

Abstract
Rationale
Excess signaling through cardiac Gβγ subunits is an important component of heart failure (HF) pathophysiology. They recruit elevated levels of cytosolic G-protein coupled receptor kinase 2 (GRK2) to agonist-stimulated β-adrenergic receptors (β-ARs) in HF, leading to chronic β-AR desensitization and down-regulation; these events are all hallmarks of HF. Previous data suggested that inhibiting Gβγ signaling and its interaction with GRK2 could be of therapeutic value in HF.
Objective
To investigate small molecule Gβγ inhibition in HF.
Methods and Results
We recently described novel small molecule Gβγ inhibitors that selectively block Gβγ binding interactions, including M119 and its highly related analog, gallein. These compounds blocked interaction of Gβγ and GRK2 in vitro and in HL60 cells. Here, we show they reduced β-AR-mediated membrane recruitment of GRK2 in isolated adult mouse cardiomyocytes. Furthermore, M119 enhanced both adenylyl cyclase activity and cardiomyocyte contractility in response to β-AR agonist. To evaluate their cardiac-specific effects in vivo, we initially utilized an acute pharmacologic HF model (30 mg/kg/day isoproterenol, 7 days). Concurrent daily injections prevented HF, and partially normalized cardiac morphology and GRK2 expression in this acute HF model. To investigate possible efficacy in halting progression of pre-existing HF, calsequestrin cardiac transgenic mice (CSQ) with extant HF received daily injections for 28 days. The compound alone halted HF progression, and partially normalized heart size, morphology and cardiac expression of HF marker genes (GRK2, ANF and β-MHC).
Conclusions
These data suggest a promising therapeutic role for small molecule inhibition of pathologic Gβγ signaling in the treatment of HF.
Keywords: G proteins, adrenergic receptor, G-protein coupled receptor kinases, cardiomyopathy, heart failure, cardiomyocyte
Introduction
Heart failure (HF) is a devastating disease with poor prognosis, and remains a leading cause of death worldwide 1, 2. Excess signaling through cardiac G-protein Gβγ subunits is an important component of HF pathophysiology. In particular, they recruit elevated levels of cytosolic G-protein coupled receptor kinase 2 (GRK2, βARK1) to agonist-stimulated β-ARs in HF 3, leading to the chronic β-AR desensitization, down-regulation and pathologic signaling that are hallmarks of HF 4, 5.
Increasing evidence suggests a critical role for Gβγ-mediated signaling in HF. In particular, GRK2 is significantly upregulated in cardiomyocytes of animal models of HF and human HF patients; this elevates Gβγ-GRK2 interactions and contributes to chronic desensitization of β-AR signaling6, 7; interestingly, levels of GRK2 appear to correlate with the severity of HF 6, 8. Enhancing Gβγ-GRK2 interaction by cardiac targeted overexpression of GRK2 (s) can directly cause HF in experimental animal models 9; its genetic ablation has generally proven to be cardioprotective 10-12.
Several studies suggest targeting Gβγ-mediated signaling as an effective treatment for HF. First, studies employing a Gβγ-sequestering peptide derived from the Gβγ-binding domain of GRK2, known as βARKct or GRK2ct, have demonstrated that interfering with Gβγ interactions in a variety of animal models of HF improves cardiac function, normalizes β-AR receptor levels and restores signaling responsiveness 13, 14. Similarly, overexpression of a truncated dominant-negative form of phosducin, another Gβγ-binding protein, can also restore cardiac function when virally delivered to failing rabbit hearts 15. Also, disruption of PI3Kγ recruitment to the membrane-associated Gβγ-GRK2 complex by βARKct or expression of the GRK2-binding PIK domain of PI3Kγ is cardioprotective in a variety of HF models 16-18. These large peptide inhibitor studies highlight the therapeutic potential of Gβγ inhibitors in HF.
We recently developed a novel small molecule targeting strategy to selectively inhibit Gβγ binding interactions. Studies from our laboratories have identified a number of small molecules that inhibit peptide binding to the protein interaction domain of the Gβγ subunit, suggesting possible salutary modulation of pathologic Gβγ-dependent signaling in HF19, 20. Here we demonstrate that two of these compounds, M119 and its highly related, similarly efficacious compound gallein 19, 20, influence β-AR-dependent, Gβγ-mediated signaling in isolated mouse cardiomyocytes. Furthermore, we show that these compounds block the progression of cardiac dysfunction and hypertrophy in two murine models of HF. We conclude that M119 and gallein, and other related Gβγ-targeting compounds, will provide valuable tools for dissecting the functional importance of different Gβγ-protein interactions and may lead to development of Gβγ-targeted small molecule therapeutics for the treatment of HF.
Materials and Methods
Animals
Isoproterenol infusion
32 12-week old C57Bl6-J wild-type male mice (Jackson Laboratories) were divided into four groups of 8 mice each: V-V (Isoproterenol (Iso) vehicle/ M119 vehicle), V-M (Iso vehicle/ M119), I-V (Iso/ M119 vehicle), and I-M (Iso/ M119). M119 or M119 vehicle (1 X PBS, pH 7.7) was delivered at 100 mg/kg/day via daily peritoneal injection (200 mL/ injection). Filtered solutions of Iso or Iso-vehicle (0.002% ascorbic acid in saline) were delivered via implantable mini-osmotic pump at a concentration of 30 mg/kg/day. All animal procedures were performed in accordance with the guidelines of the Department of Laboratory Animal Medicine and the University Committee on Animal Resources at the University of Rochester Medical Center.
Calsequestrin
Ten 8 to 12-week old CSQ overexpressing mice were divided into two groups: Gallein and vehicle. Gallein or vehicle (1 x PBS, pH 7.7) was delivered at 30 mg/kg/day via daily peritoneal injection (200 μl/ injection). All animal procedures were performed in accordance with the guidelines of the Department of Laboratory Animal Medicine and the University Committee on Animal Resources at the University of Rochester Medical Center.
Echocardiography
Transthoracic 2D and M-mode echocardiography analysis was used to assess heart function in trained, conscious mice with a VisualSonics Vevo 770 echocardiography machine equipped with a 30MHz probe (VisualSonics). At least five animals from each group were measured and the pooled data was analyzed for statistical significance.
Myocyte Isolation
Wild type male C57Bl6-J mice aged 12-16 weeks were anesthetized with 0.5 mL heparin (100 U/mL) and 0.5 mL of ketamine/midazolam in saline combination via intraperitoneal injection. Once anesthetized, the heart was removed, immediately suspended on a Langerdorff apparatus by cannulation of the aortic root and perfused at constant rate of 4 mL/min at 37°C starting with 4 min of perfusion buffer (5 mM NaHCO3, 30 mM taurine, 10 mM BDM, 5mM Glucose, pH 7.4). Subsequently, enzymatic digestion was achieved by the infusion of Calcium-free digestion buffer (120mg Collagenase type II in 50 mL perfusion buffer) for 3 min followed by 10 minutes of perfusion with calcium containing digestion buffer (digestion buffer + 40 nM CaCl2). The heart was then removed and placed in a dish filled with 2 mL of stopping buffer (10% FBS, 12.5 μM CaCl2 in perfusion buffer). Following removal of the atria, the ventricles were teased apart and pipetted into small pieces. To remove undigested tissue, the cell suspension was filtered through a 200 μm mesh and allowed to settle by gravity for 10 min at 37°C. The supernatant was discarded and the cardiomyocyte pellet was resuspended in 10 mL of stopping buffer. CaCl2 (100 mM) was added incrementally to a final concentration of 1.2 mM and the cardiomyocytes were again allowed to settle. The final pellet was resuspended in 5 mL of plating medium (MEM, 2.5% FBS, 1X pencillin/streptomycin, 2 mM L-glutamine) and plated onto laminin coated BT-CS glass chamber slides (Cell Micro Controls) for 1-2 hours in a 37°C incubator at 2% CO2 prior to contractility studies.
Myocyte Contractility
The glass chamber slides were placed on a microscope stage (Olympus IX71) connected to a field stimulator specifically designed for driving isolated cardiomyocytes (MyoPacer, IonOptix). Cardiomyocytes were stimulated at 0.5Hz and imaged with a variable field rate camera (MYO100 MyoCam, Ionoptix) using both edge detection and sarcomere length technology. Peak contraction was measured as the percentage of peak cell shortening. Cardiomyocytes were treated with one of 8 solutions: vehicle (control), Iso (1 μM), M119 (10 μM), Gallein (10 μM), propranolol (1 μM), Iso/M119 (1 μM and 10 μM, respectively), propranolol/Iso (1 μM each), or propranolol/M119 (1 μM and 10 μM, respectively) for 10 minutes each. Treatment order was varied between experiments to control for time-dependent changes in cardiomyocyte contractility. All studies were recorded and saved in graphical and numerical formats. Each data point represents cardiomyocytes isolated from a different animal with at least 7 individual cardiomyocytes averaged per treatment and an equal number of contractions (>6) included for each cardiomyocyte.
Additional information and methods for Morphometry/Histology, Protein Preparation, Immunoblotting, RNA extraction, Real-Time PCR, cAMP assay, statistical analysis and small molecule information are all included in Online methods.
Results
Small molecule Gβγ inhibition influences β-AR signaling in isolated cardiomyoctes
We recently identified compounds M119 and gallein as highly related small molecules with similar efficacy that modulate GPCR signaling by interfering with Gβγ binding interactions. M119 and gallein block peptide binding to Gβγ with equal efficacy, they block Gβγ-GRK2 association in vitro and in HL-60 leukocytes in cell culture, and produce equivalent dose-response curves in an inflammatory model in vivo 19, 20 (and Online Figure I). Previous studies employing peptide inhibitors of Gβγ indicate that targeting of β-AR-dependent, Gβγ-mediated signaling can lead to dramatic improvement in cardiac function in diverse models of HF. Here, we tested whether cell-permeant small molecules could influence β-AR signaling in cardiomyocytes and improve cardiac function in murine models of HF. To begin, we examined whether M119 affected basal and β-AR-elicited cAMP generation in adult mouse cardiomyocytes. Elevation of cAMP production elicited by the β-AR agonist isoproterenol (Iso) was significantly enhanced in the presence of M119 (Figure 1A), which also induced a slight increase in cAMP at baseline.
Figure 1. Acute M119 treatment enhances cardiomyocyte β-AR signaling.
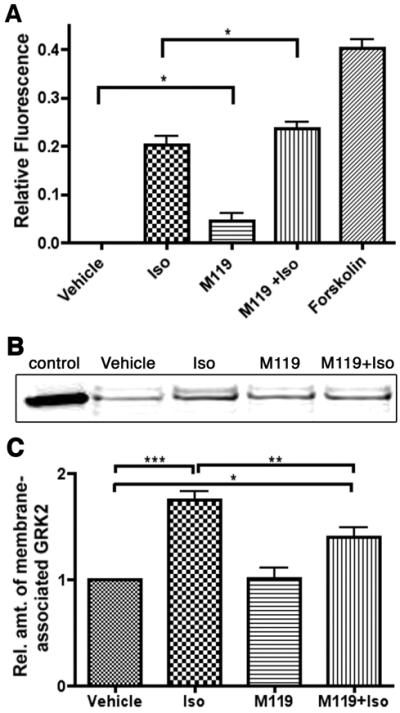
A) Isolated adult cardiomyocytes treated with M119 (10 μM) show enhanced cAMP generation at baseline and in response to Iso stimulation. B, C) M119 (10 μM) interferes with β-AR induced GRK2 membrane recruitment. Representative western blot analysis of GRK2 protein level in membrane fractions of cardiomyocytes treated with vehicle alone, Iso, M119, or M119 and Iso. Positive control lane of proteins from HEK293 cells transfected with GRK2 (B). Densitometric analysis of GRK2 membrane recruitment from four independent experiments demonstrates that M119 leads to ~50% decrease in Iso-induced GRK2 membrane recruitment (C). *P<.05, **P<.01, ***P<.001
Next we examined whether M119 or gallein influence β-AR-mediated GRK2 membrane recruitment. Adult mouse cardiomyocytes from wild type mice were pretreated with 10 μM gallein or vehicle followed by 1 μM Iso or vehicle, after which cardiomyocytes were subjected to subcellular fractionation. Immunoblot analysis of the membrane fraction demonstrated increased GRK2 with Iso alone compared to vehicle treated cells (Figure 1B,C, Online Figure IIA,B). Gallein or M119 alone did not significantly affect the subcellular distribution of GRK2 compared to control treated cells. However, both M119 and gallein reduced β-AR-mediated GRK2 membrane translocation, as cells treated with either compound as well as Iso show only ~1.3 fold increase in membrane bound GRK2 compared to control cells (Figure 1B,C, Online Figure IIA,B). Thus, consistent with our prior in vitro studies using purified proteins and in HL-60 leukocytes stimulated with fMLP19, M119 and gallein can reduce β-AR mediated GRK2 recruitment to the membrane in adult cardiomyocytes.
The specificity of M119 on Gβγ signaling was also determined by assessing electrophysiology in Xenopus oocytes expressing the cardiac G-protein coupled inward rectifying KACh channel (GIRK) subunits Kir3.1 and Kir3.4, together with the muscarinic type 2 receptor. M119 showed no affect on both the basal and ACh activated Kir3.1/Kir3.4 current (Online Figure III), supporting previous data indicating these compounds selectively interfere with a subset of Gβγ interactions.
Based on these results, we sought to determine whether M119 could influence cardiomyocyte contractility both at baseline and in response to β-AR stimulation by measuring sarcomere shortening of isolated adult mouse cardiomyocytes. Compared to untreated cardiomyocytes, Iso significantly enhanced contractility demonstrating these cells were responsive to β-AR agonist (Figure 2A,B). Combined treatment with Iso and M119 resulted in a further significant increase in contraction over Iso alone. In addition to affecting the length of sarcomere shortening, M119 alone and in combination with Iso also increased the rate of cardiomyocyte contraction (Figure 2C). The effect of M119 alone on cardiomyocyte contractility was not unexpected since M119 could enhance signaling downstream from constitutive signaling activity of β-ARs reported to account for a portion of baseline cardiomyocyte contractility 21, 22. Consistent with previous reports of this phenomenon, we found that propranolol alone slightly reduced the rate of contraction (Figure 2C). Furthermore, we found that propranolol completely inhibited the effect of M119 (and Iso) on baseline contractility (Figure 2B,C), suggesting compound specificity for β-AR-Gβγ signaling in cardiomyocytes. Collectively these data demonstrate that small molecule Gβγ inhibition enhances β-AR-mediated signaling and contractility in isolated adult cardiomyocytes.
Figure 2. M119 enhances cardiomyocyte contractility in vitro.

A) Representative tracings of untreated isolated adult cardiomyocytes and cells treated with M119, Iso, or M119 and Iso. B) Averaging of 4-7 independent experiments (1 experiment = average of ≥ 7 cardiomyocytes per condition) showing that M119 treatment significantly increased percent contractility over baseline and enhanced Iso-stimulated contractility. Pretreatment with the general β-AR antagonist, propranolol, abolishes the effects of M119 and Iso on cardiomyocyte contractility. C) M119 also increases the rate of shortening both in the presence and absence of Iso. *P<.05, **P<.01, ***P<.001
M119 improves cardiac function in an acute pharmacologic model of HF
Based on our in vitro experiments, we next examined the effect of M119 on cardiac function in vivo. Chronic stimulation of β-ARs by Iso delivered via implantable miniosmotic pumps is an established acute pharmacologic murine model of HF 12, 23 and this model was used to examine the effects of M119 on HF. Echocardiographic measurements were recorded from all mice prior to initiating the treatment regimen. Miniosmotic pumps containing Iso or saline were implanted and the mice were concurrently injected daily with M119 or vehicle for 7 days. Following 7 days of treatment, cardiac function was again analyzed by echocardiography. As shown in Figure 3, Iso-treated mice showed significant signs of cardiac dysfunction after 7 days with a dramatic reduction in percent fractional shortening compared to vehicle controls. Mice treated with M119 alone showed no significant decrease in percent fractional shortening and no alteration in heart rate compared to vehicle controls, indicating that M119 by itself does not alter cardiac function (Figure 3 and Online Table I). Importantly, Iso-treated mice that received daily injections of M119 maintained essentially normal contractile performance compared to significantly decreased cardiac function observed in Iso-pumped animals that received vehicle control injections (Figure 3 and Online Table I). Iso pumped animals treated with M119 also showed significant normalization of LV volumes and mean velocity of circumferential fiber shortening (Online Table I).
Figure 3. M119 improves cardiac function, and reduces cardiac hypertrophy and interstitial fibrosis, in an acute pharmacologic HF model.
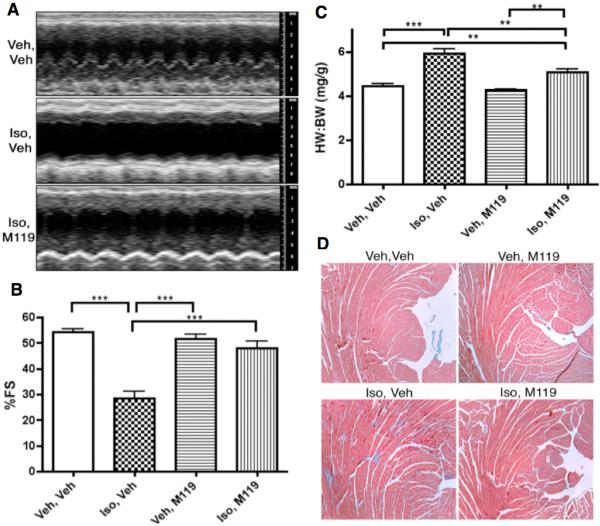
A) Representative M-mode tracings of vehicle-only, Iso-treated and M119+Iso treated animals. B) Average fractional shortening data of animals in each treatment group (n=6-8 per group) shows significant normalization of cardiac function in Iso-treated animals that also received M119. C) HW to BW ratios of animals following 7-day treatment. Iso treated animals show significant cardiac hypertrophy compared to vehicle controls. M119 treatment significantly reduces cardiac hypertrophy when administered to Iso-pumped animals. D) Masson’s Trichrome staining of tissue sections showed that Iso+M119 animals exhibit reduced interstitial fibrosis compared to Iso-only animals. **P<.01, ***P<.001.
Concurrent M119 reduces Iso-induced cardiac dysfunction and hypertrophy
Cardiac morphometry was also examined to determine whether M119 influences hypertrophic growth of the heart. Following echocardiographic analysis of treated mice, body weight (BW) and heart weight (HW) were determined for all mice. As shown in Figure 3C, Iso-treated animals exhibited a ~33% increase in mean HW:BW ratio compared to vehicle controls, indicating that chronic Iso infusion induced significant cardiac hypertrophy. By contrast, M119 treatment of Iso-pumped animals significantly reduced cardiac hypertrophy. These morphometric data were substantiated by significant normalization of ventricular wall thickness with M119 treatment of Iso-pumped animals as detected by echocardiography (Online Table I). Animals treated with M119 alone showed no significant change in HW:BW ratio, indicating that this drug does not by itself induce cardiac hypertrophy.
Isolated hearts were examined using histological staining techniques. Hematoxylin and eosin staining revealed that hearts from Iso-pumped animals were visibly larger than vehicle-pumped animals, consistent with the observed effect on HW:BW ratios (data not shown). In addition, Masson’s trichrome staining revealed that Iso-pumped, M119-treated animals demonstrate reduced interstitial and peri-vascular fibrosis compared to Iso-pumped animals (Figure 3D). No evidence of tissue pathology was found in any group upon staining of lung, liver or brain tissue (not shown).
Gallein halts the progression of HF in mice with established HF
Cardiac restricted calsequestrin overexpressing mice (CSQ) replicate many hallmarks of HF, including dysfunctional β-AR signaling 24. To investigate the efficacy of Gβγ-targeting compounds in halting HF progression, CSQ mice (a generous gift from Dr. Larry Jones) with established HF at 8 weeks of age were treated with M119’s highly related and similarly efficacious compound gallein14 administered daily for one month by intraperitoneal injection.
Echocardiographic analysis showed that gallein treatment completely prevented the progression of HF, maintaining essentially all echocardiographic measures, including functional data for both fractional shortening and ejection fraction (Figure 4 and Online Table II). Conversely, vehicle treated animals demonstrated progressive worsening of cardiac function and of numerous measures associated with the progression of eccentric hypertrophy and dilated cardiomyopathy, including chamber dilation and ventricular wall thinning (Figure 4 and Table 2). Additionally, morphometric analysis showed that gallein inhibited the progression of pathologic cardiac hypertrophy associated with this genetic HF model (Figure 5A, C). Gallein also numerically (but not significantly) enhanced cardiac β-AR cell surface expression toward normal, determined by radioligand binding (Online Figure IV). Finally, improved cardiac function in CSQ mice following gallein treatment was accompanied by a concomitant reduction in expression of both atrial natriuretic factor (ANF) and β-myosin heavy chain (β-MHC) (Figure 5B).
Figure 4. Gallein prevents progression of HF in CSQ mice, trends toward improvement.

Two groups of five male CSQ mice at 8 weeks of age were initiated on once daily injections of 30 mg/kg/day gallein for one month, and were followed by serial conscious echocardiography. A) Quantitation of fractional shortening data. B) representative M-mode echocardiographic images at 4 weeks. *P<0.05 vs. baseline
Figure 5. Gallein improves cardiac morphology and HF marker gene expression in CSQ mice.

A). Morphometric analysis of heart weight to body weight (HW:BW) ratio in CSQ mice treated with vehicle or gallein. B. Real-time PCR analysis of ANF and β-MHC RNA expression in CSQ+V or CSQ+G mice. *P<0.05 vs. vehicle.
In parallel experiments, age-, gender- and background strain (DBA)-matched nontrasngenic control animals received equivalent daily gallein or vehicle injections for one month. Compared to vehicle treatment, no histologic abnormalities were observed in the gallein treated group (heart, lung, liver, brain; not shown), nor were there any differences in cardiac morphology or morphometry (Online Figure VB). Interestingly, a slight but significant elevation of cardiac contractility (% Fractional Shortening) was observed by echocardiography in the gallein group following 1 month of treatment (Online Figure VA and Online Table III), mirroring prior reports that cardiac βARKct expression slightly elevated cardiac contractility at baseline 9, 25, 26.
Small molecule Gβγ inhibitors reduce GRK2 expression in HF
Increased expression of GRK2 is a hallmark of HF; its reduction is concomitant with improved cardiac function 6, 7, 27. Further, inducible ablation of cardiac GRK2 expression is cardioprotective 10, 11. Therefore we examined whether small molecule Gβγ inhibitor treatment altered cardiac GRK2 protein expression. As demonstrated previously 23, Iso-pumped animals show robust increase in GRK2 protein level after one week of treatment (Figure 6A). Interestingly, upregulation of GRK2, indicative of HF, was significantly reduced in Iso-pumped animals treated with M119 (Figure 6A). GRK2 expression was also significantly decreased in CSQ animals treated with Gallein (Figure 6B). Thus, in addition to the immediate effects of small molecule Gβγ inhibitor treatment on Gβγ-mediated GRK2 membrane recruitment demonstrated in cardiac myocytes in vitro, long-term small molecule Gβγ inhibitor treatment normalized cardiac GRK2 expression in two distinct animal models of HF.
Figure 6. Molecular markers of HF are reduced by small molecule Gβγ inhibitor treatment.

A) Representative immunoblotting of GRK2 in Iso pumped mice concurrently treated with M119 for seven days (above, representative image of n=6 per group) and densitometric quantitation analysis of GRK2 normalized to β-Actin (below). B) Immunoblotting of GRK2 in CSQ mice with established HF treated with daily gallein for four weeks (above) and densitometric quantitation analysis of GRK2 normalized to β-Actin (below, n=5 per group). GRK2 protein levels were significantly reduced by small molecule Gβγ inhibitor treatment as determined by densitometric analysis of western blots. *P<.05, **P<.01
Discussion
Recent studies from our laboratories identified a number of small molecules that bind Gβγ and modulate Gβγ-protein interactions 19, 20. These compounds were able to inhibit a number of Gβγ-dependent signaling events, including Gβγ-GRK2 association. GRK2 protein levels are significantly elevated in HF, and substantial evidence indicates that blocking both Gβγ and the GRK2-Gβγ interaction in diverse HF models is cardioprotective 13, 14. In this study, we tested whether M119 and its highly related and similarly efficacious Gβγ compound inhibitor gallein could be used to target Gβγ in cardiomyocytes and thereby influence aspects of β-AR signaling and improve cardiac function in animal models of HF. Our data indicate that M119 and gallein interfere with Gβγ-GRK2 interactions in cardiomyocytes, and may enhance β-AR signaling in vitro. M119 and gallein both halt HF progression and improve cardiac function, morphometry, histology and gene expression in animal models of either new onset or established HF.
Recruitment of GRK2 to the membrane by Gβγ is among the more well-established functions of Gβγ downstream of β-AR signaling. General inhibition of Gβγ, and of the Gβγ-GRK2 interaction, using various Gβγ inhibitory peptides (e.g. βARKct, nt-del-Phosducin, PI3Kγinact) has proven to be a highly effective treatment for a variety of animal HF models and in isolated failing human cardiomyocytes 28. The results of these studies have alternately been viewed as a specific effect of inhibiting GRK2 function and as a broader effect on Gβγ signaling. Consistent with the latter hypothesis, another Gβγ sequestering peptide, a truncated form of the Gβγ binding protein phosducin (nt-del-Phosducin), has also been used successfully to restore β-AR signaling in cardiomyocytes 29. More recently, a peptide inhibitor of PI3Kγ, which is recruited to Gβγ in part through association with GRK2, has also proven to be effective in treating animal models of HF 16, 17, 27. Collectively these results suggest that generally inhibiting pathologic Gβγ-dependent signaling in failing cardiomyocytes is salutary. Consistent with these observations, our novel small molecule based approach to targeting Gβγ signaling appears to function, at least in part, like peptide inhibitors of Gβγ signaling. Whether the observed effects are due specifically to Gβγ-GRK2 inhibition, or to other Gβγ signaling components, will be a target of our future studies. Importantly, the current study suggests selective interference with a subset of Gβγ interactions, as Gβγ-GIRK signaling was not affected (Online Figure III).
Our studies of isolated adult cardiomyocytes showed that small molecule Gβγ inhibition enhanced cAMP production and cardiomyocyte contractility following acute β-AR stimulation. These data are consistent with published results showing similar effects of βARKct expression in cardiac myocytes in culture 28 and in vivo 9. Since M119 and gallein were also found to reduce Iso-induced GRK2 membrane recruitment, we conclude that these small molecules influence β-AR signaling in cardiomyocytes similar to βARKct.
Gβγ inhibitory compounds M119 and gallein partially normalized cardiac morphology and gene expression and halted HF progression in the acute Iso pump model of HF and in CSQ mice with established HF. These data suggest promising utility of small molecule Gβγ inhibition in treating both new onset and extant HF. Importantly, β-blockers are a current standard therapy in the treatment of human HF. Combination of cardiac-restricted βARKct expression with the β-blocker metoprolol has previously demonstrated synergistic benefit for cardiac function in CSQ mice30, suggesting a possible synergy of small molecule Gβγ inhibition with β-blocker therapy, which will be a target of our future investigation.
In addition to its role in the cardiomyocytes, recent evidence demonstrates that dysregulation of adrenergic receptor-dependent Gβγ signaling in other tissues may also contribute to cardiac dysfunction. Lymperopoulos and coworkers recently showed a significant increase of GRK2 in the chromaffin cells of the adrenal medulla, suggesting that increased Gβγ-GRK2 association contributes to dysfunctional feedback inhibition of catecholamine release via α2-AR signaling 31. Interestingly, viral delivery of βARKct to the adrenal gland restored α2-AR feedback inhibition of catecholamine release and dramatically improved cardiac function, due at least in part to decreased chronic stimulation of cardiac β-ARs. Increased GRK2 expression is also associated with hypertension 32, 33. β-AR signaling normally promotes vasodilation but overexpression of GRK2 within vascular smooth muscle cells leads to diminished β-AR signaling and elevated resting blood pressure, a major risk factor for HF 34. Because heart disease can result from dysfunctional signaling in multiple organs, we and others 31 believe that systemic delivery of small molecule Gβγ inhibitors could simultaneously target multiple causes of this disease. This approach to modulating intracellular signaling is also consistent with the growing trend of therapeutics targeting intracellular components of pathological signaling in cardiovascular disease 35.
In summary we have demonstrated that small molecule Gβγ inhibitors function in vivo to improve cardiac function and halt HF progression in both new onset and extant HF in mice. Small molecule Gβγ inhibitors appear to maintain β-AR responsiveness, in part by interfering with GRK2 membrane recruitment. Future studies will determine whether Gβγ inhibitor compounds modulate other known Gβγ-dependent signaling events, including Gβγ-mediated signaling via novel ERK1/2 phosphorylation36, and whether these effects contribute to their effects in cardiac pathophysiology. Such studies will aim to determine whether small molecule targeting of Gβγ may be an effective therapeutic paradigm for the treatment of HF.
Novelty and Significance.
What is known?
Excess signaling through cardiac G-protein βγ (Gβγ)-subunits via β-adrenergic receptors (β-AR) is a major component of the pathologic changes associated with the progression of heart failure (HF).
Numerous reports indicate that peptide inhibitors of β-AR/Gβγ signaling can block many of the molecular changes associated with HF and can improve cardiac function.
We recently identified two membrane permeable, structurally-related small molecules that selectively target Gβγ-protein interactions that can be used effectively in vivo.
What new information does this article contribute?
Two structurally-related small molecules that target specific Gβγ signaling pathways are cardioprotective in mouse models of both new onset and extant HF.
Cardiac protection conferred by these Gβγ targeting compounds is associated with partial reversal of pathologic molecular changes known to occur in HF.
Summary.
Prior studies have demonstrated that interfering with Gβγ signaling downstream of β-AR activation is cardioprotective in HF models. Previous approaches generally used large peptides that required viral vector delivery to target cells/organs. Here we demonstrate that systemic delivery of small molecule inhibitors of Gβγ signaling can disrupt pathologic molecular changes underlying HF. Further, we demonstrate that these small molecules reduce the progression of HF in two distinct mouse models. Our study provides rationale for further development of Gβγ-targeting compounds as a therapeutic approach for HF. This could lead to new cardiac therapies that, in combination with existing drugs, may improve patient health.
Supplementary Material
Acknowledgements
The authors wish to thank Sundeep Malik for help with immunoblotting.
Sources of Funding
This work was supported in part by NIH R01s HL091475, HL084087 and HL089885 (BCB), GM081772 (AVS), a T32 (GM07356) and by an AHA postdoctoral fellowship (SLB).
Non-standard Abbreviations and Acronyms
- Iso
isoproterenol
- GRK2
g-protein coupled receptor kinase 2
- Gβγ
g-protein beta-gamma subunit
- β-AR
β-adrenergic receptor
- BW
body weight
Footnotes
Disclosures
None.
Subject Codes: [11] Other heart failure, [130] Animal models of human disease, [105] Contractile function, [108] Other myocardial biology
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith SC, Jr., Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: endorsed by the Heart Rhythm Society. Circulation. 2005;112:e154–235. doi: 10.1161/CIRCULATIONAHA.105.167586. [DOI] [PubMed] [Google Scholar]
- 2.Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O’Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y. Heart Disease and Stroke Statistics--2009 Update. A Report From the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008 doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 3.Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG, Lefkowitz RJ. Role of beta gamma subunits of G proteins in targeting the beta-adrenergic receptor kinase to membrane-bound receptors. Science. 1992;257:1264–1267. doi: 10.1126/science.1325672. [DOI] [PubMed] [Google Scholar]
- 4.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 5.Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation. 1993;87:454–463. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- 6.Hata JA, Williams ML, Schroder JN, Lima B, Keys JR, Blaxall BC, Petrofski JA, Jakoi A, Milano CA, Koch WJ. Lymphocyte levels of GRK2 (betaARK1) mirror changes in the LVAD-supported failing human heart: lower GRK2 associated with improved beta-adrenergic signaling after mechanical unloading. J Card Fail. 2006;12:360–368. doi: 10.1016/j.cardfail.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 7.Iaccarino G, Dolber PC, Lefkowitz RJ, Koch WJ. Bbeta-adrenergic receptor kinase-1 levels in catecholamine-induced myocardial hypertrophy: regulation by beta- but not alpha1-adrenergic stimulation. Hypertension. 1999;33:396–401. doi: 10.1161/01.hyp.33.1.396. [DOI] [PubMed] [Google Scholar]
- 8.Blaxall BC, Tschannen-Moran BM, Milano CA, Koch WJ. Differential gene expression and genomic patient stratification following left ventricular assist device support. J Am Coll Cardiol. 2003;41:1096–1106. doi: 10.1016/s0735-1097(03)00043-3. [DOI] [PubMed] [Google Scholar]
- 9.Koch W, Rockman H, Samama P, Hamilton R, Bond R, Milano C, Lefkowitz R. Cardiac function in mice overexpressing the β-adrenergic receptor kinase or a βARK inhibitor. Science. 1995;268:1350–1353. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- 10.Dorn GW., 2nd. GRK mythology: G-protein receptor kinases in cardiovascular disease. J Mol Med. 2009;87:455–463. doi: 10.1007/s00109-009-0450-7. [DOI] [PubMed] [Google Scholar]
- 11.Raake PW, Vinge LE, Gao E, Boucher M, Rengo G, Chen X, DeGeorge BR, Jr., Matkovich S, Houser SR, Most P, Eckhart AD, Dorn GW, 2nd, Koch WJ. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res. 2008;103:413–422. doi: 10.1161/CIRCRESAHA.107.168336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matkovich SJ, Diwan A, Klanke JL, Hammer DJ, Marreez Y, Odley AM, Brunskill EW, Koch WJ, Schwartz RJ, Dorn GW., 2nd. Cardiac-specific ablation of G-protein receptor kinase 2 redefines its roles in heart development and beta-adrenergic signaling. Circ Res. 2006;99:996–1003. doi: 10.1161/01.RES.0000247932.71270.2c. [DOI] [PubMed] [Google Scholar]
- 13.Hata JA, Koch WJ. Phosphorylation of G protein-coupled receptors: GPCR kinases in heart disease. Mol Interv. 2003;3:264–272. doi: 10.1124/mi.3.5.264. [DOI] [PubMed] [Google Scholar]
- 14.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 15.Hippe HJ, Lutz S, Cuello F, Knorr K, Vogt A, Jakobs KH, Wieland T, Niroomand F. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gbeta subunits. Specific activation of Gsalpha by an NDPK B.Gbetagamma complex in H10 cells. J Biol Chem. 2003;278:7227–7233. doi: 10.1074/jbc.M210305200. [DOI] [PubMed] [Google Scholar]
- 16.Nienaber JJ, Tachibana H, Naga Prasad SV, Esposito G, Wu D, Mao L, Rockman HA. Inhibition of receptor-localized PI3K preserves cardiac beta-adrenergic receptor function and ameliorates pressure overload heart failure. J Clin Invest. 2003;112:1067–1079. doi: 10.1172/JCI18213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perrino C, Naga Prasad SV, Patel M, Wolf MJ, Rockman HA. Targeted inhibition of beta-adrenergic receptor kinase-1-associated phosphoinositide-3 kinase activity preserves beta-adrenergic receptor signaling and prolongs survival in heart failure induced by calsequestrin overexpression. J Am Coll Cardiol. 2005;45:1862–1870. doi: 10.1016/j.jacc.2005.02.062. [DOI] [PubMed] [Google Scholar]
- 18.Perrino C, Naga Prasad SV, Schroder JN, Hata JA, Milano C, Rockman HA. Restoration of beta-adrenergic receptor signaling and contractile function in heart failure by disruption of the betaARK1/phosphoinositide 3-kinase complex. Circulation. 2005;111:2579–2587. doi: 10.1161/CIRCULATIONAHA.104.508796. [DOI] [PubMed] [Google Scholar]
- 19.Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, Font JL, Bidlack JM, Smrcka AV. Differential targeting of Gbetagamma-subunit signaling with small molecules. Science. 2006;312:443–446. doi: 10.1126/science.1120378. [DOI] [PubMed] [Google Scholar]
- 20.Lehmann DM, Seneviratne AM, Smrcka AV. Small molecule disruption of G protein beta gamma subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol Pharmacol. 2008;73:410–418. doi: 10.1124/mol.107.041780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lefkowitz RJ, Cotecchia S, Samama P, Costa T. Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. Trends Pharmacol Sci. 1993;14:303–307. doi: 10.1016/0165-6147(93)90048-O. [DOI] [PubMed] [Google Scholar]
- 22.Seifert R, Wieland T. G protein-coupled receptors as drug targets : analysis of activation and constitutive activity. Weinheim[Chichester: Wiley-VCH ;John Wiley, distributor]; 2005. [Google Scholar]
- 23.Iaccarino G, Tomhave ED, Lefkowitz RJ, Koch WJ. Reciprocal in vivo regulation of myocardial G protein-coupled receptor kinase expression by beta-adrenergic receptor stimulation and blockade. Circulation. 1998;98:1783–1789. doi: 10.1161/01.cir.98.17.1783. [DOI] [PubMed] [Google Scholar]
- 24.Cho MC, Rapacciuolo A, Koch WJ, Kobayashi Y, Jones LR, Rockman HA. Defective beta-adrenergic receptor signaling precedes the development of dilated cardiomyopathy in transgenic mice with calsequestrin overexpression. J Biol Chem. 1999;274:22251–22256. doi: 10.1074/jbc.274.32.22251. [DOI] [PubMed] [Google Scholar]
- 25.Rockman HA, Chien KR, Choi D-J, Iaccarino G, Hunter JJ, Ross J, Jr., Lefkowitz RJ, Koch WJ. Expression of a β-adrenergic receptor kinase-1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci USA. 1998;9(95):7000–5. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rockman HA, Choi DJ, Akhter SA, Jaber M, Giros B, Lefkowitz RJ, Caron MG, Koch WJ. Control of myocardial contractile function by the level of beta-adrenergic receptor kinase 1 in gene-targeted mice. J Biol Chem. 1998;273:18180–18184. doi: 10.1074/jbc.273.29.18180. [DOI] [PubMed] [Google Scholar]
- 27.Penela P, Murga C, Ribas C, Tutor AS, Peregrin S, Mayor F., Jr. Mechanisms of regulation of G protein-coupled receptor kinases (GRKs) and cardiovascular disease. Cardiovasc Res. 2006;69:46–56. doi: 10.1016/j.cardiores.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 28.Williams ML, Hata JA, Schroder J, Rampersaud E, Petrofski J, Jakoi A, Milano CA, Koch WJ. Targeted beta-adrenergic receptor kinase (betaARK1) inhibition by gene transfer in failing human hearts. Circulation. 2004;109:1590–1593. doi: 10.1161/01.CIR.0000125521.40985.28. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Laugwitz KL, Pinkernell K, Pragst I, Baumgartner C, Hoffmann E, Rosport K, Munch G, Moretti A, Humrich J, Lohse MJ, Ungerer M. Effects of two Gbetagamma-binding proteins--N-terminally truncated phosducin and beta-adrenergic receptor kinase C terminus (betaARKct)--in heart failure. Gene Ther. 2003;10:1354–1361. doi: 10.1038/sj.gt.3301995. [DOI] [PubMed] [Google Scholar]
- 30.Harding VB, Jones LR, Lefkowitz RJ, Koch WJ, Rockman HA. Cardiac beta ARK1 inhibition prolongs survival and augments beta blocker therapy in a mouse model of severe heart failure. Proc Natl Acad Sci U S A. 2001;98:5809–5814. doi: 10.1073/pnas.091102398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lymperopoulos A, Rengo G, Funakoshi H, Eckhart AD, Koch WJ. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat Med. 2007;13:315–323. doi: 10.1038/nm1553. [DOI] [PubMed] [Google Scholar]
- 32.Gros R, Chorazyczewski J, Meek MD, Benovic JL, Ferguson SS, Feldman RD. G Protein-coupled receptor kinase activity in hypertension : increased vascular and lymphocyte G-protein receptor kinase-2 protein expression. Hypertension. 2000;35:38–42. doi: 10.1161/01.hyp.35.1.38. [DOI] [PubMed] [Google Scholar]
- 33.Gros R, Tan CM, Chorazyczewski J, Kelvin DJ, Benovic JL, Feldman RD. G-protein-coupled receptor kinase expression in hypertension. Clin Pharmacol Ther. 1999;65:545–551. doi: 10.1016/S0009-9236(99)70074-3. [DOI] [PubMed] [Google Scholar]
- 34.Eckhart AD, Ozaki T, Tevaearai H, Rockman HA, Koch WJ. Vascular-targeted overexpression of G protein-coupled receptor kinase-2 in transgenic mice attenuates beta-adrenergic receptor signaling and increases resting blood pressure. Mol Pharmacol. 2002;61:749–758. doi: 10.1124/mol.61.4.749. [DOI] [PubMed] [Google Scholar]
- 35.McKinsey TA, Kass DA. Small-molecule therapies for cardiac hypertrophy: moving beneath the cell surface. Nat Rev Drug Discov. 2007;6:617–635. doi: 10.1038/nrd2193. [DOI] [PubMed] [Google Scholar]
- 36.Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat Med. 2009;15:75–83. doi: 10.1038/nm.1893. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.