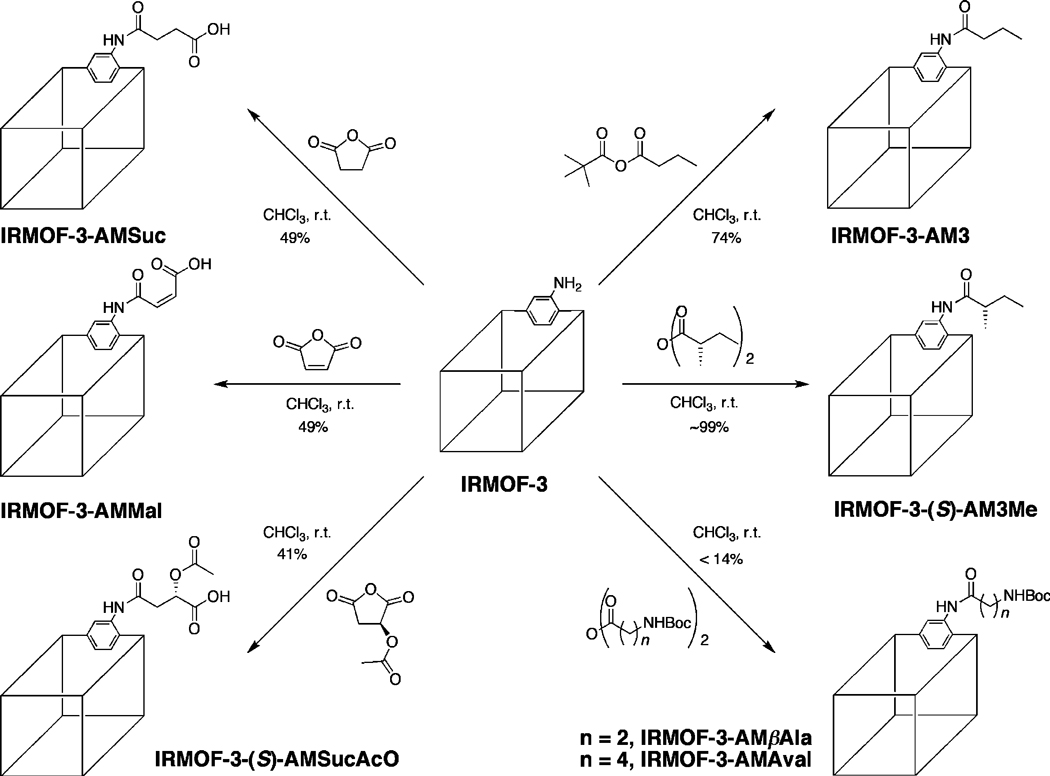
Postsynthetic Modification Reactions Performed with IRMOF-3
Table 1.
Percent Conversions of Postsynthetic Modification Reactions with IRMOF-3 and Different Anhydrides As Determined by 1H NMRa
| IRMOF-3- | reagent | reaction conditions | % conversion |
|---|---|---|---|
| AMβAla | Boc-β-alanine anhydride | 2 equiv anhydride, 2 mL CHCl3, 2 days | 7±1 |
| AMAval | Boc-aminovaleric anhydride | 2 equiv anhydride, 4 mL CHCl3, 2 days | 14±8 |
| (S)-AM3Me | (S)-(+)-2-methylbutyric anhydride | 2 equiv anhydride, 2 mL CHCl3, 5 days | 99±1 |
| AMMal | maleic anhydride | 0.5 equiv anhydride, 2 mL CHCl3, 1 day | 49±1 |
| AMSuc | succinic anhydride | 0.5 equiv anhydride, 2 mL CHCl3, 1 day | 49±1 |
| (S)-AMSucAcO | (S)-(−)-2-acetoxysuccinic anhydride | 0.5 equiv anhydride, 2 mL CHCl3, 1 day | 41±3 |
| AM3 | butyric pivalic anhydride | 2 equiv anhydride, 8 mL CHCl3, 3 days | 74±2 |
Values listed are an average (with standard deviations) of at least three independent experiments.
Figure 1.
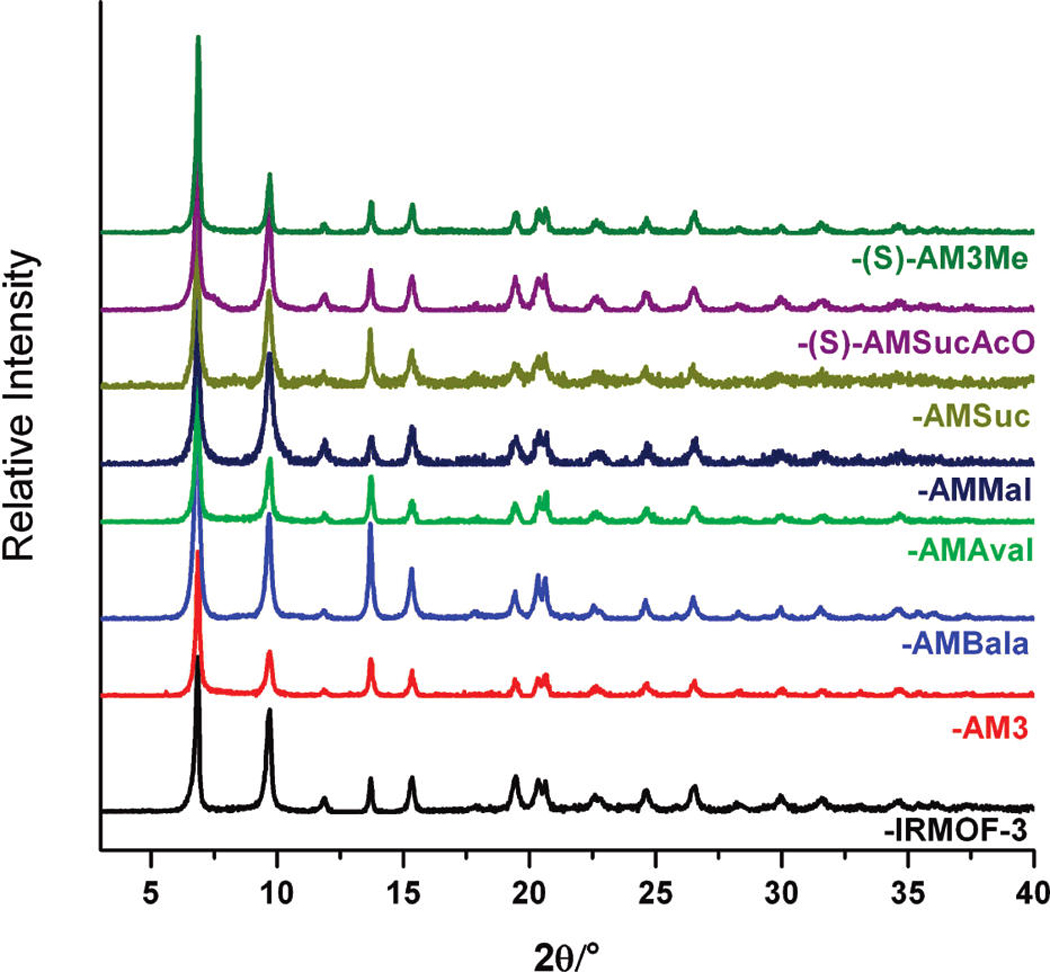
PXRD patterns of anhydride modified IRMOF-3 samples. Modified IRMOF-3 samples were soaked and exchanged with fresh CHCl3 for 3 days. All modified samples show a PXRD pattern similar to that of the starting material (IRMOF-3, bottom).
Conversion was found to be dependent on the nature of the reagent applied. Boc-protected aliphatic aminoanhydrides, β-alanine anhydride and aminovaleric anhydride, were found to have low reactivity (<15%) with IRMOF-3. The low conversion is attributed to the bulky Boc protecting groups. Under similar conditions, linear alkyl anhydrides with analogous chain lengths were found to undergo >50% conversion.46 The introduction of IR-MOF-3-AMβAla and IRMOF-3AMAval potentially provides access to aliphatic amine functionality. In contrast to the Boc-protected aminoanhydrides, (S)-(+)-2-methylbutyric anhydride displayed nearly quantitative conversion to give IRMOF-3-(S)-AM3Me, similar to small linear alkyl anhydrides that have been previously studied.44,46 While there have been several reports utilizing chiral ligands in the construction of chiral MOFs,61 this latter finding deserves particular note, as it is the first time an achiral MOF has been converted into a chiral MOF material via a covalent postsynthetic modification approach.
The cyclic anhydrides, maleic anhydride, succinic anhydride, and (S)-(−)-2-acetoxysuccinic anhydride were found to react smoothly with IRMOF-3 (using 0.5 equiv gave ~50% conversion). 1H NMR and ESI-MS analysis indicated that, upon modification, these anhydrides formed open-chain products (as opposed to cyclic imides), generating pendant carboxylic acid groups (Figures S7–S12, Supporting Information). This is consistent with postsynthetic modification studies using maleic and succinic anhydride reported by Fujita et al. that were structurally verified by single-crystal X-ray diffraction.62 1H NMR indicates that IRMOF-3 modified with (S)-(−)-2-acetoxysuccinic anhydride produces two distinct isomeric modified groups with 41% and 6% conversion. The asymmetry of acetoxysuccinic anhydride can allow ring opening in two directions, and literature studies suggest that the predominant species is the amide with the acetoxy group adjacent to the carboxylic acid (Figure S11, Supporting Information).63 The carboxylic acid functionalized MOFs are structurally stable, retain microporosity, and remain thermally stable (Figure 1, Table S1 and Table S16, Supporting Information). It was found that IRMOF-3 could be >85% converted under certain reaction conditions (2 equiv of anhydride, 5 days, 25 °C) with these cyclic anhydrides; however, these highly modified samples showed a loss of crystal quality (Figure S13, Supporting Information). This was confirmed by BET surface area measurements, which gave extremely low surface areas (e.g., 29 m2/g for IRMOF-3-AMMal at 92% conversion; Table S1, Supporting Information). Given that these modified MOFs contain large numbers of free carboxylate groups, it is proposed that the acidic nature and coordinating ability of these carboxylate groups disrupt the framework, resulting in a phase change and collapse. In view of the difficulty associated with incorporating free carboxylic acids into MOFs through conventional solvothermal synthesis, the facile preparation of IRMOF-3-AMMal, -AMSuc, and -(S)-AMSucAcO highlights the significance of the postsynthetic modification approach. Furthermore, IRMOF-3-(S)-AMSucAcO represents the introduction of both a chiral substituent and an acidic group in a single postsynthetic modification event.
The development of asymmetric anhydrides for selective covalent modification was explored with butyric pivalic anhydride. Our studies have shown that utilization of anhydrides for postsynthetic modification can be restricted by the production of strongly acidic byproducts that results in degradation of the IRMOF lattice. For example, acylation of IRMOF-3 with trifluoroacetic anhydride produces trifluoroacetic acid, which destroys the framework by catalyzing hydrolysis of the secondary building unit (unpublished results).64 We reasoned that such functional groups could be delivered to IRMOF-3 in the form of an asymmetric anhydride, where reaction of IRMOF-3 would strongly favor delivery of one substituent over another. The lack of reactivity of tert-butyl anhydride and tert-butyl isocyanate with IRMOF-3 in previous studies,47,48 ascribed to the sterics of the tert-butyl group, offered an opportunity to exploit steric effects to selectively incorporate functional groups through asymmetric anhydrides. The utilization of tert-butyl-based asymmetric anhydrides provides a synthetic route toward selective modification that circumvents degradation by generating the benign pivalic acid byproduct. Indeed, as a proof of concept, treatment of IRMOF-3 with the asymmetric anhydride produced the previously described IRMOF-3-AM3. The 1H NMR spectra of the modified sample indicate a conversion of ~74%, which is lower than that of IRMOF-3 treated with butyric anhydride (~99%) under identical reaction conditions.46 The reduced conversion likely correlates with the introduction of the bulky tert-butyl group. Nonetheless, a reasonable conversion to the modified product is obtained and shows that the mixed anhydride can be used to deliver groups to the MOF structure without degradation of the frame-work (Figure 1).
Modification of MOFs with Multiple Reagents
In an earlier study, the tandem postsynthetic modification of IRMOF-3 with two different anhydrides was reported.45 In an effort to expand upon the range of functionality within a MOF, a series of tandem postsynthetic modifications was attempted with various anhydrides and isocyanates (Scheme 2). In an initial experiment, IRMOF-3 was treated sequentially with decanoic anhydride followed by propyl isocyanate. The 1H NMR spectra of the modified MOF (upon digestion), designated IRMOF-3-AM9/UR3, shows two singlet resonances associated with the phenyl (C-3 position) protons of the corresponding amide (AM9) and urea (UR3) dicarboxylate products (Figure 2). The spectra also contain two sets of doublet resonances associated with the phenyl (C-5 and C-6 positions) protons of both the amide- and urea-modified products, clearly demonstrating modification by both reagents. The presence of both modified products was confirmed by ESI-MS analysis from a single digested crystal. Under these reaction conditions, IRMOF-3-AM9/UR3 contains ~21% of both the amide and urea modified products (Table 2), while 58% remains unmodified (i.e., NH2–BDC). Additionally, LC-UV/MS was performed on the modified samples to verify the presence of the three different dicarboxylate components. The LC-UV/MS trace indicates the presence of three species that correspond to the following: unmodified NH2–BDC, the amide modified product (AM9), and the urea modified product (UR3) (Figure 3). Crystallinity and microporosity were maintained, as confirmed by PXRD (Figure 4) and BET gas adsorption measurements (Table S2, Supporting Information). While the material does not show complete thermal degradation until ~430 °C, IRMOF-3-AM9/UR3 does display a measurable weight loss (measured, 14 wt %; calcd, ~6.4%) at ~250 °C analogous to other urea modified systems (Figure S16, Supporting Information).47 To further validate multiple modifications with different reagents and examine how the size of different reagents affects subsequent modification processes in a single MOF lattice, IRMOF-3 was treated with decanoic anhydride and phenyl isocyanate. IRMOF-3-AM9/URPh contained 24% of the alkyl amide and 32% of the phenyl urea ligands, as shown by 1H NMR (Figure S17, Supporting Information); modification was also verified by ESI-MS (Figure S19, Supporting Information). The material showed similar crystallinity (Figure 4), thermal stability (Figure S16, Supporting Information), and microporosity (Table S2, Supporting Information) to that found for IRMOF-3-AM9/UR3. The preparation of IRMOF-3-AM9/UR3 and IRMOF-3-AM9/URPh unequivocally demonstrates, for the first time, that IRMOF-3 can be modified with both anhydride and isocyanate reagents, generating materials with at least three distinct functional groups within the pores (i.e., amine, amide, and urea).
Scheme 2.
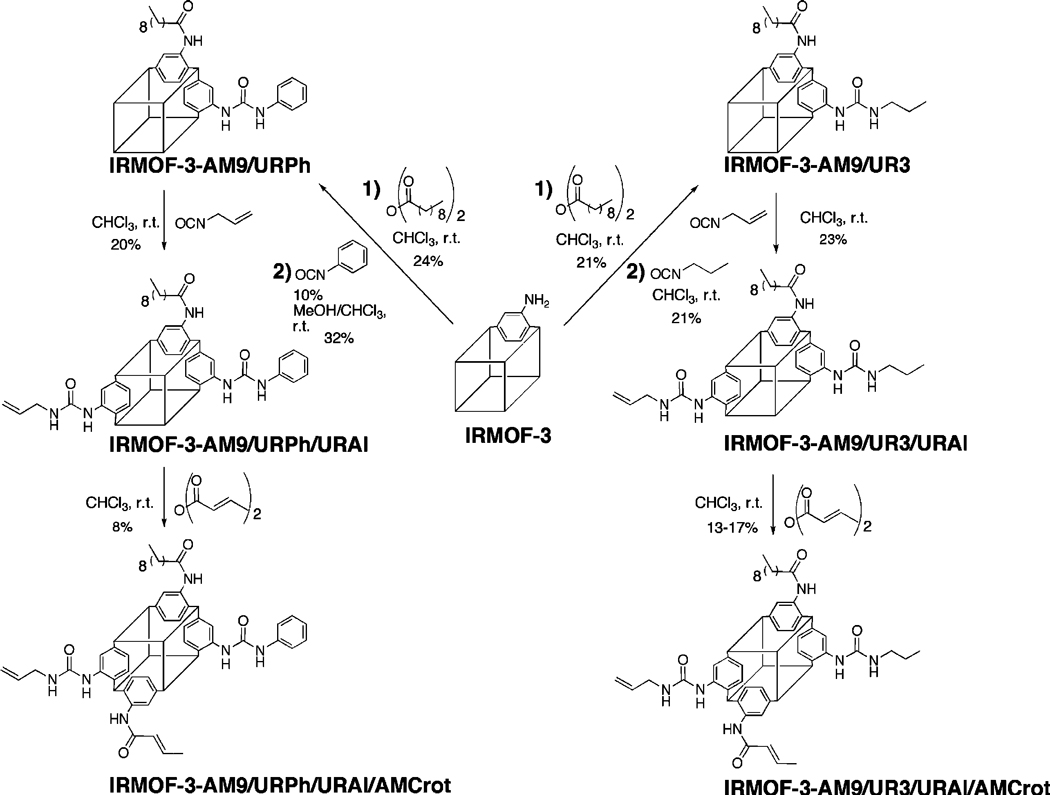
Reagents Used for Multiple Postsynthetic Modification Reactions with IRMOF-3
Figure 2.
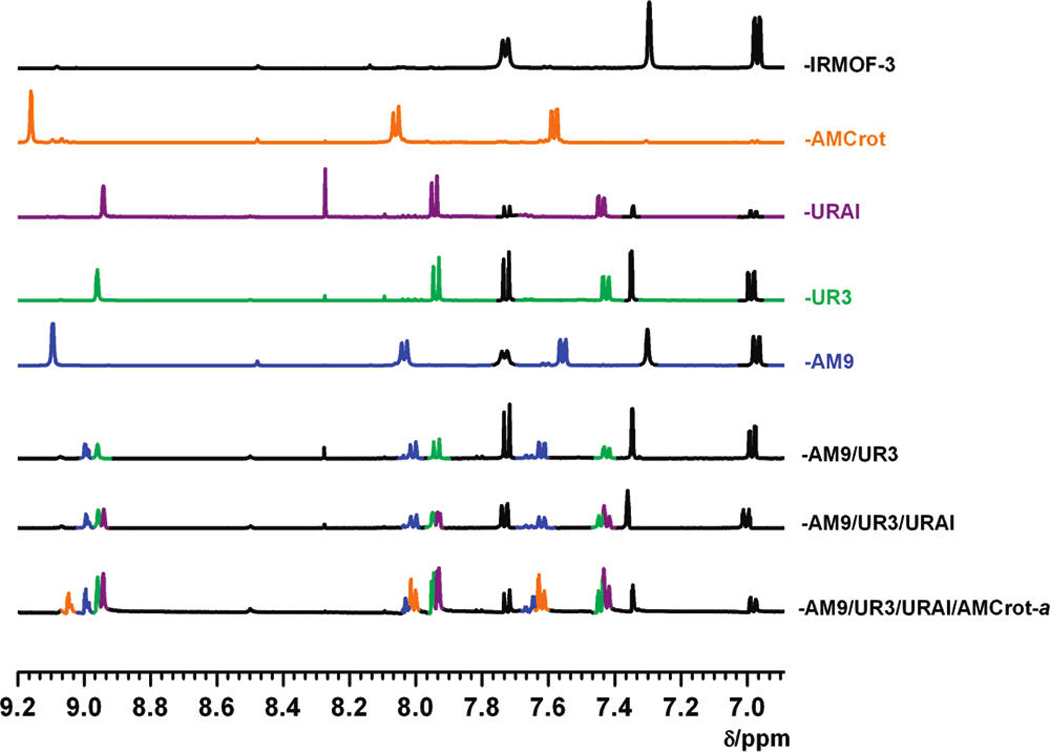
1H NMR spectra of multiple modified IRMOF-3 samples. IRMOF-3 samples modified with decanoic anhydride (blue, 51% conversion), propyl isocyanate (green, 60% conversion), allyl isocyanate (purple, 75% conversion), crotonic anhydride (orange, 100% conversion), and unmodified IRMOF-3 (black) digested in DCl/D2O and DMSO-d6. Resonances in the spectra for IRMOF-3-AM9/UR3, IR-MOF-3-AM9/UR3/URAl, and IRMOF-3-AM9/UR3/URAl/AMCrot-a are color-coded corresponding to the top five spectra. The small differences in the positions of some of these resonances is attributed to variations in solution pH after sample digestion. IRMOF-3 resonances appear black in all spectra shown.
Table 2.
Percent Conversions of Multiple Postsynthetic Modification Reactions with IRMOF-3 and Different Anhydrides or Isocyanates As Determined by 1H NMRa
| % conversion |
|||||
|---|---|---|---|---|---|
| IRMOF-3- | reagentsb | AM9 | UR3 or URPh |
URAl | AMCrot |
| AM9/UR3 | (1)DA | 21±1 | 21±3 | ||
| (2) PI | |||||
| AM9/UR3/URAl | (1)DA | 21±4 | 22±1 | 23±1 | |
| (2) PI | |||||
| (3) AI | |||||
| AM9/UR3/URAl/AMCrot-a | (1) DA | 24±4 | 18±6 | 21±3 | 17± 1 |
| (2) PI | |||||
| (3) AI | |||||
| (4) CA | |||||
| AM9/UR3/URAl/AMCrot-bc | (1) DA | 23±1 | 8±2 | 13±2 | 13±1 |
| (2) PI | |||||
| (3) AI | |||||
| (4) CA | |||||
| AM9/URPh | (1)DA | 24±2 | 32±2 | ||
| (2) PhI | |||||
| AM9/URPh/URAl | (1)DA | 26±5 | 28±2 | 20±3 | |
| (2) PhI | |||||
| (3) AI | |||||
| AM9/URPh/URAl/AMCrot | (1) DA | 20±1 | 30±1 | 20±1 | 8±1 |
| (2) PhI | |||||
| (3) AI | |||||
| (4) CA | |||||
Values listed are an average (with standard deviations) of at least three independent experiments.
AI = allyl isocyanate; CA = crotonic anhydride; DA = decanoic anhydride; PI = propyl isocyanate; PhI = phenyl isocyanate.
Propyl and allyl isocyanate reaction times reduced to 3 h each.
Figure 3.
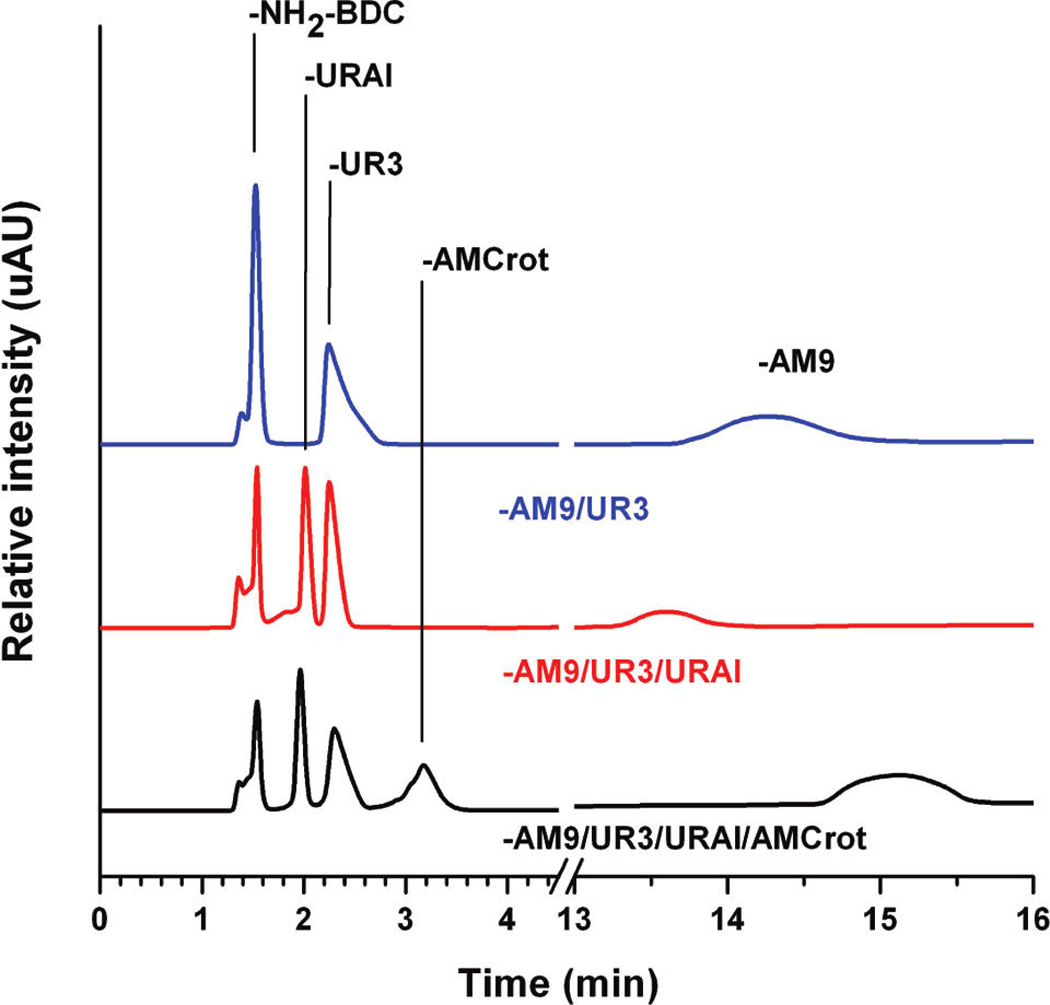
LC-UV/MS traces of IRMOF-3-AM9/UR3 (blue), IRMOF-3-AM9/UR3/URAl (red), and IRMOF-3-AM9/UR3/URAl/AMCrot-a (black). The chromatogram from 13 to 16 min is magnified 10-fold.
Figure 4.
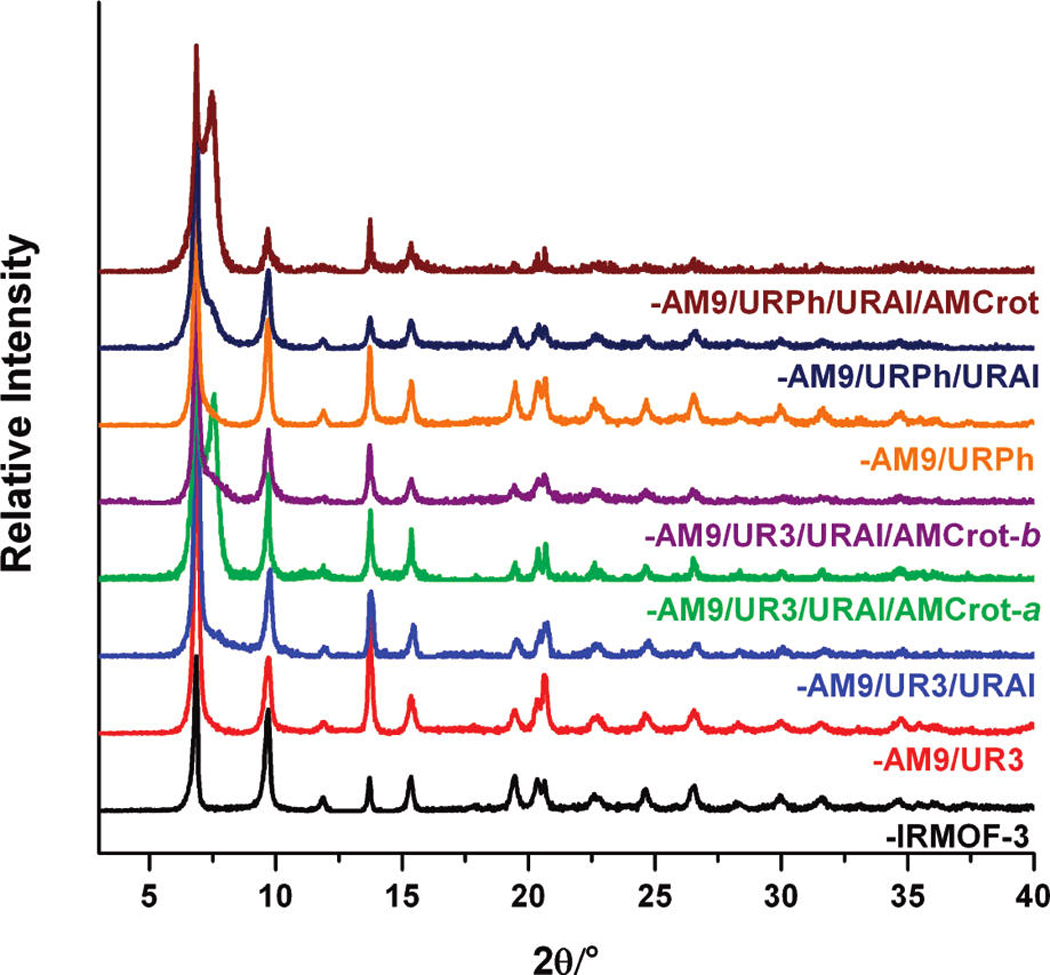
PXRD patterns of multiple modified IRMOF-3 samples. Modified IRMOF-3 samples were soaked and exchanged with fresh CHCl3 for 3 days. After the solvent was decanted off, the samples were left drying in the air for 10 min prior to PXRD analysis.
Having demonstrated that IRMOF-3 could be modified with anhydride and isocyanate reagents, the extent to which sequential postsynthetic modification could be performed by additional reagents was explored on IR-MOF-3-AM9/UR3 and IRMOF-3-AM9/URPh. Each of these MOFs was further treated with allyl isocyanate followed by crotonic anhydride. Using the aforementioned reaction conditions, IRMOF-3-AM9/UR3 was subsequently treated with allyl isocyanate. The 1H NMR spectra of digested IRMOF-3-AM9/UR3/URAl show resonances at 7.4 and 7.9 ppm that display overlap among doublet peaks associated with the phenyl (C5, C6 position) protons of modified urea (UR3, URAl) products (Figure 2). Nonetheless, modification by all three reagents can be established with three distinct singlet resonances associated with the phenyl (C-3 position) protons of the two urea (UR3, URAl) and one amide (AM9) products. IRMOF-3-AM9/UR3/URAl was found to contain 21% of the amide and 22% and 23% of the urea (UR3 and URAl, respectively) products, leaving 34% unmodified amine groups.
The additional urea functionality is also evidenced by TGA, as IRMOF-3-AM9/UR3/URAl demonstrates an increased weight loss (20 wt %) at ~250 °C relative to that of IRMOF-3-AM9/UR3 (14 wt %; Figure S16, Supporting Information). Furthermore, LC-UV/MS and ESI-MS confirm the presence of four individual species corresponding to the amine (NH2–BDC), amide (AM9), and urea (UR3, URAl) functional groups (Figure 3). The multiply modified material displays PXRD peaks consistent with the IRMOF-3 starting material (Figure 4).
The relatively bulky phenyl urea and alkyl amide groups in IRMOF-3-AM9/URPh were found not to inhibit the reactivity of allyl isocyanate in the modification process. Interestingly, unlike IRMOF-3-AM9/UR3/ URAl, the 1H NMR spectrum of digested IRMOF-3-AM9/URPh/URAl contains overlap (8.9 ppm) among the singlet phenyl protons associated with the two urea (URPh, URAl) products (Figure S17, Supporting Information); however, the doublet resonances associated with one of the phenyl (C-5 or C-6 position) protons of their respective urea products (URPh ~7.5 ppm and UR3 ~7.4 ppm) are clearly distinguishable, allowing determination of conversion. As found by 1H NMR, IRMOF-3-AM9/URPh/URAl contains 48% urea species (28% and 20% for URPh and URAl, respectively), 26% amide product (AM9), and 26% unmodified NH2–BDC. ESI-MS (Figure S20, Supporting Information) and LC-UV/ MS (Figure S21, Supporting Information) confirm the presence of four species in the two systems consistent with unmodified NH2–BDC, the amide product (AM9), and the two ureas (URPh, URAl). IRMOF-3-AM9/URPh/ URAl displayed similar PXRD peaks and thermal properties as the IRMOF-3 starting material (Figure 4, Figure S16, Supporting Information). The development of IRMOF-AM9/UR3/URAl and IRMOF-AM9/ URPh/URAl further suggests that multiple reagents can be utilized in tandem postsynthetic modification to generate multifunctional materials without MOF degradation.
Due to its relatively small size and high reactivity45, crotonic anhydride was utilized for a final, additional modification on IRMOF-3-AM9/UR3/URAl and IRMOF-3-AM9/URPh/URAl. The modified functional groups of the preceding systems significantly affected the subsequent modification with crotonic anhydride. IRMOF-3-AM9/UR3/URAl/AMCrot-a was found to contain 24% and 17% amide products (AM9 and AM-Crot, respectively) in addition to 18% and 21% urea products (UR3 and URAl, respectively), leaving ~20% unmodified (NH2–BDC). IRMOF-3-AM9/URPh/ URAl/AMCrot consists of 20% and 8% amide (AM9 and AMCrot, respectively) products along with 30% and 20% urea (URPh and URAl, respectively) products. The low conversion of the crotonic amide product (AMCrot) relative to that of the IRMOF-3-AM9/UR3/URAl/AM-Crot-a system may reflect the reduced ability for crotonic anhydride to diffuse and react with available amine groups, due to the larger phenyl urea groups already present in the material. The presence of five distinct species correlating to unmodified NH2–BDC, the amide products (AM9, AMCrot), and urea products (UR3 or URPh and URAl) was confirmed by LC-UV/MS (Figure 3, Figure S21, Supporting Information) and ESI-MS. Both systems showed a near identical weight loss at ~ 250 °C as that of their precursors and similar overall thermal stability to IRMOF-3 (Figure S16, Supporting Information). However, PXRD clearly indicates some degradation of the framework in both systems on account of the additional modification reactions, with the emergence of a shoulder at 2θ = 7.4° (Figure 4). Degradation may have arisen from the prolonged exposure to ambient moisture from the air or solvents during the sequential reactions and the additional time employed in the preparation of the multiple modified materials.
Given the facile ability to control the heterogeneous nature of postsynthetic modification through reaction time or concentration, some experiments were performed to modulate the relative abundance of the functional groups in a multiply modified MOF. Several tandem modification reactions were attempted with the aforementioned anhydrides (decanoic, crotonic) and isocyanates (propyl, allyl) using shorter reaction times. On the basis of 1H NMR, IRMOF-3-AM9/UR3/ URAl/AMCrot-b was found to contain 23% and 13% amide (AM9 and AMCrot, respectively) products along with 8% and 13% urea (UR3 and URAl, respectively) products, leaving ~53% of the framework unmodified. The reduced time circumvents structural degradation as the multiple modified sample displays PXRD peaks consistent with as-synthesized IRMOF-3 (Figure 4). Microporosity is also maintained, as BET gas adsorption measurements showed reasonable surface areas (~1330 m2/g, Table S2, Supporting Information). The development of IRMOF-3-AM9/UR3/URAl/AMCrot-b confirms that modification of multiple reagents can be tuned with varying reaction conditions. More importantly, it demonstrates the facile synthesis of multifunctional materials through a tandem postsynthetic modification approach.
Conclusions
In summary, the findings presented in this study demonstrate that postsynthetic modification is a versatile method for the functionalization of MOFs. IRMOF-3 has been postsynthetically modified with alkyl and cyclic anhydrides. The anhydrides successfully transformed IRMOF-3 into a diverse set of new IRMOFs functionalized with amine, carboxylate, and chiral groups, which will be explored for applications in enantioselective separation and catalysis. A tert-butyl-based asymmetric anhydride was shown to selectively deliver a “payload” moiety to IRMOF-3. This could potentially be utilized to deliver novel chemical groups that would otherwise generate corrosive byproducts. Through the use of sequential modification, a set of multifunctional MOFs with as many as five different substituents was successfully prepared. The presence of multiple functional groups within the MOFs was confirmed by 1H NMR spectroscopy, ESI-MS, and LC-UV/MS. Given the facile control over reaction time, reagent concentration, and other reaction parameters, tandem postsynthetic modification offers a viable route to functionalized MOFs with unprecedented chemical complexity.
Supplementary Material
Acknowledgment
We thank Dr. Y. Su (U.C.S.D.) for performing the mass spectrometry experiments and Emily Dugan for preliminary studies on IRMOF-3-(S)-AM3Me. This work was supported by U.C.S.D., the NSF (CHE-0546531), and the DOE (DE-FG02-08ER-46519). S.J.G. was supported by a supplement to NCI grant 3R01 CA095298-07S1.
Footnotes
Supporting Information Available: Figures S1–S21 and Tables S1–S2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hoskins BF, Robson R. J. Am. Chem. Soc. 1989;111:5962–5964. [Google Scholar]
- 2.Hoskins BF, Robson R. J. Am. Chem. Soc. 1990;112:1546–1554. [Google Scholar]
- 3.Gardner GB, Venkataraman D, Moore JS, Lee S. Nature. 1995;374:792–795. [Google Scholar]
- 4.Yaghi OM, Li HL, Davis C, Richardson D, Groy TL. Acc. Chem. Res. 1998;31:474–484. [Google Scholar]
- 5.Kitagawa S, Kondo M. Bull. Chem. Soc. Jpn. 1998;71:1739–1753. [Google Scholar]
- 6.Robson R. J. Chem. Soc., Dalton Trans. 2000:3735–3744. [Google Scholar]
- 7.Eddaoudi M, Moler DB, Li H, Chen B, Reineke TM, O’Keeffe M, Yaghi OM. Acc. Chem. Res. 2001;34:319–330. doi: 10.1021/ar000034b. [DOI] [PubMed] [Google Scholar]
- 8.Moulton B, Zaworotko MJ. Chem. Rev. 2001;101:1629–1658. doi: 10.1021/cr9900432. [DOI] [PubMed] [Google Scholar]
- 9.Janiak C. Dalton Trans. 2003:2781–2804. [Google Scholar]
- 10.James SL. Chem. Soc. Rev. 2003;32:276–288. doi: 10.1039/b200393g. [DOI] [PubMed] [Google Scholar]
- 11.Yaghi OM, O’Keeffe M, Ockwig NW, Chae HK, Eddaoudi M, Kim J. Nature. 2003;423:705–714. doi: 10.1038/nature01650. [DOI] [PubMed] [Google Scholar]
- 12.Kitagawa S, Kitaura R, Noro S-i. Angew. Chem., Int. Ed. 2004;43:2334–2375. doi: 10.1002/anie.200300610. [DOI] [PubMed] [Google Scholar]
- 13.Rao CNR, Natarajan S, Vaidhyanathan R. Angew. Chem. Int. Ed. 2004;43:1466–1496. doi: 10.1002/anie.200300588. [DOI] [PubMed] [Google Scholar]
- 14.Ockwig NW, Delgado-Friedrichs O, O’Keeffe M, Yaghi OM. Acc. Chem. Res. 2005;38:176–182. doi: 10.1021/ar020022l. [DOI] [PubMed] [Google Scholar]
- 15.Férey G, Mellot-Draznieks C, Serre C, Millange F. Acc. Chem. Res. 2005;38:217–225. doi: 10.1021/ar040163i. [DOI] [PubMed] [Google Scholar]
- 16.Bradshaw D, Claridge JB, Cussen EJ, Prior TJ, Rosseinsky MJ. Acc. Chem. Res. 2005;38:273–282. doi: 10.1021/ar0401606. [DOI] [PubMed] [Google Scholar]
- 17.Ferey G. Chem. Soc. Rev. 2008;37:191–214. doi: 10.1039/b618320b. [DOI] [PubMed] [Google Scholar]
- 18.Eddaoudi M, Kim J, Vodak D, Sudik A, Wachter J, O’Keeffe M, Yaghi OM. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4900–4904. doi: 10.1073/pnas.082051899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furukawa H, Kim J, Ockwig NW, O’Keeffe M, Yaghi OM. J. Am. Chem. Soc. 2008;130:11650–11661. doi: 10.1021/ja803783c. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Eddaoudi M, O’Keeffe M, Yaghi OM. Nature. 1999;402:276–279. [Google Scholar]
- 21.Rosi NL, Eckert J, Eddaoudi M, Vodak DT, Kim J, O’Keeffe M, Yaghi OM. Science. 2003;300:1127–1129. doi: 10.1126/science.1083440. [DOI] [PubMed] [Google Scholar]
- 22.Férey G, Latroche M, Serre C, Millange F, Loiseau T, Percheron-Guégan A. Chem. Commun. 2003:2976–2977. doi: 10.1039/b308903g. [DOI] [PubMed] [Google Scholar]
- 23.Rowsell JLC, Millward AR, Park KS, Yaghi OM. J. Am. Chem. Soc. 2004;126:5666–5667. doi: 10.1021/ja049408c. [DOI] [PubMed] [Google Scholar]
- 24.Millward AR, Yaghi OM. J. Am. Chem. Soc. 2005;127:17998–17999. doi: 10.1021/ja0570032. [DOI] [PubMed] [Google Scholar]
- 25.Rowsell JLC, Yaghi OM. Angew. Chem., Int. Ed. 2005;44:4670–4679. doi: 10.1002/anie.200462786. [DOI] [PubMed] [Google Scholar]
- 26.Ma S, Sun D, Simmons JM, Collier CD, Yuan D, Zhou H-C. J. Am. Chem. Soc. 2008;130:1012–1016. doi: 10.1021/ja0771639. [DOI] [PubMed] [Google Scholar]
- 27.Banerjee R, Phan A, Wang B, Knobler C, Furukawa H, O’Keeffe M, Yaghi OM. Science. 2008;319:939–943. doi: 10.1126/science.1152516. [DOI] [PubMed] [Google Scholar]
- 28.Britt D, Tranchemontagne D, Yaghi OM. Proc. Natl. Acad. Sci. U.S.A. 2008;105:11623–11627. doi: 10.1073/pnas.0804900105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang B, Cote AP, Furukawa H, O’Keeffe M, Yaghi OM. Nature. 2008;453:207–211. doi: 10.1038/nature06900. [DOI] [PubMed] [Google Scholar]
- 30.Chae HK, Siberio-Pérez DY, Kim J, Go Y, Eddaoudi M, Matzger AJ, O’Keeffe M, Yaghi OM. Nature. 2004;427:523–527. doi: 10.1038/nature02311. [DOI] [PubMed] [Google Scholar]
- 31.Koh K, Wong-Foy AG, Matzger AJ. Angew. Chem., Int. Ed. 2008;47:677–680. doi: 10.1002/anie.200705020. [DOI] [PubMed] [Google Scholar]
- 32.Fujita M, Kwon YJ, Washizu S, Ogura K. J. Am. Chem. Soc. 1994;116:1151–1152. [Google Scholar]
- 33.Seo JS, Whang D, Lee H, Jun SI, Oh J, Jeon YJ, Kim K. Nature. 2000;404:982–986. doi: 10.1038/35010088. [DOI] [PubMed] [Google Scholar]
- 34.Wu C-D, Hu A, Zhang L, Lin W. J. Am. Chem. Soc. 2005;127:8940–8941. doi: 10.1021/ja052431t. [DOI] [PubMed] [Google Scholar]
- 35.Cho S-H, Ma B, Nguyen ST, Hupp JT, Albrecht-Schmitt TE. Chem. Commun. 2006:2563–2565. doi: 10.1039/b600408c. [DOI] [PubMed] [Google Scholar]
- 36.Shultz AM, Farha OK, Hupp JT, Nguyen ST. J. Am. Chem. Soc. 2009;131:4204–4205. doi: 10.1021/ja900203f. [DOI] [PubMed] [Google Scholar]
- 37.Halder GJ, Kepert CJ, Moubaraki B, Murray KS, Cashion JD. Science. 2002;298:1762–1765. doi: 10.1126/science.1075948. [DOI] [PubMed] [Google Scholar]
- 38.Nuzhdin AL, Dybtsev DN, Bryliakov KP, Talsi EP, Fedin VP. J. Am. Chem. Soc. 2007;129:12958–12959. doi: 10.1021/ja076276p. [DOI] [PubMed] [Google Scholar]
- 39.Rieter WJ, Taylor KM, Lin W. J. Am. Chem. Soc. 2007;129:9852–9853. doi: 10.1021/ja073506r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horcajada P, Serre C, Maurin G, Ramsahye NA, Balas F, Vallet-Regi M, Sebban M, Taulelle F, Ferey G. J. Am. Chem. Soc. 2008;130:6774–6780. doi: 10.1021/ja710973k. [DOI] [PubMed] [Google Scholar]
- 41.Rieter WJ, Pott KM, Taylor KM, Lin W. J. Am. Chem. Soc. 2008;130:11584–11585. doi: 10.1021/ja803383k. [DOI] [PubMed] [Google Scholar]
- 42.Kiang Y-H, Gardner GB, Lee S, Xu Z, Lobkovsky EB. J. Am. Chem. Soc. 1999;121:8204–8215. doi: 10.1021/ja0115518. [DOI] [PubMed] [Google Scholar]
- 43.Kitaura R, Onoyama G, Sakamoto H, Matsuda R, Noro S-i, Kitagawa S. Angew. Chem., Int. Ed. 2004;43:2684–2687. doi: 10.1002/anie.200352596. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Cohen SM. J. Am. Chem. Soc. 2007;129:12368–12369. doi: 10.1021/ja074366o. [DOI] [PubMed] [Google Scholar]
- 45.Wang Z, Cohen SM. Angew. Chem., Int. Ed. 2008;47:4699–4702. doi: 10.1002/anie.200800686. [DOI] [PubMed] [Google Scholar]
- 46.Tanabe KK, Wang Z, Cohen SM. J. Am. Chem. Soc. 2008;130:8508–8517. doi: 10.1021/ja801848j. [DOI] [PubMed] [Google Scholar]
- 47.Dugan E, Wang Z, Okamura M, Medina A, Cohen SM. Chem. Commun. 2008:3366–3368. doi: 10.1039/b806150e. [DOI] [PubMed] [Google Scholar]
- 48.Wang Z, Tanabe KK, Cohen SM. Inorg. Chem. 2009;48:296–306. doi: 10.1021/ic801837t. [DOI] [PubMed] [Google Scholar]
- 49.Haneda T, Kawano M, Kawamichi T, Fujita M. J. Am. Chem. Soc. 2008;130:1578–1579. doi: 10.1021/ja7111564. [DOI] [PubMed] [Google Scholar]
- 50.Kaye SS, Long JR. J. Am. Chem. Soc. 2008;130:806–807. doi: 10.1021/ja7102108. [DOI] [PubMed] [Google Scholar]
- 51.Burrows AD, Frost CG, Mahon MF, Richardson C. Chem. Commun. 2009 doi: 10.1039/b906170c. online contents. [DOI] [PubMed] [Google Scholar]
- 52.Costa JS, Gamez P, Black CA, Roubeau O, Teat SJ, Reedijk J. Eur. J. Inorg. Chem. 2008:1551–1554. [Google Scholar]
- 53.Ingleson MJ, Barrio JP, Guilbaud JB, Khimyak YZ, Rosseinsky M. J. Chem. Commun. 2008:2680–2682. doi: 10.1039/b718367d. [DOI] [PubMed] [Google Scholar]
- 54.Burrows AD, Frost CG, Mahon MF, Richardson C. Angew. Chem. Int. Ed. 2008;47:8482–8486. doi: 10.1002/anie.200802908. [DOI] [PubMed] [Google Scholar]
- 55.Hwang YK, Hong DY, Chang JS, Jhung SH, Seo YK, Kim J, Vimont A, Daturi M, Serre C, Ferey G. Angew. Chem., Int. Ed. 2008;47:4144–4148. doi: 10.1002/anie.200705998. [DOI] [PubMed] [Google Scholar]
- 56.Morris W, Doonan CJ, Furukawa H, Banerjee R, Yaghi OM. J. Am. Chem. Soc. 2008;130:12626–12627. doi: 10.1021/ja805222x. [DOI] [PubMed] [Google Scholar]
- 57.Ahnfeldt T, Gunzelmann D, Loiseau T, Hirsemann D, Senker J, Ferey G, Stock N. Inorg. Chem. 2009;48:3057–3064. doi: 10.1021/ic8023265. [DOI] [PubMed] [Google Scholar]
- 58.Gadzikwa T, Lu G, Stern CL, Wilson SR, Hupp JT, Nguyen ST. Chem. Commun. 2008:5493–5495. doi: 10.1039/b805101a. [DOI] [PubMed] [Google Scholar]
- 59.Rowsell JLC, Yaghi OM. J. Am. Chem. Soc. 2006;128:1304–1315. doi: 10.1021/ja056639q. [DOI] [PubMed] [Google Scholar]
- 60.Li B, Martin AL, Gillies ER. Chem. Commun. 2007:5217–5219. doi: 10.1039/b713569f. [DOI] [PubMed] [Google Scholar]
- 61.Lin WB. J. Solid State Chem. 2005;178:2486–2490. [Google Scholar]
- 62.Kawamichi T, Kodama T, Kawano M, Fujita M. Angew. Chem., Int. Ed. 2008;47:8030–8032. doi: 10.1002/anie.200802545. [DOI] [PubMed] [Google Scholar]
- 63.Faucaud A. Bull, Soc. Chim. France. 1963:873. [Google Scholar]
- 64.Kaye SS, Dailly A, Yaghi OM, Long JR. J. Am. Chem. Soc. 2007;129:14176–14177. doi: 10.1021/ja076877g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.