

Abstract
Fibronectin (FN)1, a ubiquitous glycoprotein that plays critical roles in physiologic and pathologic conditions, undergoes alternative splicing which distinguishes plasma FN (pFN) from cellular FN (cFN). Although both pFN and cFN can be incorporated into the extracellular matrix, a distinguishing feature of cFN is the inclusion of an alternatively spliced exon termed EDA (for Extra Type III Domain A). The molecular steps involved in EDA splicing are well-characterized, but pathways influencing EDA splicing are less clear. We have previously found an obligate role for inhibition of the tumor suppressor phosphatase and tensin homologue on chromosome ten (PTEN), the primary regulator of the PI3K/Akt pathway, in fibroblast activation. Here we show TGF-β, a potent inducer of both EDA splicing and fibroblast activation, inhibits PTEN expression and activity in mesenchymal cells, corresponding with enhanced PI3K/Akt signaling. In pten−/− fibroblasts, which resemble activated fibroblasts, inhibition of Akt attenuated FN production and decreased EDA alternative splicing. Moreover, inhibition of mammalian target of rapamycin (mTOR) in pten−/− cells also blocked FN production and EDA splicing. This effect was due to inhibition of Akt-mediated phosphorylation of the primary EDA splicing regulatory protein SF2/ASF. Importantly, FN silencing in pten−/− cells resulted in attenuated proliferation and migration. Thus, our results demonstrate that the PI3K/Akt/mTOR axis is instrumental in FN transcription and alternative splicing, which regulates cell behavior.
Keywords: Fibronectin, alternative splicing, PTEN, fibrosis, fibroblast, rapamycin
INTRODUCTION
Fibronectin (FN) is a ubiquitously expressed, multi-domain glycoprotein that is critical for development and that is instrumental in numerous physiologic and pathologic processes. FN is found in two different forms: a circulating form produced by hepatocytes and found in the plasma compartment (“plasma FN” or pFN), and a form produced by fibroblasts and other cells (“cellular FN” or cFN); both forms may be incorporated into insoluble matrix, although cFN is not typically observed circulating in plasma. Although potentially over 20 separate isoforms of FN can be generated from a single gene, one distinctive feature of cFN is the inclusion of at least one of two extra exons, termed Extra Type III Domain A (EDA) and Extra Type III Domain B (EDB), by alternative exon splicing (reviewed in [1]). We have recently found that EDA cFN is critical for fibroblast activation and the development of experimental pulmonary fibrosis [2]. In humans, pulmonary fibrosis is a progressive and often fatal disorder with no known effective therapeutic options. Thus, identifying determinants of FN expression and EDA cFN splicing may inform possible therapeutic targets.
The phosphatidylinositol-3-kinase (PI3K) pathway is a major downstream mediator of growth factor signaling [3]. Following receptor ligation by growth factors, PI3K is phosphorylated and activated, resulting in the generation of 3′-phosphorylated inositol lipid second messengers from membrane lipids. Of the generated phospholipids, phosphatidylinositol-3,4,5-trisphosphate (PIP3) is a major constituent [4]. In fibroblasts, PIP3 binds to pleckstrin-homology (PH) domains in a number of signaling proteins, including protein kinase B (PKB)/Akt, resulting in myriad and diverse phenotypic responses. For example, PI3K/Akt activation in chick embryonic fibroblasts results in actin filament remodeling and cellular migration [5]. In normal human lung fibroblasts, PI3K/Akt activity promotes Type I collagen mRNA stabilization [6]. The activity of PI3K is antagonized directly by phosphatase and tensin homologue on chromosome 10 (PTEN), a tumor suppressor protein that dephosphorylates PIP3 at the D3 position of the inositol ring [7], thereby negating PI3K effects. In addition to growth factor stimulation, intrinsic PI3K activity is also augmented by insulin receptor substrate-1 (IRS-1). We have previously shown a role for the PI3K/Akt pathway in fibroblast activation, proliferation, and collagen production since genetic and acquired losses of PTEN (which result in unopposed PI3K activity) lead to myofibroblast differentiation [8] and fibroblast migration [9].
Evidence suggests that the mammalian target of rapamycin (mTOR) pathway functions both upstream and downstream of PI3K/Akt to regulate growth factor and nutrient-induced cellular responses [10]. In complex with rictor, mTOR phosphorylates Akt at S473, priming it for full activation by subsequent phosphorylation. In complex with raptor, mTOR can be activated by Akt and, via a feedback loop, can inhibit PI3K activity. The raptor-mTOR complex, also called mTOR complex 1 (mTORC1), is sensitive to the effects of rapamycin, an immunosuppressive macrolide antibiotic used for the prevention of solid organ transplantation rejection. In contrast, rictor-mTOR complex (mTORC2) is insensitive to acute rapamycin treatment, although chronic rapamycin treatment may inhibit its function as well.
Numerous studies demonstrate that FN stimulates growth, matrix metalloproteinase secretion, and cell survival via a PI3K/Akt/mTOR pathway [11–13]. However, evidence of PI3K/Akt-induced FN expression is less well-defined. Interestingly, experimental data also suggest that Akt activity directly influences alternative FN splicing [14]. Given the direct role of both PTEN and EDA cFN in fibroblast activation and lung fibrosis [2, 8], we undertook these experiments to test the hypothesis that PTEN influences FN production and alternative splicing via Akt/mTOR activity.
MATERIALS AND METHODS
Cells and media
Wild-type (WT) C57Bl/6 mouse embryonic fibroblasts (mEF) were from ATCC (Rockville, MD). Embryonic mouse fibroblasts lacking both pten (pten−/−) alleles were described previously [15]. IMR-90 (diploid normal human fetal lung fibroblasts) cells were obtained from the Coriell Institute for Medical Research (Camden, NJ). Cells were maintained in DMEM with 10% fetal calf serum (FCS), antibiotics, glutamine, and HEPES.
Reagents and plasmids
The PI3K inhibitor LY294002 was from Cell Signaling Technologies (Beverly, MA). Human TGF-β1 was from R&D (Minneapolis, MN). Akt inhibitor X was from Calbiochem (San Diego, CA). pGL3-FN-luc encodes a 1.2 kb fragment of the human fibronectin promoter, including several regulatory elements, and was described previously [16], and the pRL-CMV plasmid was purchased from Promega (Madison, WI). The plasmids pSVED-mEDA and pSVED-ΔA were previously described [17]. Unless otherwise noted, all other reagents were from Sigma (St. Louis, MO).
Antibodies
The following antibodies were utilized for experiments: anti-S473 phospho-Akt and anti-Akt (Cell Signaling Technology); anti-FN (H-300; Santa Cruz Biotechnology, Santa Cruz, CA); anti-EDA cFN (clone 3E2, Sigma); anti-SF2/ASF and anti-SR protein (Invitrogen, Carlsbad, CA).
Luciferase assays
Luciferase assays were performed as we have previously published [2] with modifications. Fibroblasts were co-transfected with pGL3-FN-luc and pRL-CMV using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. Transfected cells were treated under the indicated conditions for 48 hours then lysed using Dual Luciferase Assay Lysis Buffer (Promega, Madison, WI). All lysates were stored at −20 °C until assayed in a standard dual-wavelength luminometer. Results are reported as relative luminescence (± SD) compared to the control condition, and are normalized based on Renilla luciferase expression.
PTEN small interfering (si)RNA
PTEN and control siRNA was purchased from Cell Signaling Technology as a kit (Cat# 6250). IMR-90 cells were transfected with control or PTEN siRNA constructs following the manufacturer’s instructions for 48 hours prior to lysis and harvesting of cellular proteins for Western blotting, as we have previously described [18].
FN short hairpin (sh)RNA
Fibronectin 1 and control shRNA bacterial glycerol stocks (XM_129845 Mission® shRNA) were purchased from Sigma. Pten−/− cells were grown to approximately 60% confluence prior to transfection with shRNA constructs utilizing ESCORT II Transfection reagent (Sigma) following the manufacturer’s instructions. Transfected cells were selected with puromycin, and stably transfected cells were subsequently maintained in puromycin.
Proliferation Assay
Cell proliferation was assessed using a commercially available kit (CyQuant NF Proliferation Assay Kit, Invitrogen) following the manufacturer’s instructions. Briefly, 8×103 cells were plated per well in a 96-well plate. Following overnight adhesion, media were removed and dye binding solution was added to each well. The plate was covered and incubated for 60 minutes at 37 °C followed by assessing mean fluorescence intensity (excitation: 485 nm, emission: 530 nm).
Scratch Assay
Cell migration was assessed using a standard scratch assay as we have previously described [19]. Briefly, cells were plated in 6-well plates until confluent. A single scratch was made in the cell monolayer with a sterile 200 μl pipette tip and the cell layer washed thoroughly with serum-free media. The scratch area was measured every 2 hours using an inverted microscope (Nikon) and measured using ImageJ software (version 1.31, NIH). Results are reported as scratch area as a percentage of the original scratch area.
Western blot
Western blot was performed as we have previously reported [2]. Densitometry of visualized bands on gels was performed using Image J software.
Semi-quantitative and conventional RT-PCR
Semi-quantitative RT-PCR was carried out with primers and probes used to detect endogenous total FN, EDA FN, and GAPDH as reported previously [2]. Primers specific for the pSVED-mEDA mini-gene EDA+ and EDA− products were as previously published [17] and were used in conventional PCR reactions to amplify only the pSVED-mEDA construct. PTEN primers for real-time PCR are as we have previously reported [20].
Statistical Analysis
All experiments were performed a minimum of 3 times. Statistical analyses were performed using GraphPad Prism 5.02 (GraphPad Software, La Jolla, CA). Data are presented as mean ± SD unless otherwise noted. Differences between groups were evaluated using Student’s t test. For multiple comparisons, one-way ANOVA with Bonferroni’s post-test analysis was used. Data were considered significant at p<0.05.
RESULTS
PTEN downregulation promotes EDA cFN production via unopposed PI3K/Akt stimulation
Our prior data demonstrated decreased PTEN expression in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis [20] and that these PTEN-deficient cells localize to the active site of matrix synthesis, the fibroblastic focus [8]. Thus we sought to determine if PTEN downregulation would alter FN expression and/or EDA splicing.
Utilizing pten−/− murine embryonic fibroblasts or wild-type (WT) controls, we first examined FN protein expression by Western blot. Consistent with previous data suggesting a PI3K-dependent effect on FN expression [21], we found a marked increase in basal FN protein in pten−/− cells compared to WT cells (Figure 1A). That this effect was dependent on the PI3K/Akt pathway was evidenced by our observation that pharmacologic blockade of PI3K (using LY294002) or of Akt (using Akt inhibitor X) decreased FN levels in pten−/− cells (Figure 1B). Additionally, normal lung fibroblasts treated with PTEN siRNA also demonstrated enhanced FN levels when compared to control siRNA-treated fibroblasts (Figure 1C). The lack of PTEN expression in pten−/− cells, and the downregulation of PTEN in siRNA-treated cells, was confirmed by Western blot (Figure 1D). Upon reduction of PTEN activity we confirmed that Akt activity, measured by levels of S473 phospho-Akt on Western blot, was augmented (Figure 1C).
Figure 1. Enhanced FN protein levels following loss or inhibition of PTEN.
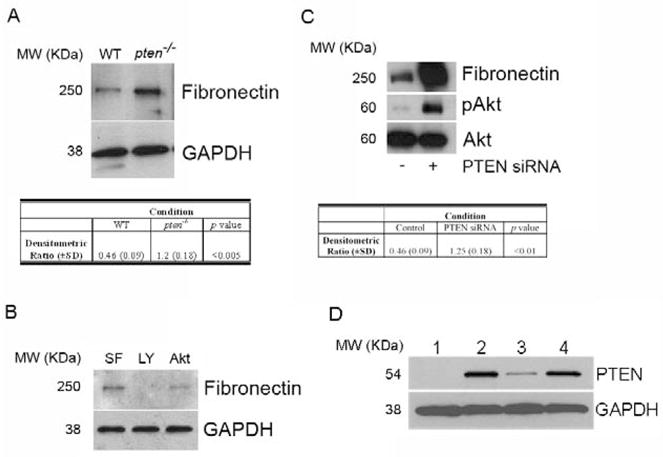
(A) FN protein level is enhanced in pten−/− cells compared to WT cells. Cells were serum-starved for 24 hours prior to analysis of FN level levels by Western blot. Blots were stripped and re-probed for GAPDH as a loading control. Results are representative of 3 separate blots. MW (KDa) refers to the molecular weight of the identified protein in KDa. Table reflects densitometric ratio (± SD) of FN to GAPDH in repeated samples. The difference is statistically significant (P<0.005). (B) The PI3K/Akt pathway mediates FN levels in pten−/− cells. Serum-starved pten−/− cells were left untreated in serum-free media (SF), or were treated with the PI3K inhibitor LY294002 (LY) or the Akt inhibitor X (Akt) for 24 hours prior to harvesting for FN levels by Western blot. Blots were stripped and re-probed for GAPDH as a loading control. Data shown are representative of 3 separate experiments. MW (KDa) refers to the molecular weight of the identified protein in KDa. (C) Pten knockdown in normal lung fibroblasts enhances FN protein level. IMR-90 cells were transiently transfected with PTEN siRNA or scrambled control construct for 24 hours, and protein lysates were assessed by Western blot. Phospho-Akt was probed to confirm functional suppression of PTEN activity. Total Akt was assessed as a loading control. Table reflects densitometric ratio (± SD) of FN to Total Akt in repeated samples. The difference is statistically significant (P<0.01). Data shown are representative blots of 3 separate experiments. MW (KDa) refers to the molecular weight of the identified protein in KDa. (D) PTEN protein level, as assessed by Western blot, in the various cells used in the experiments. The blots were stripped and re-probed for GAPDH as a loading control. Lane 1 = pten−/− cells, Lane 2 = IMR-90 cells, Lane 3 = IMR-90 with PTEN siRNA, Lane 4 = IMR-90 with control siRNA. Blot is representative of a minimum of 3 separate experiments. MW (KDa) refers to the molecular weight of the identified protein in KDa.
Pten−/− cells express markedly elevated S473 Akt phosphorylation due to a loss of PI3K activity regulation [15]. Based on previous data demonstrating a role for Akt in EDA exon splicing [14], we next assessed pten−/− cells for EDA cFN protein levels and noted an increase in EDA cFN protein by Western blotting (Figure 2A). Similar increases in EDA cFN protein levels were seen in normal lung fibroblasts treated with PTEN siRNA (Figure 2B).
Figure 2. Enhanced EDA cFN protein production following loss or downregulation of PTEN.

(A) Western blot analysis of EDA cFN protein level in serum-starved pten−/− vs. WT cells. Blots were stripped and re-probed for GAPDH as a loading control. Western blot experiments were performed 3 times. MW (KDa) refers to the molecular weight of the identified protein in KDa. Table reflects densitometric ratio (± SD) of EDA cFN to GAPDH in repeated samples. The difference is statistically significant (P<0.005). (B) PTEN knockdown in normal lung fibroblasts enhances EDA cFN protein level. IMR-90 cells were transiently transfected with PTEN siRNA or scrambled control construct for 24 hours, and protein lysates were assessed by Western blot for EDA cFN. GAPDH was assessed as a loading control. MW (KDa) refers to the molecular weight of the identified protein in KDa. Table reflects densitometric ratio (± SD) of EDA cFN to GAPDH in repeated samples. The difference is statistically significant (P<0.01). The blot is representative of 3 independently-performed experiments.
In agreement with the data from Blaustein et al [14], we found that pten−/− cells transfected with an EDA mini-gene reporter construct driven by the constitutive α-globin promoter (pSVED-mEDA) [17, 22] showed a five-fold increase in incorporation of the EDA exon into the mini-gene-derived mRNA (expressed as the EDA+/EDA− ratio) compared to WT cells in which the EDA+/EDA− ratio was 0.22 (Figure 3A). Transfection efficiency was monitored by use of a mutated mini-gene that lacks a 6 base-pair sequence of the exonic splicing enhancer (pSVED-ΔA) which renders EDA incorporation impossible ([17], not shown). Confirming the dependence of EDA cFN splicing on the PI3K/Akt pathway, we found that pharmacologic inhibition of Akt in pten−/− cells resulted in a decrease in EDA cFN splicing (Figure 3B).
Figure 3. EDA alternative splicing is regulated by PI3K/Akt.
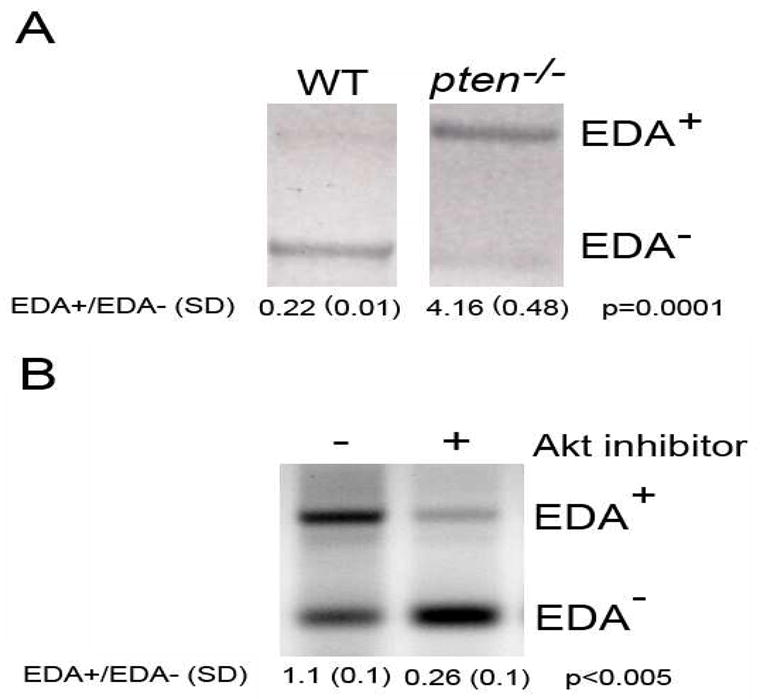
(A) Reverse transcription followed by PCR analysis of mini-gene EDA+ and EDA− mRNA isoforms in WT and pten−/− cells. Densitometric analysis reveals an EDA+/EDA− ratio of 4.18 (±0.48) in pten−/− cells compared to 0.22 (±0.01) in WT cells. Results are representative of 3 independently-performed experiments. The EDA+/EDA- ratio (±SD) is noted below the blots. The difference is statistically significant (p=0.0001). (B) Reverse transcription followed by PCR analysis of mini-gene EDA+ and EDA− mRNA isoforms in pten−/− cells treated with or without Akt inhibitor. Densitometry of EDA splicing showed that pten−/− cells receiving Akt inhibitor have significantly lower EDA splicing than control-treated cells. The EDA+/EDA- ratio (±SD) is noted below the blots. The difference is statistically significant (p<0.005). Results are representative of 3 separate experiments.
TGF-β inhibits PTEN expression and activity in fibroblasts
Evidence suggests that tissue fibrosis occurs as a result of the pro-fibrotic effects of the pleiotropic cytokine transforming growth factor-β (TGF-β). Interestingly, TGF-β also regulates PTEN expression in epithelial cells (evidenced by the alternative name for PTEN, TEP-1 [TGF-β-regulated, epithelial-cell enriched phosphatase] [23]). Therefore, we asked whether TGF-β stimulation of fibroblasts also resulted in downregulation of PTEN expression and/or activity in fibroblasts. Using real-time PCR, we found that WT mEF cells treated with TGF-β (2 ng/ml, a dose commonly used to induce myofibroblast differentiation [8]) demonstrated a significant and sustained decrease in PTEN gene expression over 48 hours (Figure 4A). Although TGF-β regulation of PTEN is well-established in epithelial cells, our data are among the first to show a direct TGF-β-stimulated regulatory effect on PTEN in mesenchymal cells [24]. Further, TGF-β-induced decrease in PTEN expression directly correlated with a marked increase in S473 phosphorylation on Akt, consistent with an associated decrease in PTEN activity (Figure 4B). These data indicate that TGF-β stimulation of fibroblasts inhibits PTEN expression and activity.
Figure 4. TGF-β suppresses PTEN mRNA and protein expression in mesenchymal cells.

(A) WT murine embryonic fibroblasts were serum-starved for 24 hours prior to treatment with TGF-β (2 ng/ml) for the indicated times, and pten mRNA expression was monitored by real-time PCR (expressed as mean ± SD). TGF-β significantly suppressed PTEN expression through 48 hours. Results represent pooled data from 3 separate experiments. *p<0.001, **p<0.01. (B) Serum-starved human lung fibroblasts were treated in the presence or absence of TGF-β (2 ng/ml) for 24 hours. Whole-cell lysates were probed for S473 phospho-Akt demonstrating a robust induction with TGF-β. The membrane was stripped and re-probed for total Akt as a loading control. MW (KDa) refers to the molecular weight of the identified protein in KDa. Results are representative of 4 separate experiments.
mTOR regulates FN expression and EDA splicing
Akt is a serine-threonine kinase that phosphorylates a number of cytoplasmic and nuclear proteins to regulate varied cellular processes such as migration [25], apoptosis [15], proliferation [26], and gene transcription [27]. Interestingly, Akt also influences EDA exon splicing by direct phosphorylation of the primary splicing regulatory (SR) protein SF2/ASF [14]. Although Akt activity is induced following PDK-1-mediated phosphorylation of Thr308 in the activation loop [28], it first requires phosphorylation of Ser473 by the mammalian target of rapamycin (mTOR)-rictor complex [29] to attain maximal activity. The mTOR-rictor complex does not appear to be rapamycin-sensitive, although another complex of mTOR with raptor [30] is susceptible to the effects of rapamycin.
We reasoned that rapamycin inhibition of mTOR would inhibit FN and EDA cFN protein expression that would be due in part to effects on EDA mRNA splicing. To test this, pten−/− and WT cells were treated with or without rapamycin (500 nM) for 24 hours prior to assessing both total and EDA cFN protein production by Western blot. Interestingly, we found that WT fibroblasts treated with rapamycin demonstrated a substantial increase in FN and EDA cFN protein levels, whereas the opposite effect was observed in pten−/− cells (Figure 5A). Similarly, normal human lung fibroblasts treated with rapamycin also showed enhanced basal total and EDA cFN levels (Figure 5B, lanes 1 and 3). In contrast, rapamycin inhibited total and EDA cFN protein levels in normal lung fibroblasts following TGF-β stimulation (Figure 5B, lanes 2 and 4). To determine whether rapamycin treatment of pten−/− and WT cells preferentially affected EDA splicing, we performed quantitative RT-PCR experiments for the endogenous EDA mRNA. When normalized to total fibronectin on the same samples, we found that rapamycin treatment resulted in a significant decrease in EDA-containing mRNA in pten−/− cells (p=0.001; Figure 5C). In contrast, WT cells responded to rapamycin treatment with a substantial and significant increase in EDA-containing mRNA transcripts (p<0.0001; Figure 5C). Consistent with our prior data (Figure 2A), we also found that basal EDA-containing mRNA transcript level in pten−/− cells was significantly greater than in WT cells (p<0.0001; Figure 5C). These data indicate that PTEN activity is a determinant of the inhibitory effects of rapamycin on FN production and on splicing of the EDA exon. Moreover, these results suggest that in the absence of enhanced Akt activity, rapamycin has a stimulatory effect on FN production and EDA exon splicing in mesenchymal cells.
Figure 5. Rapamycin inhibits FN mRNA, FN protein, and endogenous EDA-containing mRNA in pten-deficient cells, but not in WT cells.

(A) Pten−/− or WT cells were treated with or without rapamycin for 24 hours prior to lysis and analysis by Western blot. In pten−/− cells, rapamycin decreased both total and EDA cFN protein production; in contrast, rapamycin enhanced total and EDA cFN protein production in WT cells. MW (KDa) refers to the molecular weight of the identified protein in KDa. The blot is representative of 3 separate experiments. (B) Human fetal lung fibroblasts demonstrate enhanced FN and EDA cFN protein expression with rapamycin treatment (lane 3) compared to untreated cells (lane 1). TGF-β1-stimulated fibroblasts, however, demonstrate decreased FN and EDA cFN protein in the presence of rapamycin (lane 4) compared to cells stimulated with TGF-β1 alone (lane 2). MW (KDa) refers to the molecular weight of the identified protein in KDa. The blot is representative of 3 separately-performed experiments. (C) Rapamycin enhances EDA exon splicing and incorporation into full-length FN in WT cells, but not pten−/− cells. EDA-containing mRNA was normalized to total FN expression to determine the relative effect of rapamycin on EDA splicing. WT (open bars) and pten−/− (closed bars) cells were stimulated with or without rapamycin for 24 hours, and RNA harvested for real-time PCR evaluation of endogenous EDA. Whereas rapamycin treatment increased EDA-containing mRNA significantly in WT cells, it decreased EDA-containing mRNA in pten−/− cells. Note that levels of EDA-containing mRNA in pten−/− cells is approximately 2-fold higher than in WT cells, consistent with our previous findings. Data are pooled from three independent RNA samples performed in triplicate wells. *p=0.001, **p<0.0001.
SF2/ASF phosphorylation is enhanced in pten−/− cells compared to WT cells
Prior study has shown that Akt can directly phosphorylate the major EDA splicing SR protein SF2/ASF to influence EDA splicing [14]. Since pten−/− cells possess enhanced EDA splicing, we next questioned whether this was due to effects on SF2/ASF expression or activity. Thus, we first assayed pten−/− and WT cell lysates for SF2/ASF levels, both in the presence and absence of rapamycin. We found no difference in whole-cell expression of SF2/ASF between the cell types (Figure 6A), implying that SF2/ASF levels are not regulated by the PTEN/Akt axis. We next sought to determine whether SF2/ASF activity was altered between the cell types. Using immunoprecipitation of phosphorylated SR proteins using anti-pSR protein antibody (clone 1H4, which recognizes only phosphorylated SR proteins [31]) and subsequent immunoblotting with anti-SF2/ASF antibody, we noted enhanced SF2/ASF phosphorylation in pten−/− cells compared to WT cells (Figure 6B, compare lanes 1 and 3). Moreover, we confirmed that rapamycin treatment resulted in decreased phosphorylation of SF2/ASF in pten−/− cells, whereas phosphorylation was enhanced in WT cells. Immunoprecipitation of equal amounts of nuclear lysates with anti-SF2/ASF and subsequent blotting for SF2/ASF confirmed equal protein immunoprecipitation (Figure 6B).
Figure 6. Rapamycin treatment decreases SF2/ASF phosphorylation in pten-deficient cells.
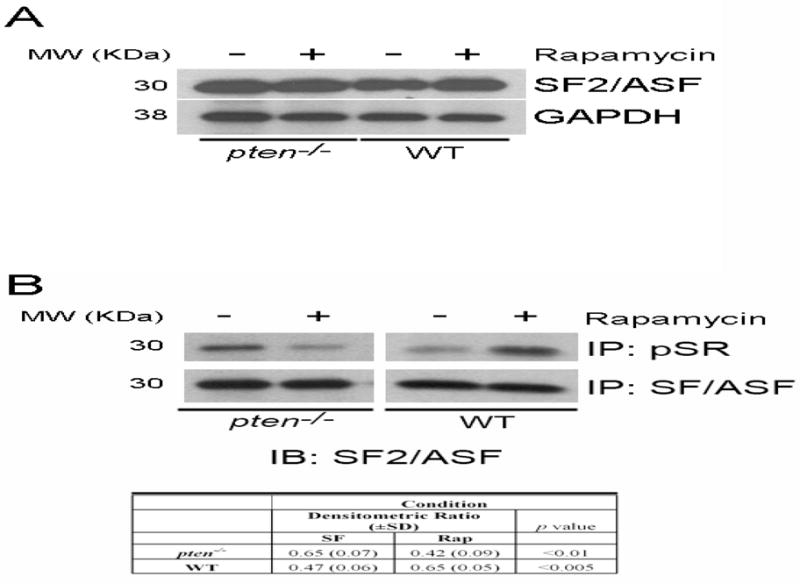
(A) Equivalent expression of SF2/ASF levels in pten−/− and WT cells, which is unaffected by rapamycin treatment. GAPDH is a loading control. MW (KDa) refers to the molecular weight of the identified protein in KDa. Results are representative of 3 independent experiments. (B) Nuclear lysates of pten−/− or WT cells treated with or without rapamycin were immunoprecipitated with anti-SF2/ASF antibody or anti-pSR antibody, which only recognizes phosphorylated SR proteins. Subsequent immunoblotting reveals decreased SF2/ASF phosphorylation in pten−/− cells treated with rapamycin, despite equal protein immunoprecipitation. Conversely, WT cells demonstrate increased phosphorylation on rapamycin stimulation. The table reflects densitometric ratio (± SD) of immunoprecipitated SR protein bands to immunoprecipitated SF2/ASF in repeated samples. The differences before and after rapamycin are statistically significant. Note also that pten−/− cells express higher basal phosphorylation of SRP compared to WT cells. MW (KDa) refers to the molecular weight of the identified protein in KDa. Data are representative of 3 separate experiments.
Fibronectin silencing in pten−/− cells attenuates proliferation and migration
In order to determine the functional effect of decreased FN production in pten−/− cells, we inhibited FN production using shRNA technology. Following stable transfection of pten−/− cells with either FN or control shRNA (as described in Materials and Methods, we first confirmed FN knockdown in our cells. As shown in Figure 7A, FN shRNA transfection resulted in robust downregulation of FN expression, whereas control shRNA had no effect. Subsequently, we evaluated the effect of FN knockdown on fibroblast proliferation and migration. As shown in Figure 7B, pten−/− cells transfected with FN shRNA demonstrated a ~43% reduction in proliferation compared to control cells (p<0.005). Moreover, FN silencing in pten−/− cells resulted in slower migratory capacity compared to control cells (Figure 7C), suggesting that in pten−/− cells FN is necessary for full effects on proliferation and migration.
Figure 7. FN knockdown inhibits pten−/− fibroblast proliferation and migration.
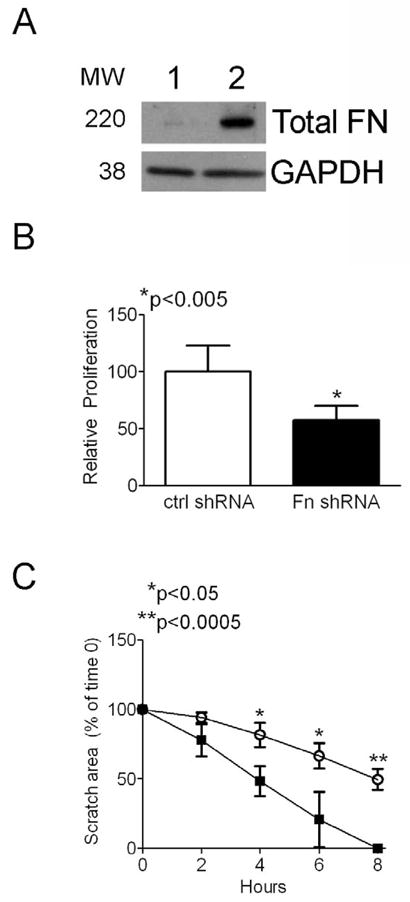
(A) FN shRNA (Lane 1) stable transfection attenuates FN production in pten−/− cells, whereas control shRNA (Lane 2) has no effect. GAPDH is a loading control. MW denotes molecular weights of bands. (B) Control and FN shRNA-transfected pten−/− cells were assessed for proliferation using a commercially-available kit as described in Materials and Methods. FN knockdown resulted in an approximately 43% reduction in fibroblast proliferation compared to control shRNA-treated cells. Results are representative of 2 separate experiments. *p<0.005. (C) pten−/− fibroblast migration is attenuated following FN knockdown. Cells plated to confluence were scratched with a sterile 200 μl pipette tip and the wound area was measured every 2 hours. FN shRNA-treated fibroblasts (open circles) migrated significantly more slowly than control shRNA-transfected cells (closed squares). Results are representative of 2 separate experiments performed in triplicate wells. *p<0.05. **p<0.0005.
DISCUSSION
In this manuscript, we demonstrate that the PI3K/Akt/mTOR pathway is capable of FN regulation by two distinct methods: by controlling FN protein levels as well as by influencing alternative splicing of the EDA exon. Although published data have firmly established that cells plated on FN activate the PI3K/Akt pathway resulting in numerous downstream effects [32, 33], our data now suggest that a reciprocal enhancement of FN production and EDA alternative splicing downstream of PI3K/Akt signaling also occurs, suggesting a mechanism whereby mitogenic agents such as FN that enhance PI3K/Akt activity induce further production of FN, thus amplifying the effects on cellular behavior.
We have previously found that in idiopathic pulmonary fibrosis, a lung disorder with no effective therapy, fibroblasts primarily responsible for active collagen synthesis are relatively PTEN deficient [8, 20] and that this accounts for the development of a myofibroblast phenotype [8]. Moreover, evidence in both animals and humans suggests that EDA cFN is deposited locally at sites of active fibrogenesis prior to collagen deposition [34, 35]. In the current series of experiments, we found that relatively enhanced Akt activity due to loss or inhibition of PTEN corresponds with enhanced FN production with an even greater proportion of FN molecules containing the EDA domain, and that both are likely to be important in mediating downstream effects. Further, we found that pharmacologic inhibition of Akt suppresses EDA cFN and total FN protein levels, but only in cells with relatively augmented basal Akt activity. Thus, we speculate that the pro-fibrotic growth factor TGF-β, by inhibiting PTEN expression/activity, results in Akt activation and subsequently enhanced EDA cFN production in fibroblasts. The local production of EDA cFN, which then feeds back in an autocrine/paracrine fashion, is necessary for fibroblast activation following TGF-β stimulation [2, 36].
Previously, Blaustein and colleagues demonstrated that Akt is capable of directly phosphorylating the SR protein SF2/ASF [14], resulting in enhanced alternative splicing of the FN EDA exon. Although the mechanism for this observation was not elucidated, data suggests that SF2/ASF activates mTORC1, resulting in phosphorylation of S6K and elongation initiation factor 4EBP-1 (eIF4EBP-1) [37]. Subsequently, phosphorylation and inactivation of eIF4EBP-1 promotes protein translation by allowing eIF4E to interact with other cap-binding elements to generate an active translation initiation complex [38]. Our data are in line with these findings, and extend it in two important ways. First, we now show that Akt phosphorylation of SF2/ASF is associated with enhanced EDA splicing, and that this effect can be negatively regulated by rapamycin. Second, and perhaps more importantly, we show that rapamycin treatment of WT cells (in which PTEN activity is replete) enhances FN protein levels and EDA-containing mRNA levels, likely by virtue of enhanced SF2/ASF phosphorylation and activity. This finding, although seemingly antithetical, may be explained by the well-described action of rapamycin to enhance PI3K/Akt activity in some cells. It is possible that, in the absence of PTEN, robust constitutive PI3K activity is relatively insensitive to the regulatory effects of S6-kinase 1 (S6K) and insulin receptor substrate-1 (IRS-1) such that rapamycin treatment primarily blocks S6K-mediated FN translation. However, in PTEN-expressing cells with lower basal Akt activity, rapamycin inhibition of S6K enhances IRS-1 activity resulting in augmented PI3K/Akt activity, FN production, and EDA splicing (Figure 8). Based on prior studies showing that SF2/ASF-overexpressing cells are inhibited by rapamycin but not Akt inhibition [37], our data imply that direct inhibition of mTORC1 by rapamycin is the mechanism most likely responsible for our findings. This hypothesis will need to be tested in subsequent studies.
Figure 8. Schematic of the PTEN/PI3K/Akt/mTOR axis in fibroblasts proposed in this manuscript.

mTOR, in complex with rictor and gβL, phosphorylates Akt on S473. Akt is subsequently phosphorylated on T308 by the actions of PDK1 downstream of PI3K. PTEN negatively regulates this activity. Akt directly phosphorylates and activates SF2/ASF to enhance EDA splicing and, through a series of steps involving the tuberous sclerosis complex and Rheb (depicted as the broken arrow), promotes mTOR/raptor/gβL activity. mTOR/raptor/gβL activates p70S6K, which promotes FN protein translation as well as inhibiting insulin receptor substrate-1 (IRS-1) thereby inhibiting PI3K activity. In the absence of PTEN, high basal Akt activity results in augmented EDA splicing and p70S6K-mediated FN expression. The FN expression is blocked by rapamycin. In cells with normal PTEN expression, PI3K activity is more sensitive to IRS-1 regulation and rapamycin in this case augments PI3K/Akt activity.
Our studies indicate that total and EDA cFN protein production, and EDA-containing mRNA level, is attenuated by rapamycin in pten−/− cells. These data are consistent with previous reports showing inhibition of FN production by both inhibitors of PI3K and mTOR in human non-small cell lung cancer [39]. It is unclear whether our observation of increased total and EDA cFN protein production and EDA-containing mRNA transcripts induced by rapamycin in normal mesenchymal cells would have any detrimental biologic effect; however, this should certainly be considered. A recent study demonstrated that a related mTOR inhibitor, everolimus, promoted progression of interstitial fibrosis in the remnant kidney rat model of renal injury [40], further supporting this possibility. Although this particular study did not specifically evaluate lesional FN expression, augmented ECM deposition was observed [40].
Our data also indicate that PI3K/Akt-mediated FN production and EDA-containing mRNA levels are inhibited by rapamycin in pten−/− cells. Since EDA fibronectin is necessary for TGF-β-induced myofibroblast differentiation [36] and for in vivo lung fibrogenesis [2], rapamycin therapy may be beneficial in disorders characterized by TGF-β overexpression, enhanced myofibroblast differentiation, and enhanced ECM production. Concordant with our observations, previous study has demonstrated that the rapamycin analogue SDZ RAD is efficacious in preventing collagen deposition in a rodent model of pulmonary fibrosis [41]. Whether the effect of SDZ RAD was due to a decrease in bleomycin-induced inflammation that predisposes to fibrosis in this model is not clear, although this is known to occur [42]. Based on our data, another possible mechanistic explanation that could be explored is whether SDZ RAD in the aforementioned study reduced FN and EDA splicing that led to diminished collagen production and tissue fibrosis. Based on our previous studies [2] and the data presented herein, we propose that the PI3K/Akt/mTOR pathway may represent a viable therapeutic target for diseases characterized by excessive ECM production downstream of enhanced Akt activation.
Acknowledgments
This work was supported by National Institutes of Health Grant R01 HL085083 and by the Martin E. Galvin Fund for Idiopathic Pulmonary Fibrosis Research and the Quest for Breath Foundation (to E.S.W.).
Footnotes
Abbreviations: FN – fibronectin, EDA – Extra Type III Domain ‘A’, PI3K – phosphatidylinositol-3′-kinase, PKB/Akt – Protein Kinase B/AKT, PTEN – phosphatase and tensin homologue on chromosome 10, mTOR – mammalian target of rapamycin, PIP3 – phosphatidylinositol-3,4,5-trisphosphate, PH – pleckstrin homology, mEF – murine embryonic fibroblast, SR protein – splicing regulatory protein
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.White ES, Baralle FE, Muro AF. New insights into form and function of fibronectin splice variants. J Pathol. 2008;216:1–14. doi: 10.1002/path.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muro AF, Moretti FA, Moore BB, Yan M, Atrasz RG, Wilke CA, Flaherty KR, Martinez FJ, Tsui JL, Sheppard D, Baralle FE, Toews GB, White ES. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177:638–645. doi: 10.1164/rccm.200708-1291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kinashi T, Escobedo JA, Williams LT, Takatsu K, Springer TA. Receptor tyrosine kinase stimulates cell-matrix adhesion by phosphatidylinositol 3 kinase and phospholipase C-gamma 1 pathways. Blood. 1995;86:2086–2090. [PubMed] [Google Scholar]
- 4.Seminario MC, Precht P, Bunnell SC, Warren SE, Morris CM, Taub D, Wange RL. PTEN permits acute increases in D3-phosphoinositide levels following TCR stimulation but inhibits distal signaling events by reducing the basal activity of Akt. Eur J Immunol. 2004;34:3165–3175. doi: 10.1002/eji.200425206. [DOI] [PubMed] [Google Scholar]
- 5.Qian Y, Corum L, Meng Q, Blenis J, Zheng JZ, Shi X, Flynn DC, Jiang BH. PI3K induced actin filament remodeling through Akt and p70S6K1: implication of essential role in cell migration. Am J Physiol Cell Physiol. 2004;286:C153–163. doi: 10.1152/ajpcell.00142.2003. [DOI] [PubMed] [Google Scholar]
- 6.Ricupero DA, Poliks CF, Rishikof DC, Cuttle KA, Kuang PP, Goldstein RH. Phosphatidylinositol 3-kinase-dependent stabilization of alpha1(I) collagen mRNA in human lung fibroblasts. Am J Physiol Cell Physiol. 2001;281:C99–C105. doi: 10.1152/ajpcell.2001.281.1.C99. [DOI] [PubMed] [Google Scholar]
- 7.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 8.White ES, Atrasz RG, Hu B, Phan SH, Stambolic V, Mak TW, Hogaboam CM, Flaherty KR, Martinez FJ, Kontos CD, Toews GB. Negative regulation of myofibroblast differentiation by PTEN (Phosphatase and Tensin Homolog Deleted on chromosome 10) Am J Respir Crit Care Med. 2006;173:112–121. doi: 10.1164/rccm.200507-1058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White ES, Atrasz RG, Dickie EG, Aronoff DM, Stambolic V, Mak TW, Moore BB, Peters-Golden M. Prostaglandin E(2) inhibits fibroblast migration by E-prostanoid 2 receptor-mediated increase in PTEN activity. Am J Respir Cell Mol Biol. 2005;32:135–141. doi: 10.1165/rcmb.2004-0126OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Yue P, Kim YA, Fu H, Khuri FR, Sun SY. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 2008;68:7409–7418. doi: 10.1158/0008-5472.CAN-08-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee BH, Ruoslahti E. alpha5beta1 integrin stimulates Bcl-2 expression and cell survival through Akt, focal adhesion kinase, and Ca2+/calmodulin-dependent protein kinase IV. J Cell Biochem. 2005;95:1214–1223. doi: 10.1002/jcb.20488. [DOI] [PubMed] [Google Scholar]
- 12.Thant AA, Nawa A, Kikkawa F, Ichigotani Y, Zhang Y, Sein TT, Amin AR, Hamaguchi M. Fibronectin activates matrix metalloproteinase-9 secretion via the MEK1-MAPK and the PI3K-Akt pathways in ovarian cancer cells. Clin Exp Metastasis. 2000;18:423–428. doi: 10.1023/a:1010921730952. [DOI] [PubMed] [Google Scholar]
- 13.Han S, Khuri FR, Roman J. Fibronectin stimulates non-small cell lung carcinoma cell growth through activation of Akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways. Cancer Res. 2006;66:315–323. doi: 10.1158/0008-5472.CAN-05-2367. [DOI] [PubMed] [Google Scholar]
- 14.Blaustein M, Pelisch F, Tanos T, Munoz MJ, Wengier D, Quadrana L, Sanford JR, Muschietti JP, Kornblihtt AR, Caceres JF, Coso OA, Srebrow A. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol. 2005;12:1037–1044. doi: 10.1038/nsmb1020. [DOI] [PubMed] [Google Scholar]
- 15.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 16.Michaelson JE, Ritzenthaler JD, Roman J. Regulation of serum-induced fibronectin expression by protein kinases, cytoskeletal integrity, and CREB. Am J Physiol Lung Cell Mol Physiol. 2002;282:L291–301. doi: 10.1152/ajplung.00445.2000. [DOI] [PubMed] [Google Scholar]
- 17.Muro AF, Iaconcig A, Baralle FE. Regulation of the fibronectin EDA exon alternative splicing. Cooperative role of the exonic enhancer element and the 5′ splicing site. FEBS Lett. 1998;437:137–141. doi: 10.1016/s0014-5793(98)01201-0. [DOI] [PubMed] [Google Scholar]
- 18.Sagana RL, Yan M, Cornett AM, Tsui JL, Stephenson DA, Huang SK, Moore BB, Ballinger MN, Melonakos J, Kontos CD, Aronoff DM, Peters-Golden M, White ES. Phosphatase and tensin homologue on chromosome 10 (PTEN) directs prostaglandin E2-mediated fibroblast responses via regulation of E prostanoid 2 receptor expression. J Biol Chem. 2009;284:32264–32271. doi: 10.1074/jbc.M109.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lazar MH, Christensen PJ, Du M, Yu B, Subbotina NM, Hanson KE, Hansen JM, White ES, Simon RH, Sisson TH. Plasminogen activator inhibitor-1 impairs alveolar epithelial repair by binding to vitronectin. Am J Respir Cell Mol Biol. 2004;31:672–678. doi: 10.1165/rcmb.2004-0025OC. [DOI] [PubMed] [Google Scholar]
- 20.White ES, Thannickal VJ, Carskadon SL, Dickie EG, Livant DL, Markwart S, Toews GB, Arenberg DA. Integrin alpha4beta1 regulates migration across basement membranes by lung fibroblasts: a role for phosphatase and tensin homologue deleted on chromosome 10. Am J Respir Crit Care Med. 2003;168:436–442. doi: 10.1164/rccm.200301-041OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H, Quah SY, Dong JM, Manser E, Tang JP, Zeng Q. PRL-3 down-regulates PTEN expression and signals through PI3K to promote epithelial-mesenchymal transition. Cancer Res. 2007;67:2922–2926. doi: 10.1158/0008-5472.CAN-06-3598. [DOI] [PubMed] [Google Scholar]
- 22.Muro AF, Caputi M, Pariyarath R, Pagani F, Buratti E, Baralle FE. Regulation of fibronectin EDA exon alternative splicing: possible role of RNA secondary structure for enhancer display. Mol Cell Biol. 1999;19:2657–2671. doi: 10.1128/mcb.19.4.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- 24.Kato M, Putta S, Wang M, Yuan H, Lanting L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, Rossi JJ, Natarajan R. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11:881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian Y, Zhong X, Flynn DC, Zheng JZ, Qiao M, Wu C, Dedhar S, Shi X, Jiang BH. ILK mediates actin filament rearrangements and cell migration and invasion through PI3K/Akt/Rac1 signaling. Oncogene. 2005 doi: 10.1038/sj.onc.1208525. [DOI] [PubMed] [Google Scholar]
- 26.Zhao H, Dupont J, Yakar S, Karas M, LeRoith D. PTEN inhibits cell proliferation and induces apoptosis by downregulating cell surface IGF-IR expression in prostate cancer cells. Oncogene. 2004;23:786–794. doi: 10.1038/sj.onc.1207162. [DOI] [PubMed] [Google Scholar]
- 27.Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene. 2004;23:8571–8580. doi: 10.1038/sj.onc.1207902. [DOI] [PubMed] [Google Scholar]
- 28.Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol. 1997;7:776–789. doi: 10.1016/s0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- 29.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 30.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 31.Tuma RS, Stolk JA, Roth MB. Identification and characterization of a sphere organelle protein. J Cell Biol. 1993;122:767–773. doi: 10.1083/jcb.122.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han SW, Roman J. Fibronectin induces cell proliferation and inhibits apoptosis in human bronchial epithelial cells: pro-oncogenic effects mediated by PI3-kinase and NF-kappa B. Oncogene. 2006;25:4341–4349. doi: 10.1038/sj.onc.1209460. [DOI] [PubMed] [Google Scholar]
- 34.Kuhn C, McDonald JA. The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am J Pathol. 1991;138:1257–1265. [PMC free article] [PubMed] [Google Scholar]
- 35.Hernnas J, Nettelbladt O, Bjermer L, Sarnstrand B, Malmstrom A, Hallgren R. Alveolar accumulation of fibronectin and hyaluronan precedes bleomycin-induced pulmonary fibrosis in the rat. Eur Respir J. 1992;5:404–410. [PubMed] [Google Scholar]
- 36.Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. 1998;142:873–881. doi: 10.1083/jcb.142.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karni R, Hippo Y, Lowe SW, Krainer AR. The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc Natl Acad Sci U S A. 2008;105:15323–15327. doi: 10.1073/pnas.0801376105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michlewski G, Sanford JR, Caceres JF. The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol Cell. 2008;30:179–189. doi: 10.1016/j.molcel.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 39.Zheng Y, Ritzenthaler JD, Roman J, Han S. Nicotine stimulates human lung cancer cell growth by inducing fibronectin expression. Am J Respir Cell Mol Biol. 2007;37:681–690. doi: 10.1165/rcmb.2007-0051OC. [DOI] [PubMed] [Google Scholar]
- 40.Vogelbacher R, Wittmann S, Braun A, Daniel C, Hugo C. The mTOR inhibitor everolimus induces proteinuria and renal deterioration in the remnant kidney model in the rat. Transplantation. 2007;84:1492–1499. doi: 10.1097/01.tp.0000282866.92367.99. [DOI] [PubMed] [Google Scholar]
- 41.Simler NR, Howell DC, Marshall RP, Goldsack NR, Hasleton PS, Laurent GJ, Chambers RC, Egan JJ. The rapamycin analogue SDZ RAD attenuates bleomycin-induced pulmonary fibrosis in rats. Eur Respir J. 2002;19:1124–1127. doi: 10.1183/09031936.02.00281602. [DOI] [PubMed] [Google Scholar]
- 42.Wang Q, Wang Y, Hyde DM, Gotwals PJ, Lobb RR, Ryan ST, Giri SN. Effect of antibody against integrin alpha4 on bleomycin-induced pulmonary fibrosis in mice. Biochem Pharmacol. 2000;60:1949–1958. doi: 10.1016/s0006-2952(00)00491-3. [DOI] [PubMed] [Google Scholar]