

Abstract
Current GWAS have primarily focused on testing association of single SNPs. To only test for association of single SNPs has limited utility and is insufficient to dissect the complex genetic structure of many common diseases. To meet conceptual and technical challenges raised by GWAS, we suggest gene and pathway-based GWAS as complementary to the current single SNP-based GWAS. This publication develops three statistics for testing association of genes and pathways with disease: linear combination test, quadratic test and decorrelation test, which take correlations among SNPs within a gene or genes within a pathway into account. The null distribution of the suggested statistics is examined and the statistics are applied to GWAS of rheumatoid arthritis in the Wellcome Trust Case–Control Consortium and the North American Rheumatoid Arthritis Consortium studies. The preliminary results show that the suggested gene and pathway-based GWAS offer several remarkable features. First, not only can they identify the genes that have large genetic effects, but also they can detect new genes in which each single SNP conferred a small amount of disease risk, and their joint actions can be implicated in the development of diseases. Second, gene and pathway-based analysis can allow the formation of the core of pathway definition of complex diseases and unravel the functional bases of an association finding. Third, replication of association findings at the gene or pathway level is much easier than replication at the individual SNP level.
Keywords: GWAS, gene association analysis, pathway association analysis, complex diseases
Introduction
Substantial progress in GWAS of complex diseases has been made and at least 300 loci have been found to be significantly associated with as many as 120 diseases and traits in these studies.1 In spite of the great success of GWAS, current GWAS continue to be primarily focused on testing associations of a single SNP with a disease one at a time. As common diseases are often caused by multiple genes and environments that are organized into a myriad of complex networks, to only test for association of a single SNP has limited utility2 and is insufficient to dissect the complex genetic structure of common diseases for the following reasons. First, the common approach to the current GWAS is to select dozens of the most significant SNPs in the list for further investigations. However, the set of most significant SNPs often accounts for only a small proportion of the genetic variants associated with disease and offers limited understanding of complex diseases.3 Common diseases often arise from the joint action of multiple loci within a gene or joint action of multiple genes within a pathway. Although each single SNP may confer only a small disease risk, their joint actions are likely to have a significant role in the development of disease. If one only considers the most significant SNPs, the genetic variants that jointly have significant risk effects, individually making only a small contribution, will be missed. Second, locus heterogeneity, in which alleles at different loci cause disease in different populations, will increase the difficulty in replicating associations of a single marker with a disease.4 The list of significant SNPs from several studies may have little overlap. Therefore, replication of association findings at the SNP level can be difficult if redundant genes have roles. Third, the ultimate purpose of genetic studies of complex diseases is to decipher the path from genotype to phenotype. In spite of the conduct of extensive studies in search of genes causing complex diseases, connections between DNA variation and complex phenotypes, which are essential for unraveling pathogeneses of complex diseases and predicting variation in human health, still have been elusive. Health states of individuals are a complex, multidimensional phenomenon. Clinical manifestations arise from integrated actions of multiple genetic and environmental factors, through dynamic, epigenetic and regulatory mechanisms.5, 6, 7 What has been generally missing in the current GWAS is the context in which DNA variation occurs. It was reported that a gene location within a cellular network may have significant effect on the results of the given gene mutation.8 The genetic variation occurring at multiple loci often perturbs signal, regulatory and metabolic pathways, resulting in complex changes in phenotype. SNPs and genes carry out their functions through intricate pathways of reactions and interactions. Knowing the list of risk, SNPs is not sufficient to understand disease mechanisms.9
To overcome these limitations, recently, Wang et al10 suggested to extend gene set enrichment analysis for gene expression data, which intend to identify subtle, but coordinated expression variations of gene groups to GWAS. The challenge for extension is how to represent a gene in GWAS. Wang et al10 suggested to choose the most significant SNP from each gene as a representative. But, in GWAS, a gene often contains a variable number of SNPs. The genes that contain a number of SNPs jointly having significant risk effects, but individually making only a small contribution, will be missed in such representation. Another issue is how to deal with correlations among SNPs and genes. Owing to linkage disequilibrium (LD), there may be high correlations among some SNPs. In Wang et al's publication, the statistics that were used for testing association of a pathway with the disease did not take correlations among SNPs into account.
To solve these problems, we consider three basic units of association analysis: SNP, gene and pathway and suggest gene and pathway-based GWAS. In gene and pathway-based GWAS, each gene is represented by all SNPs, which are either located within the gene or are not >500 kb away from the gene.10 Unlike gene set enrichment analysis in which one examines whether significantly associated genes are overrepresented in the set of genes to be analyzed, we formulate the gene and pathway-based GWAS as the problem to jointly test for association of multiple SNPs within the gene or multiple genes within the pathway with disease. This allows us to holistically unravel complex genetic structure of common disease to gain insight into the biological processes and disease mechanism.
The purpose of this report is to develop a general framework for gene and pathway-based GWAS of complex diseases and novel statistics for testing association of a gene or pathway with the disease. To accomplish this, we first formulate the null hypothesis for testing association of the gene or pathway with the disease. Then, we develop three statistics to combine a set of dependent P-values of SNPs into an overall significance level for a gene or a set of dependent P-values of genes into an overall significance level for a pathway. We validate the null distribution and calculate type 1 error rates of the three developed statistics for testing association of the gene or pathway with the disease using extensive simulation studies. To illustrate how to perform the gene and pathway-based GWAS, we examine GWAS of rheumatoid arthritis (RA) in two independent studies: Wellcome Trust Case–Control Consortium (WTCCC) and the North American Rheumatoid Arthritis Consortium (NARAC) studies. Our results show that the suggested new paradigm for GWAS not only can identify the genes that have large genetic effects and can be found by single SNP association analysis, but also can detect new genes in which each single SNP confers a small disease risk, but their joint actions can be implicated in the development of diseases.
A program for implementation can be downloaded from our website http://www.sph.uth.tmc.edu/ hgc/faculty/xiong/.
Materials and methods
Gene-based association and its formal null hypothesis testing
A gene-based association analysis uses a gene as the basic unit of analysis. The gene-based association jointly considers all common variation within a gene.4 Instead of testing association of single SNPs with the disease, gene-based association jointly tests for association of all the SNPs within the gene. Formally, suppose that there are k SNPs in the gene. The null hypothesis for testing association of the ith SNP in the gene is represented by
where θi denotes the parameter, for example, the difference in allele frequencies between cases and controls. Then, the null hypothesis for testing association of a gene with disease is defined as testing for the combined null hypothesis:
The goal of testing association of the gene is to test all SNPs in the gene as a whole. Testing for association of the gene with disease is to test an overall effect of all SNPs in the gene, which combines evidence. Each SNP in the gene may confer small disease risk, and jointly they make a large contribution.
Statistics for testing association of a gene with disease
A general framework for testing association of a gene with the disease is to combine evidence from all the markers within the gene. In general, correlations among P-values of SNPs within the gene exist because of LD among SNPs. Correlations among SNPs will invalidate the existing methods for combining independent P-values. Therefore, the methods for combining independent P-values cannot be directly applied to combining P-values of SNPs within the gene. We need to develop methods for combining dependent P-values, which take correlations among SNPs into account. We suggest three statistics for combining dependent P-values. In the following discussion, we assume that Pi is the P-value of the statistic with a normal or asymptotic normal distribution.
Before presenting statistics, we introduce some notations. Consider SNP Mi with two alleles Bi and bi, and SNP Mj with two alleles Bj and bj. For cases, we define the indicator variables for alleles: 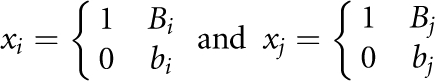 or the indicator variables for the genotypes:
or the indicator variables for the genotypes:
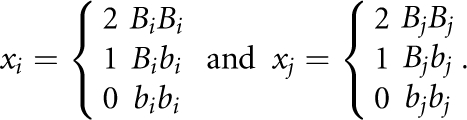 |
We similarly define the indicator variables yi and yj for controls.
Linear combination test (LCT)
The first suggested statistic is to take a linear combination of P-values for all SNPs within the gene, which is referred to as the LCT. Let e=(1, 1, …, 1)T. A statistic based on linear combination of the vector Z is defined as
where Zi=Φ−1(1−Pt), Z=(Z1, …, Zk)T, Rg is the correlation matrix of Z. A key issue is how to calculate the correlation matrix Rg. In general, Rg is difficult to calculate. However, if the P-value for each SNP is calculated by the t statistic, we have the following results. Let Zk=Φ−1(1−Pi)=Φ−1(FT(tk)), where tk is a t statistic for testing association of the k-th SNP. When the sample size is large enough, FT can be approximated by a standard normal distribution, which implies Zk≈tk. Therefore, under the null hypothesis the correlation matrix of Z among all the SNPs within a gene can be given by the sampling correlation matrix of the data: corr(Zk, Zl)≈corr(xk−yk, xl−yl). Therefore, the correlation matrix Rg can be approximated by
where xi and yi are indicator variables for either alleles or genotypes in cases and controls, respectively, and Φ is the standard normal distribution. Under the null hypothesis, TL is the standard normal distribution.
Quadratic Test (QT)
A QT that is based on the quadratic form of Z is defined as
where Z and Rg are previously defined. Under the null hypothesis, TQ is asymptotically distributed as a central χ(k)2 distribution, where k is the number of SNPs within the gene.
Decorrelation Test (DT)
Another way to combine dependent P-values is that we first transform dependent variables into independent variables and then combine independent variables. Let the correlation matrix Rg be decomposed as
where C is a nonsingular matrix. Then, the correlated random variables Zi(i=1, … , k) can be decorrelated by the following transformation:
It can be easily observed that
Thus, the variables in W are independent, which implies that the decorrelated statistics W are asymptotically distributed as a vector of independent standard normal variables. For each Wi, we calculate the P-value P*i, resulting in
All the methods for combining independent P-values can be applied to P*. For example, we can use the Fisher's combination test11 to combine P*:
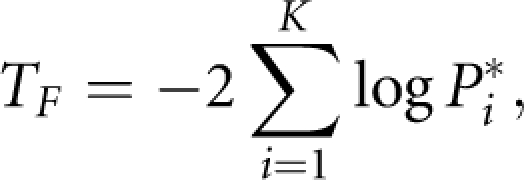 |
which follows a χ(2k)2 distribution, or Sidak, Simes, false discovery rate (FDR) method.12
Pathway-based association test
A general framework for testing association of a pathway with disease that is similar to gene-based association analysis is to combine P-values of the genes within the pathway from gene-based association analysis into an overall significant level of the pathway.
Correlation structure among genes within a pathway
Consider m genes within a pathway. Suppose that the i-th gene has ki SNPs. Let xiu, xjv, yjv and yjv be the indicator variables for the u-th allele in the i-th gene, v-th allele in the j-th gene in cases and controls, respectively. The correlation between the u-th marker in the i-th gene and the v-th marker in the j-th gene is defined as riu,jv=corr(xiu−yiu,xjv−yjv). Let Zij=Φ−1(1−Pij), where Pij is the P-value for testing association of the j-th SNP in the i-th gene. Define
Define the correlation matrix between vectors Zi and Zj as
 |
Let Ri be the correlation matrix of the vector Zi for the i-th gene in the pathway, which is defined in Equation (2), and the correlation matrix of the vector Z for the whole pathway be defined as
Recall that the statistic TLi for the i-th gene defined in Equation (1) is given by
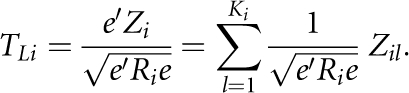 |
By simple algebra, we have
Let TL=(TL1, … ,TLm)T, rgij=corr(TLi,TLj) be the correlation between the test statistic for the i-th gene and the test statistic for the j-th gene. Then, its corresponding correlation matrix Rp for the whole pathway is given by
 |
Statistics for testing association of a pathway with disease
Similar to testing for association of a gene with the disease, the basic idea for testing association of a pathway with the disease is to combine P-values of genes within the pathway. We have three statistics for testing association of a pathway with the disease.
Linear combination test
Taking a linear combination of statistics for testing association of the genes within the pathway leads to a statistic for testing association of the pathway with the disease. Formally, we define the statistic for testing association of the pathway with the disease as
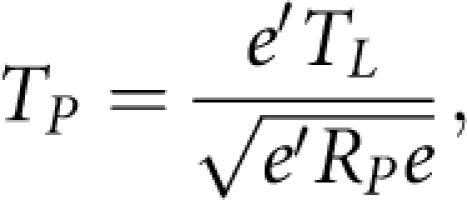 |
where TL=(TL1, … ,TLm)T and RP is defined in Equation (6). Then, under the null hypothesis, TP is asymptotically distributed as the standard normal distribution.
Quadratic test
Similar to the gene-based analysis, we can also define the following QT
Under the null hypothesis, TPQ is asymptotically distributed as a central χ(m)2 distribution.
Decorrelation test
The vector of the statistics for testing gene association TL can also be decorrelated by
where RP=CPCTP Then, TPD consists of m independent standard normal variables. Let PD=(PD1, … ,PDm)T be the vector of P-values corresponding to TPD. We can use the Fisher's combination test to combine PD:
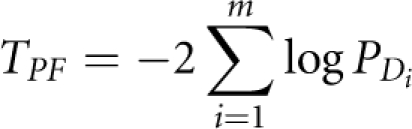 |
which follows a χ(2m)2 distribution. Other methods for combining independent P-values such as Sidak, Simes and the FDR method can also be used to combine P-values for individual genes within the pathway.
Results
Type 1 error rates of test statistics
To validate the statistics presented for testing association of genes and pathways with the disease in this publication, first verify the standard normal distribution of the Z statistic that is obtained by an inverse normal distribution transformation of the t statistic. For simplicity, here we only present results for indicator variables with alleles. The results for the genotypes were similar (data not shown). SNaP software13 was used to generate a population of 1 000 000 chromosomes. We sampled 2000 individuals as cases and 2000 individuals as controls from the population and performed 10 000 simulations. Figure 1 plots the empirical distribution of the Z statistic, which is very close to the standard normal distribution. We then calculate the type 1 error rates of the developed statistics. For calculation of type 1 error rates of the statistics for testing association of the gene with the disease, SNaP software was used to generate 1 000 000 chromosomes, each having a gene with 20 SNPs. For calculation of type 1 error rates of the statistics for testing association of the pathway with disease, SNaP software was used to generate 1 000 000 chromosomes, each having 5 blocks that are representative of genes and each block having 20 SNPs. We randomly sampled individuals from the population that were equally divided as cases and controls. The number of sampled controls range from 1000 to 3000, and 10 000 simulations were performed. Table 1 and Supplementary Table 1 show that type 1 error rates of the statistics for testing association of the gene and pathway with the disease were not appreciably different from the nominal levels (α=0.05, α=0.01 and α=0.001), respectively.
Figure 1.

Empirical distribution of the Z statistic.
Table 1. Type 1 error rates of the statistics for testing association of the gene with the disease.
| Sample size | LCT | QT | DT |
|---|---|---|---|
| 1000 | |||
| α=0.001 | 0.0012 | 0.0023 | 0.0019 |
| α=0.01 | 0.0086 | 0.0119 | 0.0124 |
| α=0.05 | 0.0455 | 0.0542 | 0.0540 |
| 1500 | |||
| α=0.001 | 0.0008 | 0.0009 | 0.0008 |
| α=0.01 | 0.011 | 0.0108 | 0.011 |
| α=0.05 | 0.0537 | 0.0535 | 0.0543 |
| 2000 | |||
| α=0.001 | 0.001 | 0.0014 | 0.0011 |
| α=0.01 | 0.0097 | 0.0122 | 0.0124 |
| α=0.05 | 0.0477 | 0.0528 | 0.0525 |
| 2500 | |||
| α=0.001 | 0.0007 | 0.0014 | 0.0014 |
| α=0.01 | 0.0096 | 0.0122 | 0.0128 |
| α=0.05 | 0.0482 | 0.0545 | 0.0542 |
| 3000 | |||
| α=0.001 | 0.0009 | 0.0015 | 0.0014 |
| α=0.01 | 0.0107 | 0.0107 | 0.0107 |
| α=0.05 | 0.049 | 0.0504 | 0.0514 |
RA in the WTCCC and NARAC studies
To evaluate the performance of the gene and pathway-based GWAS, the developed statistics were applied to RA in the WTCCC14 and NARAC15 studies to identify significantly associated genes and pathways with RA. A total of 459 653 SNPs were typed for 1860 RA patients and 2938 controls in the WTCCC studies and 545 080 SNPs were typed for 866 RA patients and 1194 controls in the NARAC studies. The total number of genes involved in the WTCCC and NARAC studies were 15 732 and 17 773, respectively.
The current GWAS are limited to taking a SNP as the basic unit for association testing. The results, wherein taking a gene or a pathway as a basic unit of association test are presented below. We assembled 465 pathways from KEGG16 and Biocarta (http://www.biocarta.com). The assignment of SNPs to a gene was obtained from the NCBI human9606 database (version b129) (ftp://ftp.ncbi.nlm.nih.gov/snp/organisms/human_96 06/database/organism_data/b129/b129_SNPContigLocusId_36_3.bcp.gz). The P-values for declaring association of the gene with RA after performing a Bonferroni correction in the WTCCC and NARAC studies were 3.2 × 10−6 and 2.8 × 10−6, respectively. All 465 pathways were involved in the WTCCC and NARAC studies. Thus, the P-value for declaring association of the pathway with RA was 1.1 × 10−4.
Table 2 summarizes all 19 replicated genes by the LCT method with their P-values. Supplementary Tables 2, 3 and 4 list 49, 47 and 45 replicated genes by the QT, DT(FDR) and DT(Fisher) methods, respectively. The QT method identified 90 and 92% of the replicated genes and they are included in the list of replicated genes identified by the DT(FDR) method and the DT(Fisher) method, respectively. Association of the genes human leukocyte antigen (HLA)-DPB1,17, 18 HLA-DQR1,18 HLA-DQB1,19, 20 and MICA21, 22 with RA were previously reported. MICA is a cell stress-induced glycoprotein and localized in the HLA region. Its reaction with T cells and natural killer cells suggest that MICA gene may have an important role in the development of autoimmune disease. The gene AIF1 (an allograft inflammatory factor 1) that is encoded within the HLA class III genomic region on chromosome 6p21 and has an important role in inflammation was reported to be associated with systemic sclerosis23 and atherosclerosis.24 RDRNA-binding protein that is located in the major histocompatibility complex (MHC) class III region on chromosome 6p21.3 was reported to be involved in the immune response and systemic inflammatory stimulation.25 The genes BAT3, BAT4 and AGPAT1 are within the human MHC class III region. The gene ZFP57 that is located on chromosome 6p22 and encodes a zinc-finger transcription factor is involved in hypomethylation of several imprinted loci in transient neonatal diabetes patients.26 The SNP rs6679677, which is in complete LD with the SNP rs2476601 in the PTPN22 gene belongs to the gene RSBN1 in the NCBI database. The PTPN22 gene that has been reported to be associated with RA several times14 also showed strong association with RA in the NARAC studies in our analysis.
Table 2. Genes with significant association with RA in both WTCCC and NARAC studies that were identified by the LCT method.
| P-value | ||
|---|---|---|
| Gene | NARAC | WTCCC |
| PTPN22 | 8.10E-08 | 2.44E-15(RSBN1)a |
| AIF1 | 4.44E-16 | 8.22E-15 |
| CREBL1 | <1E-17 | 5.91E-09 |
| HLA-DPA1 | 2.63E-11 | 2.72E-11 |
| HLA-DPB1 | 2.83E-07 | 2.34E-11 |
| HLA-DQA1 | 8.92E-12 | 1.49E-11 |
| HLA-DQA2 | 1.31E-07 | 4.84E-11 |
| HLA-DQB1 | 6E-15 | 6.55E-11 |
| MICA | 6.83E-11 | 5.82E-09 |
| RPS18 | 1.13E-08 | 2.80E-06 |
| BAT3 | 8.97E-11 | 5.16E-07 |
| BAT4 | 3.14E-10 | <1E-17 |
| RDBP | 2.24E-14 | <1E-17 |
| AGPAT1 | 9.55E-15 | 3.68E-12 |
| EHMT2 | 1.65E-09 | 7.01E-11 |
| BTNL2 | 2.97E-12 | 1.55E-07 |
| GPSM3 | <1E-17 | 5.20E-09 |
| ZFP57 | 4.69E-09 | 3.78E-07 |
| LOC731881 | 1.37E-10 | <1E-17 |
WTCCC typed SNP rs6679677 that is close to the gene PTPN22 belongs to the gene RSBN1 in the NCBI database.
To show that the strategy for considering only the most significant SNPs in the association studies may lead to missing the genetic variants that jointly have significant risk effects, but individually make only a small contribution, see Table 3. Five different markers were typed for the gene ZFP57 in both the WTCCC and NARAC studies. Table 3 shows that none of the SNPs in the gene ZFP57 showed significant association, but the gene ZFP57 itself has strong association with RA in both the WTCCC and NARAC studies. We also observe that although typed SNPs within the gene ZFP57 in two studies were different, we still can replicate association of the gene ZFP57 with RA in the two independent studies.
Table 3. P-values of SNPs in the gene ZFP57.
| NARAC | WTCCC | ||
|---|---|---|---|
| Method | P-value ZFP57 | Method | P-value ZFP57 |
| LCT | 4.69E-09 | 3.78E-07 | |
| QT | 6.70E-06 | 4.16E-06 | |
| DT(FDR) | 9.11E-06 | 1.92E-05 | |
| DT(Fisher) | 2.38E-06 | 6.04E-06 | |
| SNP | P-value | SNP | P-value |
| rs2535238 | 0.018526 | rs378596 | 0.0005011 |
| rs2747430 | 0.007419 | rs387603 | 0.0005158 |
| rs3129054 | 7.42E-05 | rs387642 | 0.007956 |
| rs9257936 | 0.024268 | rs3129063 | 0.07998 |
| rs9257940 | 0.046082 | rs3131847 | 0.006112 |
Attempting to understand and interpret a number of significant SNPs without any unifying biological theme can be challenging and demanding. SNPs and genes carry out their functions through intricate pathways of reactions and interactions. The function of many SNPs may not be well characterized, but the function of pathways, on the contrary, are much better analyzed. Pathway-based association analysis can help unravel the mechanism of complex diseases. Next we present the results of pathway-based GWAS of RA. Supplementary Table 5, Table 4, Supplementary Tables 6 and 7 list significantly associated pathways with RA in both the WTCCC and NARAC studies, which were identified by LCT, QT, DT(FDR) and DT(Fisher) methods, respectively. Figures 2 and 3 plot a MAPK signaling pathway, which was associated with RA in the WTCCC and NARAC studies, respectively. These tables and figures showed several remarkable features that can be used to extract biological insight from GWAS. First, functional pathway analysis is a key to unraveling the mechanism of complex diseases and opens a way for a pathway definition of complex diseases. Biological pathways are sets of genes that work in concert to perform particular cellular functions or biological processes. RA is an autoimmune disease characterized by chronic inflammation of the joints, the tissues around the joints and other organs in the body.27 Associated pathways identified in the WTCCC and NARAC studies can be classified into three groups. The first group consists of three pathways: antigen processing and presentation, cell adhesion molecules and type I diabetes mellitus pathways. Results of all tests (LCT, QT and DT) have shown that these three pathways were significantly associated with RA in two studies. The second group includes six pathways: MAPK signaling pathway, complement pathway, complement and coagulation cascades, alternative complement pathway, cytokines and inflammatory pathway and ether lipid metabolism pathways, which were in common in the lists of associated pathways identified by QT and DT methods. The third group includes B lymphocyte cell surface molecules, IL 5 signaling pathway, Th1/Th2 differentiation pathway, glycerophospholipid metabolism, cell communication, focal adhesion, glycerolipid metabolism, Jak-STAT signaling pathway, bystander B-cell activation pathway and antigen-dependent B-cell activation pathway. The pathways in the third group were identified by either the QT method or DT method.
Table 4. Significant pathways in both WTCCC and NARAC studies that were identified by the QT method.
| WTCCC | NARAC | |||
|---|---|---|---|---|
| Name of pathway | No. of genes | P-value (QT) | No. of genes | P-value (QT) |
| Complement and coagulation cascades pathway | 53 | 5.94E-13 | 62 | <1E-17 |
| Jak-STAT signaling pathway | 109 | 1.19E-10 | 122 | <1E-17 |
| Natural killer cell-mediated cytotoxicity pathway | 94 | 1.66E-09 | 111 | <1E-17 |
| Cytokines and inflammatory response pathway | 23 | 1.83E-07 | 23 | <1E-17 |
| Focal adhesion pathway | 175 | 4.06E-07 | 190 | <1E-17 |
| Th1/Th2 differentiation pathway | 17 | 4.62E-07 | 17 | <1E-17 |
| The role of eosinophils in the chemokine network of allergy pathway | 4 | 1.02E-05 | 5 | <1E-17 |
| Bystander B-cell activation pathway | 6 | 4.40E-05 | 7 | <1E-17 |
| B lymphocyte cell surface molecules pathway | 8 | 4.89E-05 | 10 | <1E-17 |
| Antigen-dependent B-cell activation pathway | 10 | 8.79E-05 | 10 | <1E-17 |
| IL 5 signaling pathway | 7 | 0.000103 | 8 | <1E-17 |
| MAPK signaling pathway | 203 | <1E-17 | 235 | <1E-17 |
| Cytokine–cytokine receptor interaction pathway | 175 | <1E-17 | 203 | <1E-17 |
| Cell adhesion molecules pathway | 109 | <1E-17 | 117 | <1E-17 |
| Antigen processing and presentation pathway | 47 | <1E-17 | 53 | <1E-17 |
| Type I diabetes mellitus pathway | 36 | <1E-17 | 38 | <1E-17 |
| Alternative complement pathway | 11 | <1E-17 | 12 | 5.54E-06 |
| Lysine degradation pathway | 38 | 0.000109 | 44 | 1.76E-08 |
| Glycerophospholipid metabolism pathway | 54 | 7.32E-07 | 61 | 1.93E-10 |
| Gap junction pathway | 73 | 2.08E-06 | 81 | 1.31E-10 |
| Glycerolipid metabolism pathway | 48 | 1.10E-06 | 54 | 7.9E-11 |
| Toll-like receptor signaling pathway | 74 | 1.29E-08 | 83 | 5.9E-12 |
| Ether lipid metabolism pathway | 27 | 3.26E-09 | 29 | 1.51E-13 |
| Cell communication pathway | 110 | 8.15E-11 | 119 | 8.33E-14 |
| Tight junction pathway | 115 | 7.09E-10 | 121 | 1.68E-14 |
| Complement pathway | 17 | <1E-17 | 21 | 2.22E-16 |
Figure 2.

P-values for testing association of the genes within the MAPK signaling pathway with RA in WTCCC studies. Blocks including significant genes are in red color, blocks including mild significant genes are in light red color and blocks including no significant genes are in green color.
Figure 3.

P-values for testing association of the genes within the MAPK signaling pathway with RA in NARAC studies. Blocks including significant genes are in red color, blocks including mild significant genes are in light red color and blocks including no significant genes are in green color.
In the first group, the antigen processing and presentation pathway mainly consists of MHC molecules, which are shown on cell surfaces and responsible for lymphocyte recognition and antigen presentation. The antigen processing and presentation pathway and the cell adhesion pathway are crucial for controlling inflammatory and immune responses and involved in the RA.28, 29 Close contact between different populations of cells is fundamental for inflammatory and immune responses. The type I diabetes mellitus pathway induces an uncontrolled immune attack against the insulin producing β-cells.30 These three pathways form the core pathway definition for RA.
The relationships between the second group of pathways and RA consist of the MAPK pathway, which is a key signal transduction pathway of inflammation and reported to be involved in the development of RA.31 The complement pathway helps clear pathogens from an organism and has a key role in determining the fate of immune status.32 The complement and coagulation cascades pathway is a partner of inflammation33 and involved in the pathogenesis of RA.34 The pathways in the third group such as IL 5 signaling pathway,35 Th1/Th2 differentiation pathway,36 B lymphocyte cell surface molecules pathway,37 lysine degradation,38 antigen-dependent B-cell activation pathway,39 cell communication,40 bystander B-cell activation pathway41 and focal adhesion42 are involved in inflammation and immune responses and hence are related to RA in some degree.
Second, replication of the results of pathways in independent samples is much easier than replication of genes or SNPs. Replications can be performed at the level of the SNP, the gene and pathway. As Figures 2 and 3 show, the WTCCC and NARAC studies shared no common significantly associated genes within the MAPK pathway, in other words, we failed to replicate significantly associated genes within the MAPK pathway in two independent studies. However, Table 4 and Supplementary Tables 6 and 7 show that the MAPK pathway in both studies were significantly associated with RA. This example shows that replication at the pathway level is easier than replication at the gene level.
Third, the number of genes showing significant association with RA within the pathway may be very small, but the number of genes showing mild association with RA within the pathway may be quite large. In Figures 2 and 3 shown, we can only observe two and four significantly associated genes, but we can observe 19 (9.4% of total genes within the pathway) and 29 (12.7% of total genes within the pathway) genes showing mild association with RA within the MAPK pathway in the WTCCC and NARAC studies, respectively. It is interesting that these mildly associated genes were proinflammatory cytokine, stress gene, growth factors, MAPKKK, MAPKK, MAPK and transcription factors, which were distributed among all stages, from upstream to downstream, of inducing the MAPK pathway. We also observe that even if the gene CACNA2D3 showed significant association with RA using the LCT test, the P-value of the best SNP in the gene CACNA2D3 was 0.000432, in the NARAC studies. This shows that if we consider only the most significant SNPs, the genetic variants that jointly have significant risk effects, but individually make only a small contribution, will be missed. This example also shows that each gene may confer a small contribution, but their joint actions may affect the function of the pathway, which in turn will cause disease.
Discussion
In spite of the great success of large-scale GWAS, the current approach to GWAS has mainly focused on testing association of single SNPs with disease and selected the best SNPs for further studies. However, single SNP association analysis will miss many SNPs with moderate genetic effects. Separate association finding from biological interpretation offer limited understanding of the functional basis of complex diseases. To overcome these limitations, in this report we suggest gene and pathway-based GWAS in which we take a gene and a pathway as basic units of association analysis in addition to single SNP association studies. Gene and pathway-based GWAS assess the significance of the genes and the predefined pathways, and intend to identify biological pathways with subtle but coordinated genetic variants that confer risk contributions.
To shift the paradigm from single SNP-based GWAS to gene and pathway-based GWAS, we addressed the following issues. First, unlike the extension of gene set enrichment analysis to GWAS in which we analyze whether significantly associated genes are overrepresented in the set of genes, which are of interest, we formulate the gene and pathway-based GWAS as the traditional hypothesis testing problem. In other words, to test the association of a gene or a pathway with the disease is to jointly test for association of multiple SNPs within the gene or multiple genes within the pathway with the disease. Second, the challenge facing us is how to develop statistics for testing association of a gene or a pathway with the disease. A simple approach to joint analysis of multiple SNPs within the gene and multiple genes within the pathway is to combine their P-values into an overall P-value to represent the significance of a gene or a pathway. We analyzed correlations among SNPs within the gene and correlations among genes within the pathway and found that correlations among SNPs and genes cannot be ignored (owing to space limitation, data were not shown). However, the current popular statistical methods are designed for only combining independent P-values and hence are not appropriate for gene and pathway-based GWAS. Therefore, we developed three novel statistics, which are able to combine dependent P-values of SNPs within the gene or genes within the pathway. We examined the distribution of the suggested statistics under the null hypothesis of no association of the gene or pathway with the disease and calculated their type 1 error rates by simulations. Our results have shown that type 1 error rates were close to nominal significance levels. Third, to assess their merit and limitations, we applied the developed statistical methods for gene and pathway-based association analysis to GWAS of RA in the WTCCC and NARAC studies. The results have shown that the new paradigm of GWAS not only confirmed previous association findings, but also discovered a number of new genes and pathways that were significantly associated with RA. Although the results were preliminary, they indeed showed that identification of pathways associated with disease allows us to much easier uncover pathogenesis of disease.
Gene and pathway-based GWAS offer several remarkable features. First, the new paradigm not only can identify the genes that have large genetic effects and can be found by single SNP association analysis, but also can detect new genes in which each single SNP confers small disease risk, but their joint actions can be implicated in the development of diseases. Second, the results of application of pathway analysis to RA strongly show that pathway-based analysis can add structure to genomic data and allows us to gain deep understanding of cellular processes as intricate networks of functionally related genes and to unravel the functional bases of the association finding. Third, replication of association findings at the gene or pathway level is much easier than replication at the individual SNP level. Risk SNPs (or genes) for different individuals may be different, but may be in the same gene (or pathway). Fourth, the new paradigm for GWAS will open a novel avenue to integrate GWAS with other functional analyses such as gene set enrichment analysis for gene expression data and hence will facilitate uncovering the mechanism of complex diseases. Our results strongly challenge the paradigm of GWAS that only tests the association of single SNPs.
The developed statistics for testing association of genes or pathways also have serious limitations. First, presence of both positive and negative correlations among SNPs will dramatically reduce the power to discover association of genes or pathways. Second, when the number of SNPs within the gene or number of genes within the pathway is large, numeric instability will increase the error in calculation of the inverse matrix of the correlation matrix, which in turn will increase the false-positive rate of association finding. We should overcome these limitations in the future.
Millions of dollars are spent for GWAS. Data from GWAS are very expensive, but also contain rich information. Simple statistical methods based on single SNP association analysis might not be the best strategy for deciphering the path from genomic information to clinical phenotypes. Taking full advantage of rich information and huge opportunities provided by GWAS raises great conceptual and technical challenges. To unravel the true nature of complex diseases, we need to integrate multiple approaches and multiple types of data. In the coming years, we will witness the development of a variety of novel methods for GWAS, rapid progress in GWAS and their great success.
Acknowledgments
L Luo and M Xiong are supported by grants from the National Institutes of Health NIAMS P01 AR052915-01A1, NIAMS P50 AR054144-01 CORT and NIAMS 1 R01 AR057120-01. G Peng, H Dong and Y Zhu are supported by a grant from the National Institutes of Health Tech Research and Development Program of China(863) (2007AA02Z312). CI Amos is supported by grants from the National Institutes of Health ES09912, AK44422 and CA13479.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Zhernakova A, van Diemen CC, Wijmenga C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet. 2009;10:43–55. doi: 10.1038/nrg2489. [DOI] [PubMed] [Google Scholar]
- Schadt EE, Lum PY. Thematic review series: systems biology approaches to metabolic and cardiovascular disorders. Reverse engineering gene networks to identify key drivers of complex disease phenotypes. J Lipid Res. 2006;47:2601–2613. doi: 10.1194/jlr.R600026-JLR200. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Sham PC. The future of association studies: gene-based analysis and replication. Am J Hum Genet. 2004;75:353–362. doi: 10.1086/423901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olden K, Wilson S. Environmental health and genomics: visions and implications. Nat Rev Genet. 2000;1:149–153. doi: 10.1038/35038586. [DOI] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Kruglyak L, Nickerson DA. Mapping complex disease loci in whole-genome association studies. Nature. 2004;429:446–452. doi: 10.1038/nature02623. [DOI] [PubMed] [Google Scholar]
- Benfey PN, Mitchell-Olds T. From genotype to phenotype: systems biology meets natural variation. Science. 2008;320:495–497. doi: 10.1126/science.1153716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman I, Rzhetsky A, Vitkup D. Network properties of genes harboring inherited disease mutations. Proc Natl Acad Sci USA. 2008;105:4323–4328. doi: 10.1073/pnas.0701722105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi AL. Network medicine--from obesity to the ‘diseasome'. N Engl J Med. 2007;357:404–407. doi: 10.1056/NEJMe078114. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Bucan M. Pathway-based approaches for analysis of genomewide association studies. Am J Hum Genet. 2007;81:1278–1283. doi: 10.1086/522374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA.Statistical Methods for Research Workers4th edn: London: Oliver and Boyd 1932
- Pounds S, Cheng C. Robust estimation of the false discovery rate. Bioinformatics. 2006;22:1979–1987. doi: 10.1093/bioinformatics/btl328. [DOI] [PubMed] [Google Scholar]
- Nothnagel M. Simulation of LD block-structured SNP haplotype data and its use for the analysis of case-control data by supervised learning methods. Am J Hum Genet. 2002;71 (Suppl:A2363. [Google Scholar]
- The Wellcome Trust Case-Control Consortium Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis--a genomewide study. N Engl J Med. 2007;357:1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthy D, MacGregor A, Awomoi A, et al. HLA-DPB1*0201 is associated with particular clinical features of rheumatoid arthritis. Rev Rhum Engl Ed. 1995;62:163–168. [PubMed] [Google Scholar]
- Gao X, Fernandez-Vina M, Olsen NJ, Pincus T, Stastny P. HLA-DPB1*0301 is a major risk factor for rheumatoid factor-negative adult rheumatoid arthritis. Arthritis Rheum. 1991;34:1310–1312. doi: 10.1002/art.1780341016. [DOI] [PubMed] [Google Scholar]
- Hadj Kacem H, Kaddour N, Adyel FZ, Bahloul Z, Ayadi H. HLA-DQB1 CAR1/CAR2, TNFa IR2/IR4 and CTLA-4 polymorphisms in Tunisian patients with rheumatoid arthritis and Sjogren's syndrome. Rheumatology (Oxford) 2001;40:1370–1374. doi: 10.1093/rheumatology/40.12.1370. [DOI] [PubMed] [Google Scholar]
- Seidl C, Donner H, Petershofen E, et al. An endogenous retroviral long terminal repeat at the HLA-DQB1 gene locus confers susceptibility to rheumatoid arthritis. Hum Immunol. 1999;60:63–68. doi: 10.1016/s0198-8859(98)00095-0. [DOI] [PubMed] [Google Scholar]
- Mok JW, Lee YJ, Kim JY, et al. Association of MICA polymorphism with rheumatoid arthritis patients in Koreans. Hum Immunol. 2003;64:1190–1194. doi: 10.1016/j.humimm.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Singal DP, Li J, Zhang G. Microsatellite polymorphism of the MICA gene and susceptibility to rheumatoid arthritis. Clin Exp Rheumatol. 2001;19:451–452. [PubMed] [Google Scholar]
- Alkassab F, Gourh P, Tan FK, et al. An allograft inflammatory factor 1 (AIF1) single nucleotide polymorphism (SNP) is associated with anticentromere antibody positive systemic sclerosis. Rheumatology (Oxford) 2007;46:1248–1251. doi: 10.1093/rheumatology/kem057. [DOI] [PubMed] [Google Scholar]
- Arvanitis DA, Flouris GA, Spandidos DA. Genomic rearrangements on VCAM1, SELE, APEG1and AIF1 loci in atherosclerosis. J Cell Mol Med. 2005;9:153–159. doi: 10.1111/j.1582-4934.2005.tb00345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Yano M, Doki Y, et al. Injection of LPS causes transient suppression of biological clock genes in rats. J Surg Res. 2008;145:5–12. doi: 10.1016/j.jss.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Callaway JL, Marks SM, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40:949–951. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- Yamada R, Yamamoto K. Mechanisms of disease: genetics of rheumatoid arthritis--ethnic differences in disease-associated genes. Nat Clin Pract Rheumatol. 2007;3:644–650. doi: 10.1038/ncprheum0592. [DOI] [PubMed] [Google Scholar]
- Kanazawa S, Ota S, Sekine C, et al. Aberrant MHC class II expression in mouse joints leads to arthritis with extraarticular manifestations similar to rheumatoid arthritis. Proc Natl Acad Sci USA. 2006;103:14465–14470. doi: 10.1073/pnas.0606450103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JM, Watanabe K. Cell adhesion molecules in inflammation and immunity: relevance to periodontal diseases. Crit Rev Oral Biol Med. 1994;5:91–123. doi: 10.1177/10454411940050020301. [DOI] [PubMed] [Google Scholar]
- Vreugdenhil GR.Enteroviruses and type 1 diabetes mellitus putative pathogenic pathways Dissertation 2001 , http://hdl.handle.net/2066/19000 .
- Schett G, Zwerina J, Firestein G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis. 2008;67:909–916. doi: 10.1136/ard.2007.074278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JM, Moore TL. A role for the complement system in rheumatoid arthritis. Curr Pharm Des. 2005;11:655–670. doi: 10.2174/1381612053381936. [DOI] [PubMed] [Google Scholar]
- Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime. Trends Immunol. 2007;28:184–192. doi: 10.1016/j.it.2007.02.006. [DOI] [PubMed] [Google Scholar]
- van der Pouw Kraan TC, Wijbrandts CA, van Baarsen L, et al. Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: assignment of a type I interferon signature in a subpopulation of patients. Ann Rheum Dis. 2007;66:1008–1014. doi: 10.1136/ard.2006.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorska MM, Cen O, Liang Q, Stafford SJ, Alam R. Differential regulation of interleukin 5-stimulated signaling pathways by dynamic. J Biol Chem. 2006;281:14429–14439. doi: 10.1074/jbc.M512718200. [DOI] [PubMed] [Google Scholar]
- Bouros D. Sexy and 17: two novel pathways in immune regulation. Pneumon. 2007;20:216–218. [Google Scholar]
- Chang SK, Mihalcik SA, Jelinek DF. B lymphocyte stimulator regulates adaptive immune responses by directly promoting dendritic cell maturation. J Immunol. 2008;180:7394–7403. doi: 10.4049/jimmunol.180.11.7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara T, Kato T. Nutritional biochemistry: a new redox-cofactor vitamin for mammals. Nature. 2003;422:832. doi: 10.1038/422832a. [DOI] [PubMed] [Google Scholar]
- Cariappa A, Pillai S. Antigen-dependent B-cell development. Curr Opin Immunol. 2002;14:241–249. doi: 10.1016/s0952-7915(02)00328-x. [DOI] [PubMed] [Google Scholar]
- Ullrich O, Schneider-Stock R, Zipp F. Cell-cell communication by endocannabinoids during immune surveillance of the central nervous system. Results Probl Cell Differ. 2006;43:281–305. doi: 10.1007/400_015. [DOI] [PubMed] [Google Scholar]
- Quah BJ, Barlow VP, McPhun V, Matthaei KI, Hulett MD, Parish CR. Bystander B cells rapidly acquire antigen receptors from activated B cells by membrane transfer. Proc Natl Acad Sci USA. 2008;105:4259–4264. doi: 10.1073/pnas.0800259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koukouritaki SB, Tamizuddin A, Lianos EA. Enhanced expression of the cytoskeleton-associated proteins paxillin and focal adhesion kinase in glomerular immune injury. J Lab Clin Med. 1999;134:173–179. doi: 10.1016/s0022-2143(99)90123-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.