

Abstract
Histone deacetylase (HDAC) inhibitors increase histone acetylation and enhance both memory and synaptic plasticity. The current model for the action of HDAC inhibitors assumes that they alter gene expression globally and thus affect memory processes in a nonspecific manner. Here, we show that the enhancement of hippocampus-dependent memory and hippocampal synaptic plasticity by HDAC inhibitors is mediated by the transcription factor cAMP response element-binding protein (CREB) and the recruitment of the transcriptional coactivator and histone acetyltransferase CREB-binding protein (CBP) via the CREB-binding domain of CBP. Furthermore, we show that the HDAC inhibitor trichostatin A does not globally alter gene expression but instead increases the expression of specific genes during memory consolidation. Our results suggest that HDAC inhibitors enhance memory processes by the activation of key genes regulated by the CREB:CBP transcriptional complex.
Keywords: histone deacetylase inhibitors, hippocampus-dependent memory, CREB-binding protein, KIX domain, Nr4a1, Nr4a2
Introduction
Gene transcription is required for long-lasting forms of synaptic plasticity and memory storage (for review, see Johnston et al., 2003), but the precise mechanisms that regulate these transcriptional events remain unclear. Much recent research emphasizes that transcriptional regulation involved in memory requires the coordinated interaction of multiple transcription factors and transcriptional coactivators in the context of chromatin (Felsenfeld and Groudine, 2003; Korzus 2003;Harbison et al., 2004). Chromatin provides a rich environment for transcriptional regulation. Within the nucleosomal units of chromatin, DNA is packaged around a core of histone proteins. Histones are subject to a variety of posttranslational modifications, of which the best studied is acetylation (for review, see Turner, 2005). Histone acetylation alters chromatin structure (Norton et al., 1989) and increases accessibility for transcriptional regulatory proteins (Vettese-Dadey et al., 1996). The steady-state levels of histone acetylation are the product of proteins with histone acetyltransferase (HAT) activity that add acetyl groups to histones and proteins with histone deacetylase (HDAC) activity that remove acetyl groups.
cAMP response element-binding protein (CREB)-binding protein (CBP) is a transcriptional coactivator with HAT activity (for review, see Goodman and Smolik, 2000; Kalkhoven, 2004) that we and others have demonstrated is involved in synaptic plasticity and long-term memory, using different genetically modified mice in which CBP activity was impaired (Oike et al., 1999; Bourtchouladze et al., 2003; Alarcon et al., 2004; Korzus et al., 2004; Wood et al., 2005, 2006). Several strains of cbp (Crebbp) mutant mice exhibited deficits in memory and synaptic plasticity, and drugs that inhibit HDAC activity (and thus increase histone acetylation) ameliorated impairments in hippocampal long-term potentiation (LTP) and memory in two of these cbp mutants (Alarcon et al., 2004; Korzus et al., 2004). Interestingly, HDAC inhibition was also capable of enhancing LTP in wild-type mice (Alarcon et al., 2004), consistent with two other studies showing that HDAC inhibitors facilitated both memory and LTP in rats (Levenson et al., 2004; Yeh et al., 2004).
These findings suggest that chromatin modification via histone acetylation is a major molecular pathway involved in the regulation of transcription underlying memory storage, but the molecular mechanisms by which increased histone acetylation affects memory and synaptic plasticity remain unknown. The exploration of this open area is critical to our understanding of the transcriptional processes underlying memory storage and to the development of novel therapeutic reagents. Chromatin modification is emerging as a fundamental molecular mechanism for the regulation of transcription involved in neurodevelopmental disorders (such as Rubinstein-Taybi syndrome and Rett syndrome), neurodegenerative diseases, epilepsy, and drug addiction (Steffan et al., 2001; Shahbazian and Zoghbi, 2002; Hockly et al., 2003; Levenson and Sweatt, 2005; Levine et al., 2005). Furthermore, HDAC inhibitors are being considered as a therapeutic agent to treat cognitive aspects of these diseases. Here, we use a combined neuropharmacological, genetic, and electrophysiological approach to demonstrate that a specific transcription factor/coactivator interaction between CREB and CBP is required for enhancing memory and synaptic plasticity by HDAC inhibition.
Materials and Methods
Subjects.
Male C57BL/6J mice bred in our animal facility from mice originally obtained from The Jackson Laboratory (Bar Harbor, ME) were used in most experiments. For studies using CREBαΔ mutant mice, experimental mice were male and female wild-type and homozygous products of F1 crosses of CREBαΔ+/− C57BL/6J with CREBαΔ+/− 129/SvEvTac mice (as by Graves et al., 2002). The CREBαΔ mutation (Hummler et al., 1994) had been backcrossed in a heterozygous state in each parental strain for 9–11 generations. Genotyping was performed as described previously (Graves et al., 2002). CbpKIX/KIX knock-in mice were generated as described previously (Kasper et al., 2002; Wood et al., 2006) and were generously provided by Dr. Paul Brindle (St. Jude Children's Research Hospital, Memphis, TN). Briefly, the targeting vector for CBP contained the following point mutations: Tyr650Ala, Ala654Gln, and Tyr658Ala. This triple mutation was introduced into the Cbp locus of embryonic day 14 stem cells derived from 129P2/OlaHsd mice via homologous recombination. Mice carrying the triple-mutation allele of CBP were bred and backcrossed in a heterozygous state onto the C57BL/6J background for at least 10 generations. Experimental animals were generated from heterozygous matings, and wild-type littermates were used as control for homozygous mutant mice (designated cbpKIX/KIX). The generation of calcium/calmodulin-dependent kinase IIα (CaMKIIα)-CBPΔ1 transgenic mice has been described previously (Wood et al., 2005). These mice have been bred and backcrossed in a hemizygous state onto the C57BL/6J background for eight generations. Mice were 8- to 16-weeks-old and had access to food and water in their home cages ad libitum. Lights were maintained on a 12 h light/dark cycle, with all behavioral testing performed during the light portion of the cycle. All experiments were conducted according to National Institutes of Health guidelines for animal care and use and were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. The investigator was blind to the genotype and/or drug treatment of the mice during behavioral or electrophysiological testing.
Cannula placement.
Bilateral 22 gauge guide cannulas were used to guide an injection cannula into the dorsal hippocampus. The guide cannula placement was as follows: anteroposterior, −1.7 mm; mediolateral, ±1.5 mm; dorsoventral, 1.5 mm below pedestal. Injection cannula extended an additional 0.7 mm below the guide cannula (total depth, 2.2 mm).
Injections.
Immediately after conditioning, mice received bilateral intrahippocampal injections of 1.0 μl of 16.5 mm trichostatin A (TSA) (A. G. Scientific, San Diego, CA) dissolved in 50% ethanol or 50% ethanol per side [vehicle (VEH)] from a 5.0 μl Hamilton syringe operated by a Harvard Apparatus (Holliston, MA) Pump II Dual Syringe micropump. Injections occurred over 1 min, and injection cannulas were left in place an additional 30 s to allow the fluid to diffuse. Each side was injected individually, one immediately after the other. The entire injection process took ∼4 min.
Immunohistochemistry.
Four hours after intrahippocampal injection with TSA or vehicle, mice were anesthetized with isoflurane and transcardially perfused with PBS, followed by 4.0% paraformaldehyde in PBS using a peristaltic perfusion pump (Rainin Instruments, Oakland, CA). Fixed brains were dissected, postfixed overnight, and then cryoprotected in 30% sucrose at 4°C. Brains were flash frozen in 2-methylbutane on dry ice and mounted on cryostat chucks using OCT. Coronal sections were cut at a thickness of 30 μm and collected in PBS. Floating sections were blocked for 1 h at room temperature in 8% normal goat serum (NGS) (Vector Laboratories, Burlingame, CA) with 0.3% Triton X-100 (Fisher Scientific, Houston, TX) in PBS and then incubated overnight at 4°C in 2% NGS, 0.3% Triton X-100 in PBS with anti-acetyl histone H3 antibody (1:1000; Upstate Biotechnology, Lake Placid, NY) and for 1 h at room temperature with FITC-conjugated anti-rabbit IgG secondary antibody (1:1000; Chemicon, Temecula, CA). Sections were washed three times for 5 min each in PBS before and after each incubation step. 4′,6′-Diamidino-2-phenylindole (DAPI) was added during the last wash for 20 min. Sections were mounted on slides and imaged using a Zeiss (Oberkochen, Germany) Axiovert 200M epifluorescence microscope with CARVII spinning disc confocal unit (BD Biosciences, San Jose, CA) and digitized by using an ORCA-ER CCD camera (Hamamatsu, Bridgewater, NJ). Wide-field (2.5×) and confocal (40×) images of DAPI (cell nuclei) and FITC (acetylated histone) were acquired and combined by using IPLab 4.0 software (BD Biosciences).
Fear conditioning.
Fear conditioning experiments were performed in chambers using the methods described previously (Wood et al., 2005). Mice were handled for 3 consecutive days for 1 min each day. For contextual fear conditioning, C57BL/6J mice were placed into the conditioning chamber and received a 2 s, 0.75 mA scrambled footshock 2.5 min after placement into the chamber. Mice were removed from the chamber after 3 min. During testing, mice received one 5 min exposure to the same conditioned context in the absence of shock 24 h after conditioning. For cued fear conditioning, C57BL/6J mice were placed into the chamber, and the cue (white noise) was presented from 2 to 2.5 min after placement into the chamber and coterminated with a 2 s, 0.75 mA footshock. Mice were removed from the chamber after a total of 3 min. On testing day, mice in the cued group received one 5 min exposure to a novel context (another conditioning chamber with smooth flat floor, altered dimensions, and a novel odorant) for 0–2 min [pre-conditioning stimulus (CS)], followed by exposure to the cue from 2 to 5 min (CS), 24 h after conditioning. Conditioning was assayed by measuring freezing behavior, the complete absence of movement (Fanselow, 1980). Freezing was scored during conditioning as well as testing. The behavior of each mouse was sampled at 5 s intervals, and the percentage of those intervals in which the mouse froze was calculated. Different sets of mice were used for contextual and cued conditioning experiments.
For the contextual fear conditioning experiments using CREBαΔ mutant mice, the conditions were similar to those described above except the shock intensity was lowered to 0.40 mA because our preliminary experiments revealed that the wild-type B6/129 F1 hybrid littermates were reaching a freezing ceiling using the 0.75 mA shock intensity (data not shown).
Electrophysiological analysis.
To analyze the effect of the HDAC inhibitor TSA on early-phase LTP (E-LTP), 6- to 10-week old C57BL/6J mice were killed by cervical dislocation, and hippocampi were rapidly dissected in iced oxygenated artificial CSF (ACSF). Transverse hippocampal slices, 400 μm thick, were placed in an interface chamber (Fine Science Tools, Foster City, CA) and continuously perfused with oxygenated ACSF containing either TSA (1.65 μm) or vehicle (same volume of 50% EtOH) while they equilibrated for at least 1.5 h at 30°C before starting electrophysiological recordings (Wood et al., 2005). To perform two-pathway experiments, two bipolar stimulating electrodes (0.002-inch-diameter nichrome wire; A-M Systems, Sequim, WA) placed in the stratum radiatum were used to elicit action potentials in independent sets of CA3 axons. The independence of these inputs was determined by the absence of paired-pulse facilitation across inputs using a 50 ms interpulse interval. An ACSF-filled glass microelectrode (1.5 × 0.85 mm; A-M Systems) with a resistance between 0.5 and 3 MΩ was placed between the two stimulating electrodes in the stratum radiatum region of CA1 and was used to record the resulting field potentials (fEPSPs). Data were acquired using Clampex 8.2 (Molecular Devices, Palo Alto, CA) and analyzed using Clampfit 8.2 (Molecular Devices). Peak fEPSP amplitudes from both stimulators were required to be at least 3 mV, and stimulus intensity was set to produce 40% of the maximal response. A protocol was used in which stimulations occurred every minute, with a 200 ms delay between the two stimulators. Baseline responses were recorded for 20 min, and E-LTP was then induced by applying one train of stimuli at 100 Hz for 1 s through one of the two stimulators. Recordings continued for 60 min after LTP induction. TSA or vehicle remained present throughout the recording session. Initial fEPSP slopes were normalized against the average of the 20 baseline traces and were expressed as percentage.
To analyze the effect of TSA on E-LTP in CREBαΔ mutant mice and cbpKIX/KIX mice, homozygous mutants and wild-type littermates were used. However, in this protocol, only one stimulating electrode was used, and, after recording baseline CA1 fEPSPs for 20 min in the presence of control ACSF, slices were perfused with ACSF containing either TSA (1.65 μm) or vehicle (0.05% EtOH) for another 20 min before single-train stimulation as above. Recordings continued for 120 min after tetanus, and drug remained present throughout the recording period. We also examined the effect of the transcriptional inhibitor actinomycin D (ActD) (Tocris, Ellisville MO), dissolved in 100% DMSO, on the enhancement of E-LTP by TSA in C57BL/6J mice. This experiment was performed as above, except with either ActD (25 μm) or vehicle (0.05% DMSO) added to the ACSF containing TSA. We also performed experiments using another HDAC inhibitor, sodium butyrate (NaBu) (Upstate Biotechnology) in CREBαΔ mutant and wild-type mice. NaBu was bath applied at 300 μm for 40 min beginning 20 min before tetanization, as by Levenson et al. (2004).
Preparation and purification of mRNA.
Hippocampi were harvested from each animal and placed in 500 μl of RNAlater (Ambion, Austin, TX). Aerosol Barrier ART tips, DEPC-treated sterile water, and autoclaved 1.5 ml microcentrifuge tubes were used for all subsequent steps. Tissue was homogenized in 1 ml of Trizol (Invitrogen, Carlsbad, CA) and transferred to 1.5 ml tubes, in which samples were incubated at room temperature for 5 min. Phenol–chloroform (300 μl) addition was followed by vigorous mixing and room temperature incubation for 3 min. Samples were transferred to phase-lock gel tubes (Eppendorf, Westbury, NY) and were centrifuged at 4°C and full speed for 15 min. The aqueous phase was transferred to new tubes, in which 2 vol of ethanol, 1:10 vol of NaOAc, and 1 μl of glycogen were added. After gentle mixing, the samples were incubated at −20°C for 10 min. Centrifugation at 4°C and full speed was followed by aspiration of the supernatant. Pellets were washed with 300 μl of 70% ethanol and then centrifuged at 4°C and full speed for 5 min. The supernatant was aspirated and samples were air dried for ∼10 min. After resuspension in 100 μl of water, RNA was purified using the RNeasy system (Qiagen, Valencia, CA) according to the protocol of the manufacturer. Residual DNA was removed by treatment with DNA-free (Ambion). Optical density measurement at 260 and 280 nm by the SmartSpec 3000 (Bio-Rad, Hercules, CA) spectrophotometer was used to assess the concentration and quality of mRNA in each sample.
Preparation of cDNA.
Generation of cDNA was performed by the RETROscript (Ambion) kit. Briefly, each reaction was performed with 1 μg of RNA in a total volume of 20 μl composed as follows: 50 mm Tris-HCl, pH 8.3, 75 mm KCl, 3 mm MgCl2, 5 mm DTT, 500 μm each dNTP, 5 μm random decamers, 10 U of RNase inhibitor, and 100 U of Moloney murine leukemia virus reverse transcriptase. As controls, additional reaction mixtures were generated that lacked either reverse transcriptase or template mRNA. Subsequent reactions were allowed to proceed at 44°C for 1 h, followed by heat inactivation at 100°C for 10 min.
Quantitative real-time reverse transcription-PCR.
The cDNA samples were diluted to 2 ng/μl in nuclease-free water. Reactions were prepared in 96-well optical reaction plates (Applied Biosystems, Foster City, CA) with optical adhesive covers (Applied Biosystems). Each well contained 11.5 μl of cDNA, 1 μl of 5 μm primer solution, and 12.5 μl of Quantitect SYBRgreen PCR mastermix (Qiagen). Four biological and three technical replicates were used in the TSA treatment experiments. Reactions were performed in the Applies Biosystems ABI Prism 7000 with an initial activation by incubation at 50°C for 2 min followed by incubation at 95°C for 15 min and 40 subsequent cycles of 95°C for 15 s, 56°C for 30 s, and 72°C for 30 s. For a list of primers and their corresponding sequences, see supplemental Table S1 (available at www.jneurosci.org as supplemental material). Data was normalized to actin G (Actg), hypoxanthine phosphoribosyltransferase (Hprt), and Tubulin before calculation of differences. PCR products were cloned using the TA cloning kit (Invitrogen) following the protocol of the manufacturer. Cloned fragments were then sequenced to confirm the identity of the amplified gene. The Icer primers are the same as described previously (Conti et al., 2004). Relative quantitation of gene expression was performed according to Applied Biosystems User Bulletin #2. Fold change was calculated from the ΔCt cycle threshold values with corrections for standard curve data from each gene and housekeeping gene expression levels for each sample based on the relative standard curve method described in the Applied Biosystems manual. For each sample, the ΔCt was calculated against the mean for the sample set of that gene. Next, each of these ΔCt values was corrected with the slope of the standard curve for the relevant primer to account for any variation in primer amplification efficiency. The efficiency-corrected ΔCt value was normalized to the similarly corrected ΔCt from housekeeping genes for each sample to account for variability in mRNA input. The difference between corrected ΔCt for each sample was then grouped according to condition (TSA or vehicle) and the average ΔCt calculated. Because Ct values are on a logarithmic scale, fold change is equal to two raised to the difference between experimental and control ΔCt values. Because correction has been made for primer efficiency, we presented the data as fold change. The data presented are the calculated mean for the biological replicates, with n being equal to the number of biological replicates (i.e., the number of mice examined).
Chromatin immunoprecipitation.
Animals were cannulated and fear conditioned as described for behavioral experiments. Two hours after fear conditioning, hippocampi were dissected onto a dry ice-cooled cutting surface. The tissue was finely chopped and incubated in freshly prepared 4% paraformaldehyde in PBS for 20 min with gentle rocking. Crosslinking was halted by addition of 2.5 m glycine and an additional 5 min of gentle rocking. Samples were washed three times in PBS plus protease inhibitors (Sigma, St. Louis, MO), frozen on dry ice, and stored at −80°C. Tissue was thawed on ice, resuspended in 1 ml of cell lysis buffer (10 mm Tris, 10 mm NaCl, and 0.2% NP-40), and Dounce homogenized to release nuclei. Nuclear lysis buffer (1% SDS, 10 mm EDTA, and 50 mm Tris-HCl, pH 8.1) was added to release chromatin. Chromatin was sheared into 400–600 bp fragments using the Sonic Dismembrator 100 (Fisher Scientific) by five 10 s pulses at 10 W output with 1 min on ice between pulses. Samples were diluted fivefold in chromatin immunoprecipitation (ChIP) dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8.1, and 167 mm NaCl) and precleared with Protein-G plus (Santa Cruz Biotechnology, Santa Cruz, CA). Samples were incubated overnight at 4°C with primary antibodies for acetylated H3 (Upstate Biotechnology) and acetylated H4 (Upstate Biotechnology). Mock immunoprecipitation was performed in parallel with normal rabbit IgG (Upstate Biotechnology). Immunoprecipitation was performed with 100 μl of blocked protein-G plus agarose beads at 4°C for 2 h. Beads were washed twice each with low-salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 150 mm NaCl, and 20 mm Tris-HCl, pH 8.1), high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 500 mm NaCl, and 20 mm Tris-HCl, pH 8.1), LiCl buffer (250 mm LiCl, 1% NP-40, 1% deoxycholate, 1 mm EDTA, and 10 mm Tris-HCl, pH 8.1), and Tris–EDTA. Chromatin fragments were released from beads in elution buffer (1% SDS and 0.1 m NaHCO3). Formaldehyde crosslinking was reversed by overnight incubation at 95°C in the presence of 200 mm NaCl, followed by treatment with proteinase K for 1 h at 55°C. DNA was extracted using Qiaquick spin columns (Qiagen) and analyzed by gel electrophoresis and spectrophotometry (Nanodrop, Wilmington, DE) to confirm DNA quality. Quantitative real-time PCR was performed as described above for mRNA quantification with primers specific to the Nr4a1 (nuclear receptor subfamily 4, group A, member 1) and Nr4a2 promoters (supplemental Table S1, available at www.jneurosci.org as supplemental material).
Data analysis.
ANOVAs were performed in all experiments using SigmaStat (version 2.03; Systat Software, Port Richmond, CA), Systat (version 7.0.1), or Statistica 7 (StatSoft, Tulsa, OK). Repeated-measures ANOVA were performed when appropriate. For fear conditioning data analysis, Student–Newman–Keuls post hoc tests were used. For analysis of electrophysiology experiments, Tukey's post hoc tests were used. Simple planned comparisons were made using Student's t test. For the quantitative real-time reverse transcription (RT)-PCR data, we used a Kruskal–Wallis statistic test to determine significance. Experimenters were blind to genotype, and genotypes were confirmed after behavioral and electrophysiological tests. ChIP data analysis was performed using a factorial ANOVA and Newman–Keuls post hoc test.
Results
HDAC inhibition selectively in the hippocampus enhances memory consolidation for contextual fear conditioning but not cued fear conditioning
Previous studies have found that intracerebroventricular and intraperitoneal injections of HDAC inhibitors increase histone acetylation and have beneficial effects on memory (Alarcon et al., 2004; Korzus et al., 2004; Levenson et al., 2004). Because intracerebroventricular and intraperitoneal methods of drug administration lack spatial specificity, these studies do not define the specific brain regions that mediate the effects of histone acetylation on memory. We were therefore interested in examining the effects of direct hippocampal delivery of the HDAC inhibitor TSA on fear conditioning. TSA is a member of the hydroxamate class of HDAC inhibitors, which also includes suberoylanilide hydroxamic acid, pyroxamide, and Scriptaid (Marks et al., 2004). TSA is a naturally occurring molecule that reversibly inhibits HDACs and is effective at millimolar concentrations in vivo (Korzus et al., 2004; Levenson et al., 2004). To determine whether direct delivery of TSA to the hippocampus produces selective changes in hippocampus-dependent memory storage, we examined the effects of TSA on memory for contextual and cued fear conditioning. In these two learning tasks, mice learn to associate a footshock with either a context or cue in a single training session (for review, see Maren and Quirk, 2004). Cued fear conditioning depends on the amygdala but not the hippocampus, whereas contextual fear conditioning is dependent on both the hippocampus and the amygdala (for review, see Maren, 2001). Memory for the conditioned stimulus was measured as freezing, an absence of visible movement, when presented with the conditioned context or cue in a 24 h retention test. We first examined the effect of TSA on memory for contextual fear. Previous studies examining the effects of HDAC inhibitors on memory have used pretraining drug treatment (Alarcon et al., 2004; Korzus et al., 2004; Levenson et al., 2004), and, as a result, it is not known whether HDAC inhibition affects memory by altering acquisition, consolidation, or retrieval. We administered injections of TSA immediately after conditioning (1 μl of 16.5 mm per side) and found that TSA-treated mice showed significantly higher levels of freezing when reexposed to the conditioned context (Fig. 1A) (41.7 ± 5.6%; t(20) = −2.20; p < 0.05) than mice receiving vehicle (Fig. 1A) (1 μl of 50% EtOH per side; 25.5 ± 4.7% freezing). This experiment demonstrated that HDAC inhibition is capable of enhancing memory consolidation. We also delivered TSA injections 4 h before a 24 h retention test to evaluate the effect of TSA on memory retrieval. This time point was chosen because we observed in our Western blot analysis that acetylation of histone H3 reached a peak 4 h after TSA injection (Fig. 1C). Mice receiving TSA injections 4 h before testing showed similar levels of freezing (21.1 ± 4.2%; t(14) = −0.47; p > 0.05) to littermates receiving vehicle (23.7 ± 3.7%) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Together, these results demonstrate that intrahippocampal delivery of an HDAC inhibitor enhances contextual fear conditioning by affecting the consolidation stage of contextual fear memory.
Figure 1.

Intrahippocampal injection of TSA enhances the consolidation of contextual fear memory and increases histone H3 acetylation in the hippocampus. A, C57BL/6J male mice were fitted with intrahippocampal cannulas, fear conditioned, and injected with either VEH or TSA (16.5 mm TSA). Mice receiving TSA immediately after conditioning (n = 11) exhibited a significant increase in levels of freezing in a 24 h retention test, performed in the same conditioned context compared with mice receiving vehicle (n = 11). B, Mice receiving either TSA (n = 5) or vehicle (n = 6) immediately after conditioning showed no difference in levels of freezing during a cued fear conditioning test 24 h after conditioning, either before (Pre CS) or during (CS) presentation of the tone cue. C, Western blot analysis of acid-extracted nuclear lysates shows increased histone H3 acetylation in the hippocampus taken from mice injected with TSA (16.5 mm TSA) compared with mice injected with vehicle. Samples were obtained 2, 4, or 24 h after injection and normalized by Bradford assay. Anti-β-actin signal shows equivalent loading of protein samples in each lane. D, Intrahippocampal injection of TSA locally increases histone H3 acetylation in the hippocampus. Perfusion-fixed and immunostained coronal brain sections were prepared from mice killed 4 h after receiving intrahippocampal injections of vehicle or TSA (16.5 mm TSA). A representative section from a mouse treated with vehicle shows negligible histone H3 acetylation (VEH, top right). Bottom right shows higher-magnification 40× image of CA1. A representative section from a mouse treated with TSA shows increased histone H3 acetylation in hippocampal area CA1 (TSA, top left). Bottom left shows higher-magnification 40× image of CA1. Nuclear staining with DAPI demonstrates normal hippocampal morphology in sections from both vehicle- and TSA-treated mice. DG, Dentate gyrus. *p < 0.05.
Because injections were delivered in a site-specific manner to the hippocampus, we predicted that there would be no effect on memory for cued fear conditioning, a hippocampus-independent task. This experiment was identical to the contextual fear conditioning experiment, except that mice were tested for memory of the cue–shock association by presenting the conditioned noise in a novel environment. No effect of TSA was observed in the cued fear conditioning 24 h retention test, either before or after presentation of the conditioned noise (Fig. 1B) (pre-CS: TSA-treated mice exhibited 10.0 ± 3.9% freezing; vehicle-treated mice exhibited 8.3 ± 3.2% freezing; t(9) = −0.33, p > 0.05; CS: TSA-treated mice exhibited 64.6 ± 7.3% freezing; vehicle-treated mice exhibited 55.7 ± 7.8% freezing; t(9) = −0.82, p > 0.05).
Our current cued fear conditioning experiments used a relatively low 0.75 mA footshock compared with the 1.5 mA footshock that we normally use. The stronger shock results in 78.9% freezing in the same C57BL/6J animals (Wood et al., 2005), and we therefore do not believe that a ceiling in freezing was reached in TSA-treated animals that exhibited 64.6% freezing. We also examined freezing minute by minute during the cued fear conditioning retention test and did not observe any differences (data not shown). Thus, we believe that we did not miss a period of time within the retention test during which TSA-treated animals did exhibit significantly higher freezing. These results demonstrate that site-specific administration of an HDAC inhibitor into the hippocampus enhances memory for contextual fear but not cued fear conditioning, suggesting that intrahippocampal delivery of an HDAC inhibitor selectively affects the hippocampus and not other memory-related systems.
To determine whether the enhancement in contextual fear conditioning induced by hippocampus-specific administration of TSA correlated with increased histone acetylation, C57BL/6J mice were fitted with intrahippocampal cannulas, injected with TSA (1 μl of 16.5 mm per side) or vehicle (1 μl of 50% EtOH per side), and killed at different time points after injection. Acid-extracted histones were prepared from hippocampal nuclear lysates and separated by SDS-PAGE, and the level of acetylated histone H3 was determined by Western blot analysis using acetylation state-specific antibodies. As shown in Figure 1C, acetylation of histone H3 is increased 2 and 4 h after administration of TSA but not after treatment with vehicle. Twenty-four hours after injection, histone acetylation returned to normal levels. This time-dependent histone acetylation pattern was consistently observed in two additional replicate experiments (data not shown). Similar results were obtained for acetylation of histone H4 (data not shown). To rule out the possibility that the effects of TSA were attributable to changes in CREB phosphorylation state (Michael et al., 2000), we examined the effect of TSA on CREB phosphorylation at site Ser133 after contextual fear conditioning. Mice were subjected to contextual fear conditioning and injected with TSA (1 μl of 16.5 mm per side) or vehicle (1 μl of 50% EtOH per side). No differences in CREB Ser133 phosphorylation were observed 0.5, 2, or 4 h after conditioning between TSA- and vehicle-treated mice (data not shown).
To examine the spatial range of effect of TSA on histone acetylation after injection, acetylation of histone H3 was evaluated 4 h after injection in perfusion-fixed coronal brain slices using immunohistochemistry with an antibody against acetylated H3. HDAC inhibition increased acetylation of histone H3 mainly in area CA1 of the hippocampus as well as in the upper blade of the dentate gyrus (Fig. 1D). Nuclear staining with Hoechst dye demonstrated that there was no observable damage to the hippocampus after injection in both vehicle- and TSA-treated mice. We did not observe histone H3 acetylation in other surrounding brain regions, including the cortex (except along the cannulas tracks), striatum, and amygdala (data not shown). Together, these results show that administration of TSA via intrahippocampal cannulas increases histone acetylation in a site-specific manner, suggesting that increasing histone acetylation by HDAC inhibition within the hippocampus enhances the consolidation of memory for contextual fear conditioning.
HDAC inhibition enhances LTP induced by one-train stimulation via a transcription-dependent mechanism
To examine the cellular mechanisms by which increases in histone acetylation influence hippocampal function, we studied the effects of TSA on LTP in hippocampal area CA1. LTP, defined as the activity-dependent change in the strength of neuronal connections, is a form of long-lasting synaptic plasticity that has been proposed as a cellular model for learning and memory (for review, see Malenka, 2003; Lynch, 2004). At least two mechanistically distinct forms of LTP have been demonstrated in hippocampal area CA1, an E-LTP, which does not require transcription or translation, and a late-phase LTP, which requires translation and transcription (Davis and Squire, 1984; Krug et al., 1984) (for review, see Huang and Kandel, 1996; Kandel, 2001). Previous studies examining the effect of HDAC inhibition on LTP used induction protocols with two 100 Hz tetanizations (Levenson et al., 2004) or four 100 Hz tetanizations (Alarcon et al., 2004). LTP after both of these protocols is dependent on transcription. This confounds studying the molecular mechanisms underlying HDAC inhibition because inhibitors of transcriptional mechanisms will disrupt the underlying electrically induced LTP as well as the enhancement of LTP by HDAC inhibition. Here, we studied the effects of TSA on transient E-LTP induced by a single 1 s, 100 Hz train. Importantly, this form of LTP is independent of transcription or translation, protein kinase A, and CREB (for review, see Pittenger et al., 2002; Nguyen and Woo, 2003). Therefore, this approach allows us to identify genetic and pharmacological manipulations that selectively disrupt the enhancement in LTP attributable to TSA treatment without affecting the underlying E-LTP. These experiments can in turn define the specific mechanisms mediating the enhancement of LTP by HDAC inhibition.
LTP induced by a single 1 s, 100 Hz train in C57BL/6J hippocampal slices was significantly enhanced by TSA (fEPSP slope, 160.9 ± 25.4%) compared with slices treated with vehicle (fEPSP slope, 93.8 ± 8.5%) at 1 h after tetanic stimulation (Fig. 2A) (tetanization × treatment interaction, F(1,22) = 4.88, p < 0.05; post hoc analysis, TSA vs VEH within tetanized groups, q(2,22) = 4.54, p < 0.01). A two-pathway protocol (Fig. 2C) was used to control for the effects of TSA on baseline responses. Both pathways received baseline stimulation every minute, but LTP was only elicited in one pathway, with the other serving as a control. As shown in Figure 2B, in the untetanized pathway, TSA had no effect on baseline responses (fEPSP slope at last minute of recording, 100.5 ± 6.3%) compared with vehicle (98.7 ± 13.4%; post hoc analysis, TSA vs VEH within untetanized groups, q(2,22) = 0.12, p > 0.05). Thus, only in the tetanized LTP-inducing pathway did we observe that TSA significantly enhanced E-LTP (Fig. 2A). Furthermore, we and others have not observed changes in baseline electrophysiological properties in HDAC inhibitor-treated slices (Alarcon et al., 2004; Levenson et al., 2004). These findings are important because they demonstrate that TSA alone does not induce synaptic plasticity (Yeh et al., 2004) and demonstrate that TSA does not affect basal synaptic function. Instead, TSA appears to act by modulating LTP occurring in the tetanized pathway.
Figure 2.

TSA enhances one-train E-LTP via a transcription-dependent mechanism. A, Long-term potentiation (E-LTP) was induced by a single 1 s, 100 Hz train in the CA1 region of hippocampal slices from C57BL/6J mice in the presence of VEH or TSA (1.65 μm). Slices treated with vehicle (n = 8 slices from 7 mice) showed a transient potentiation that decays to baseline by 60 min, whereas slices treated with TSA (n = 12 slices from 6 mice) showed a significantly more robust and longer-lasting potentiation (p < 0.05). Vehicle or TSA was administered throughout the experiment. B, There was no effect of TSA (n = 12 slices from 6 mice) compared with vehicle (n = 8 slices from 7 mice) on baseline responses in a second pathway that did not receive tetanization (p > 0.05). Insets in A and B show superimposed sample sweeps from the first 5 and last 5 min of the recording for VEH- and TSA-treated slices. Calibration: 5 mV, 5 ms. C, Schematic diagram showing two-pathway LTP induction protocol (adapted from Kandel et al., 2000). Both pathways received alternating baseline stimulation every minute, but LTP was only elicited in one pathway, with the other serving as a control. D, Treatment with the transcription inhibitor ActD (25 μm) blocks the enhancement of E-LTP by TSA. In this experiment, a single stimulating electrode was used, and solutions containing TSA+VEH or TSA+ActD were perfused for 20 min before tetanization and throughout the 2 h recording period. TSA+VEH-treated slices (n = 4 slices from 3 mice) have significantly enhanced LTP 100–120 min after tetanic stimulation (p < 0.05) compared with slices treated with TSA+ActD (n = 4 slices from 3 mice). Insets show superimposed average sample sweeps from the first 5 and last 5 min of the recording for TSA+VEH- and TSA+ActD-treated slices. Calibration: 5 mV, 5 ms.
To determine whether the TSA-enhanced LTP is dependent on transcription, we examined LTP induced by a single 1 s, 100 Hz train in the presence of TSA (1.65 μm TSA) or TSA plus actinomycin D (1.65 μm TSA and 25 μm ActD). ActD is a transcriptional inhibitor that has been shown to have no effect on basal synaptic transmission or on E-LTP (Nguyen et al., 1994). As shown in Figure 2D, C57BL/6J hippocampal slices treated with TSA (fEPSP slope at last minute of recording, 169.0 ± 8.6%) have significantly enhanced LTP 100–120 min after tetanic stimulation (F(1,76) = 27.5, p < 0.05) compared with slices treated with TSA and ActD (113.6 ± 7.3%). These results demonstrate that HDAC inhibition generates a transcription-dependent long-lasting form of LTP.
CREB is required for the enhancement of memory and synaptic plasticity by HDAC inhibition
We next set out to identify the specific transcriptional mechanisms that mediate the enhancements in both memory and LTP induced by TSA. The transcription factor CREB is thought to activate transcription through the coactivator CBP and its associated HAT activity (for review, see Goodman and Smolik, 2000), and the significant phenotypic overlap between memory and synaptic plasticity deficits attributable to genetic disruption of CREB or CBP suggests that they function together during memory processes (Pittenger et al., 2002; Josselyn, 2005; Wood et al., 2005). Given this potential modulation of CREB-mediated transcription by histone acetylation, we hypothesized that CREB might have a role in the enhancement of memory and E-LTP after HDAC inhibition. We examined mice with targeted deletions of the α and Δ isoforms of Creb (CREBαΔ mice) on a defined F1 hybrid genetic background (C57BL/6J and 129/SvEvTac) (Walters and Blendy, 2001; Graves et al., 2002). CREBαΔ mutant mice and wild-type CREB+/+ littermates were fitted with intrahippocampal cannulas and subjected to contextual fear conditioning. In agreement with a previous study examining fear conditioning in CREBαΔ mutant mice on this genetic background (Graves et al., 2002), CREBαΔ mutant mice had significantly reduced levels of freezing (Fig. 3) (vehicle, 14.0 ± 3.7%; main effect of genotype, F(1,29) = 64.24, p < 0.001; genotype × treatment interaction, F(2,29) = 3.22, p < 0.05; post hoc analysis, wild-type vs CREBαΔ within VEH groups q(2,29) = 3.76, p < 0.05) compared with wild-type CREB+/+ littermates (Fig. 3) (vehicle, 32.2 ± 3.9%). Similar to our observations in Figure 1A, wild-type CREB+/+ mice treated with 16.5 mm TSA showed significantly higher levels of freezing than wild-type CREB+/+ mice receiving vehicle (post hoc analysis, 16.5 mm TSA vs VEH within wild-type groups, q(3,29) = 4.36, p < 0.05). In contrast, CREBαΔ mutant mice treated with 16.5 mm TSA showed similar levels of freezing to CREBαΔ mutant mice receiving vehicle. Additionally, for the 33 mm TSA treatment, wild-type CREB+/+ mice treated with 33 mm TSA showed significantly higher levels of freezing than wild-type CREB+/+ mice receiving vehicle (post hoc analysis, 33 mm TSA vs VEH within wild-type groups, q(2,29) = 3.82, p < 0.05), whereas CREBαΔ mutant mice treated with 33 mm TSA showed similar levels of freezing to CREBαΔ mutant mice receiving vehicle. Together, these results suggest that the enhancement of memory for contextual fear conditioning by HDAC inhibition requires the transcription factor CREB.
Figure 3.

Enhancing histone acetylation by treatment with TSA does not enhance memory for contextual fear in CREBαΔ mutant mice. CREBαΔ mutant mice and wild-type (WT) littermates were fitted with intrahippocampal cannulas, subjected to contextual fear conditioning, and then immediately injected with either VEH or TSA (16.5 or 33 mm TSA). Injected mice were given a retrieval test 24 h later. Similar to a previous study examining contextual fear conditioning in CREBαΔ mutant mice (Graves et al., 2002), CREBαΔ mutant mice treated with vehicle (n = 6) showed significantly lower levels of freezing than wild-type littermates treated with vehicle (n = 6). Wild-type mice treated with either 16.5 mm TSA (n = 6) or 33 mm TSA (n = 6) showed significantly higher levels of freezing than wild-type mice treated with vehicle (n = 6), recapitulating results observed in C57BL/6J mice (Fig. 1). In contrast, no differences in levels of freezing were observed between CREBαΔ mutant mice treated with 16.5 mm TSA (n = 5) or 33 mm TSA (n = 6) and CREBαΔ mutant mice treated with vehicle (n = 6). *p < 0.05.
Our data demonstrate that the HDAC inhibitor TSA enhances LTP in area CA1 of the hippocampus and that this enhancement relies on transcription (Fig. 2). Given our results showing that TSA enhances memory for contextual fear via the transcription factor CREB, we next examined whether HDAC inhibition enhances LTP in hippocampal slices from CREBαΔ mutant mice. As mentioned previously, one-train E-LTP has been shown to be independent of CREB function (Pittenger et al., 2002) (for review, see Nguyen and Woo, 2003). Therefore, if disruption of CREB impairs the enhancement of LTP by HDAC inhibition, we can conclude that this is because the action of TSA is dependent on CREB and not because the disruption of CREB affects the underlying electrically induced E-LTP. Slices from CREBαΔ mutant mice still exhibit the transient potentiation characteristic of E-LTP (compare vehicle-treated slices in Fig. 4A,B), demonstrating that this form of LTP is not altered in these mutant mice. Hippocampal slices from wild-type CREB+/+ F1 B6/129 hybrids treated with TSA show significantly enhanced LTP (fEPSP slope at last minute of recording, 147.8 ± 10.6%) compared with vehicle (109.1 ± 5.4%) treated slices (Fig. 4A) (genotype × treatment interaction, F(1,19) = 5.02, p < 0.05; post hoc analysis, VEH vs TSA within wild-type groups, p < 0.05). In contrast, hippocampal slices from CREBαΔ mutant littermate mice failed to exhibit enhanced LTP in the presence of TSA (104.1 ± 5.3%) compared with vehicle (107.8 ± 10.4%) treated slices (Fig. 4B) (post hoc analysis, VEH vs TSA within CREBαΔ groups, p > 0.05). These results suggest that the enhancement of LTP by TSA requires the transcription factor CREB. We also examined whether a structurally dissimilar HDAC inhibitor, sodium butyrate, would also depend on CREB to exert its effects on LTP. We found that sodium butyrate paired with one train of electrical stimuli produced a robust, long-lasting potentiation in hippocampal slices from CREB wild-type mice and that this form of LTP was significantly impaired in CREBαΔ mutant mice (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). This finding suggests that the CREB pathway may be a common route by which multiple HDAC inhibitors influence synaptic plasticity. Together with the observation that administration of TSA via intrahippocampal cannula does not enhance memory for contextual fear conditioning in CREBαΔ mutant mice, our results suggest that CREB-mediated transcription is an underlying mechanism for the enhancement of memory storage and synaptic plasticity by HDAC inhibition. In our model, histone deacetylases may be functioning as memory suppressor genes (Abel et al., 1998), and it is necessary to overcome HDAC repression of transcription via a sufficiently strong activity-dependent stimulus (e.g., four-train LTP induction) or by removing the repression via HDAC inhibitors.
Figure 4.

Increasing histone acetylation by treatment with TSA does not enhance one-train E-LTP in CREBαΔ or cbpKIX/KIX mutant mice. LTP was induced by a single 1 s, 100 Hz train in the CA1 region of hippocampal slices in the presence of VEH or TSA (1.65 μm). Hippocampal slices were treated with vehicle or TSA (indicated by black line) for 20 min before tetanization (indicated by an arrow) and throughout the recording period. A, Hippocampal slices from wild-type (WT) CREB+/+ mice treated with vehicle (n = 9 slices from 7 mice) showed a transient potentiation that gradually decreased to baseline. In contrast, slices treated with TSA (n = 9 slices from 9 mice) showed a significantly more robust and longer-lasting potentiation. Insets in A show superimposed sample sweeps from the first 5 and last 5 min of the recording for VEH- and TSA-treated slices. B, Hippocampal slices from CREBαΔ mutant mice treated with TSA (n = 9 slices from 9 mice) failed to produce robust and longer-lasting potentiation induced by one train paired with TSA compared with vehicle-treated slices (n = 11 slices from 9 mice). Insets in B show superimposed sample sweeps from the first 5 and last 5 min of the recording for VEH- and TSA-treated slices. Calibration: 5 mV, 5 ms. C, Hippocampal slices from wild-type cbp+/+ mice treated with TSA (n = 6 slices from 6 mice) showed a significant enhancement in E-LTP compared with VEH-treated slices (n = 5 slices from 5 mice). D, In contrast, hippocampal slices from cbpKIX/KIX homozygous mutant mice showed no enhancement in E-LTP induced in the presence of TSA (n = 7 slices from 5 mice) as opposed to VEH (n = 7 slices from 6 mice). Insets in C and D show averaged sample sweeps from the first 5 and last 5 min of the recording. Calibration: 5 mV, 5 ms.
Genetic disruption of the CREB-binding KIX domain of CBP blocks the ability of HDAC inhibition to enhance E-LTP
We next tested the effects of HDAC inhibition on LTP in mutant mice in which the archetype interaction between CREB and the transcriptional coactivator CBP is disrupted. Phosphorylation of CREB at Ser133 induces the association of CREB with CBP via their KID and KIX domains, respectively (Parker et al., 1996). Mice carrying mutations in three highly conserved residues (Tyr650Ala, Ala654Gln, and Tyr658Ala) within the CBP KIX domain (cbpKIX/KIX) (Kasper et al., 2002) are essentially normal, apart from a modest decrease in thymus size (Kasper et al., 2002). However, mouse embryonic fibroblast derived from cbpKIX/KIX mice are compromised in their ability to support CREB-mediated transcription in transient transfection assays (Kasper et al., 2002), and we have shown recently that cbpKIX/KIX mice have deficits in long-term memory for contextual fear conditioning and novel object recognition (Wood et al., 2006). Thus, the interaction between CREB and CBP is critical for long-term memory storage, but it is not known whether this interaction is necessary for the enhancement of synaptic plasticity by HDAC inhibition. We tested the importance of the CREB:CBP interaction by examining whether TSA was capable of enhancing LTP in cbpKIX/KIX mutant mice.
Hippocampal slices from wild-type cbp+/+ mice treated with TSA show significantly enhanced LTP (last minute, 186.5 ± 7.4%) compared with slices treated with vehicle (last minute, 92.3 ± 10.7%) (Fig. 4C) (genotype × treatment interaction, F(1,19) = 38.61, p < 0.0001; post hoc analysis, VEH vs TSA within wild-type groups, p < 0.01). In contrast, hippocampal slices from cbpKIX/KIX homozygous mutant mice failed to exhibit enhanced E-LTP in the presence of TSA (last minute, 103.9 ± 5.8%) compared with vehicle (last minute, 105.3 ± 10.3%) treated slices (Fig. 4D) (post hoc analysis, VEH vs TSA within cbpKIX/KIX groups, p > 0.05). As in CREBαΔ knock-outs, slices from cbpKIX/KIX mutant mice still exhibit the transient potentiation characteristic of E-LTP (compare vehicle-treated slices in Fig. 4C,D), demonstrating that this form of LTP is not altered in these mutant mice. These results further support the hypothesis that CREB-mediated transcription is involved in the effects of TSA on memory and synaptic plasticity and highlight the interaction of CREB and CBP as a key step in the regulation of TSA-induced transcription underlying enhancement of E-LTP. Based on our findings in CREBαΔ and cbpKIX/KIX mice, we therefore predicted that CREB target genes would be affected by TSA, which we examined next using quantitative real-time RT-PCR.
Expression of the CREB target genes Nr4a1 and Nr4a2 are selectively increased by HDAC inhibition after contextual fear conditioning
To determine whether CREB-mediated transcription is affected by TSA, we used quantitative real-time RT-PCR to examine the expression of several CRE-containing genes that have been shown to be regulated by CREB. C57BL/6J mice were fitted with intrahippocampal cannulas, subjected to contextual fear conditioning, and immediately injected with either TSA (1 μl of 16.5 mm per side) or vehicle (1 μl of 50% EtOH per side). At 2 and 4 h after conditioning, mice were killed, hippocampi were removed, and total RNA was purified for conversion into cDNA (for details, see Materials and Methods). We examined the expression of the following genes: Egr1 (early growth response 1), Fos, Dusp1 (dual specificity phosphatase 1) Nr4a1, Jun, Icer, Nr4a3, 14-3-3ε, Bdnf4 (brain-derived neurotrophic factor, promoter 4), Dynorphin, Gadd45b (growth arrest and DNA-damage-inducible 45 β), and Nrn1 (neuritin 1), which have all been shown to contain one or more CRE motifs and many of which are regulated by histone acetylation or involved in memory storage (Nedivi et al., 1996; Schmidt-Kastner et al., 1996; Tao et al., 1998; Tischmeyer and Grimm, 1999; Chen et al., 2002; Kida et al., 2002; Davis et al., 2003; Fass et al., 2003). Surprisingly, we found that, of these 12 genes, only Nr4a1 [also known as nur77 (nuclear receptor 77) and NGFI-B (nerve growth factor inducible-B)] had significantly increased expression 2 h after conditioning and administration of TSA (Table 1). By 4 h after conditioning, Nr4a1 expression was back to normal baseline levels. TSA treatment alone had no effect on the expression of any of the examined genes (data not shown). This finding is in agreement with our LTP experiments showing that TSA treatment has no effect on an untetanized pathway (Fig. 2C) and indicates that increasing histone acetylation via intrahippocampal administration of TSA modulates the induction of CREB-mediated gene expression but alone is not capable of changing the expression of these genes. We performed a similar experiment in CREBαΔ mutant and wild-type littermate mice. Because expression of the closely related Nr4 family member Nr4a2 is frequently regulated in parallel with Nr4a1 expression (Kovalovsky et al., 2002; Klopotowska et al., 2005) and because Nr4a1 and Nr4a2 can heterodimerize to activate transcription (Maira et al., 1999), we added Nr4a2 to our analysis (see Discussion). TSA-enhanced expression of Nr4a1 and Nr4a2 was observed 2 h after conditioning in wild-type CREB+/+ littermates (Fig. 5A). In contrast, we found that TSA treatment after contextual fear conditioning in CREBαΔ mutant mice did not alter the expression of Nr4a1 and Nr4a2 2 h after conditioning (Fig. 5A), showing that the effects of TSA on Nr4a1 and Nr4a2 expression are CREB dependent. These results demonstrate that HDAC inhibition has a selective effect on hippocampal gene expression and suggest that the CREB-mediated increase in the hippocampal expression of Nr4a1 and Nr4a2 after contextual fear conditioning may contribute to the enhancement of memory and LTP by HDAC inhibitors.
Table 1.
Quantitative real-time RT-PCR results for all genes examined 2 and 4 h after contextual fear conditioning in C57BL/6J mice
| Gene | Treatment | 2 h |
4 h |
||||
|---|---|---|---|---|---|---|---|
| Fold change | +SEM | −SEM | Fold change | +SEM | −SEM | ||
| Egr1 | VEH | 1 | 0.08 | 0.07 | 1 | 0.08 | 0.08 |
| TSA | 0.98 | 0.14 | 0.13 | 1.39 | 0.25 | 0.22 | |
| Fos | VEH | 1 | 0.19 | 0.16 | 1 | 0.07 | 0.07 |
| TSA | 0.92 | 0.05 | 0.05 | 0.94 | 0.11 | 0.1 | |
| Dusp1 | VEH | 1 | 0.31 | 0.24 | 1 | 0.15 | 0.13 |
| TSA | 0.88 | 0.09 | 0.09 | 0.78 | 0.13 | 0.12 | |
| Nr4a1 | VEH | 1 | 0.01 | 0.01 | 1 | 0.03 | 0.03 |
| TSA | 1.23* | 0.02 | 0.02 | 1.08 | 0.07 | 0.06 | |
| Jun | VEH | 1 | 0.09 | 0.08 | 1 | 0.16 | 0.14 |
| TSA | 0.85 | 0.09 | 0.08 | 1.05 | 0.21 | 0.18 | |
| Icer | VEH | 1 | 0.08 | 0.07 | 1 | 0.09 | 0.08 |
| TSA | 0.78 | 0.14 | 0.12 | 0.95 | 0.11 | 0.1 | |
| Nor-1 (Nr4a3) | VEH | 1 | 0.07 | 0.06 | 1 | 0.18 | 0.15 |
| TSA | 0.82 | 0.16 | 0.14 | 1.07 | 0.23 | 0.19 | |
| 14-3-3ε | VEH | 1 | 0.07 | 0.06 | 1 | 0.31 | 0.24 |
| TSA | 0.95 | 0.09 | 0.08 | 1.13 | 0.03 | 0.03 | |
| Bdnf4 | VEH | 1 | 0.16 | 0.14 | 1 | 0.31 | 0.24 |
| TSA | 0.69 | 0.07 | 0.06 | 1 | 0.41 | 0.29 | |
| Dynorphin | VEH | 1 | 0.34 | 0.25 | 1 | 0.31 | 0.23 |
| TSA | 0.83 | 0.14 | 0.12 | 1.45 | 0.57 | 0.41 | |
| Gadd45b | VEH | 1 | 0.13 | 0.12 | 1 | 0.15 | 0.13 |
| TSA | 0.89 | 0.05 | 0.05 | 0.78 | 0.14 | 0.12 | |
| Nrn1 | VEH | 1 | 0.1 | 0.09 | 1 | 0.19 | 0.16 |
| TSA | 0.93 | 0.08 | 0.08 | 1.14 | 0.13 | 0.12 | |
Only Nr4a1 had significantly higher expression in the hippocampi of mice treated with TSA compared with vehicle (see Fig. 5). n = 4 for each group (TSA or VEH).
Figure 5.

TSA treatment after contextual fear conditioning affects the expression of only a subset of CREB target genes. A, CREBαΔ mutant mice and wild-type (WT) littermates were fitted with intrahippocampal cannulas, subjected to contextual fear conditioning, and then immediately injected with either vehicle (50% EtOH) or TSA (16.5 mm TSA). At 2 h after conditioning, mice were killed, hippocampi were removed, and total RNA was purified for conversion into cDNA. Expression levels were normalized to the following house keeping genes: Hprt, Actg, and Tubulin. There were no significant differences in gene expression for Nr4a1 and Nr4a2 in CREBαΔ mutant mice treated with TSA compared with those receiving vehicle. In contrast, the expression of Nr4a1 and Nr4a2 was significantly increased in CREB wild-type littermates treated with TSA. *p < 0.05. B, TSA treatment after contextual fear conditioning enhances histone acetylation at Nr4a1 and Nr4a2 promoters. Animals were cannulated and injected with TSA or vehicle. At 2 h after conditioning, mice were killed, and hippocampi were removed and crosslinked with formaldehyde. Chromatin was immunoprecipitated with anti-acetylated histone H3 and H4 antibodies (AcH3, AcH4). Immunoprecipitated DNA was quantified for enrichment of Nr4a1 and Nr4a2 promoter, normalized to input chromatin, and expressed as a ratio to vehicle-injected animals. *p < 0.05 for comparison of histone acetylation (H3 and H4) between drug- and vehicle-treated animals.
Nr4a1 expression is upregulated in the hippocampus immediately or shortly (0.5 h) after contextual fear conditioning (von Hertzen and Giese, 2005a,b; Keeley et al., 2006). Interestingly, we did not observe an increase in Nr4a1 or Nr4a2 in the hippocampus at a later time point (2 h) after contextual fear conditioning (data not shown), suggesting that the normal induction of Nr4a1 or Nr4a2 is usually short-lived. TSA administration immediately after contextual fear conditioning caused the expression of Nr4a1 and Nr4a2 to be elevated 2 h after training, whereas TSA treatment alone had no effect on the expression of either gene (data not shown). Therefore, TSA may act to augment the initial expression of Nr4a1 and Nr4a2 and/or to prolong their expression, which would explain why we observe their enhanced expression 2 h after contextual fear conditioning and TSA treatment.
There are several possible explanations for how the relatively small changes in the expression of Nr4a1 and Nr4a2 could produce such large effects on memory. The first is that our gene expression experiments were performed on RNA isolated from the entire hippocampus. Although many neurons in area CA1 show increased histone acetylation after TSA injection, only a small fraction of cells in area CA3 and the dentate gyrus are affected (Fig. 1D). Second, our work shows that these genes are induced only when TSA was paired with fear conditioning: TSA treatment alone did not result in increases in expression of these genes at this time point after conditioning. Recent research has suggested that ∼40% of hippocampal neurons are recruited during learning (Guzowski et al., 2006). These factors suggest that only a fraction of cells within the hippocampus is coactivated by both formation of the contextual fear conditioning memory and TSA treatment. Therefore, in that subset of neurons, the change in gene expression that results from the combined effects of TSA and fear conditioning is likely to be much higher. It is also important to note that, apart from Nr4a1 and Nr4a2, there may be other CRE-containing genes regulated via CBP and histone acetylation that play a role in the effects of TSA on memory and plasticity.
In cell culture, TSA has been shown to induce recruitment of both RNA polymerase II and TFIIB (transcription factor IIB) at the Nr4a1 promoter, suggesting that histone acetylation controls access of the basal transcription machinery to the promoter (Fass et al., 2003). Therefore, we used ChIP assays to examine the effect of HDAC inhibition on histone acetylation at the promoter regions of Nr4a1 and Nr4a2. C57BL/6J mice were fitted with intrahippocampal cannulas and subjected to contextual fear conditioning followed by injection of TSA or vehicle. ChIP assays were performed on hippocampal samples taken 2 h after training (a time point at which TSA increases Nr4a1 and Nr4a2 gene expression) (Fig. 5A). Acetylation of both histone H3 and H4 was significantly increased at the promoter regions of Nr4a1 and Nr4a2 by TSA treatment after contextual fear conditioning (Fig. 5B). These results support the hypothesis that TSA-mediated increases in histone acetylation at Nr4a1 and Nr4a2 promoter regions facilitate their expression during memory consolidation.
Because this manuscript is focused on the mechanisms by which TSA affects hippocampal function, we did not assess the effects of TSA in other brain regions. CREB and CBP may indeed act in other parts of the brain to mediate memory formation. However, we have seen that CBPKIX/KIX mice, in which the domain of CBP that mediates the interaction with CREB is mutated, have deficient hippocampus-dependent contextual fear memory but normal hippocampus-independent cued fear memory (Wood et al., 2006). A similar pattern has also been observed by Alarcon et al. (2004) in their heterozygous null CBP mice. These findings suggest that the CREB:CBP interaction may be of particular importance for hippocampus-dependent memory formation or that the hippocampus is especially sensitive to alterations in CBP function or histone acetylation.
Discussion
The key issue addressed in our study was the molecular mechanism by which HDAC inhibitors enhance memory storage. This is a timely question considering the clinical use of HDAC inhibitors for cancer therapy (for review, see Lund and van Lohuizen, 2004) and their potential use for treatment of mental retardation and neurodegenerative diseases (Steffan et al., 2001; Hockly et al., 2003). The use of HDAC inhibitors has rapidly emerged from the literature examining the role of chromatin modification for transcriptional regulation underlying memory processes (Oike et al., 1999; Bourtchouladze et al., 2003; Alarcon et al., 2004; Korzus et al., 2004; Levenson et al., 2004; Yeh et al., 2004; Wood et al., 2005, 2006; Chwang et al., 2007). However, this is the first study to identify a transcription factor/coactivator complex and particular genes that are associated with HDAC inhibitor-mediated enhancement of memory and synaptic plasticity.
A CREB:CBP KIX domain complex is required for the effects of increased histone acetylation on hippocampal synaptic plasticity and hippocampus-dependent memory
In this study, we used techniques that allowed us to identify mechanisms that may mediate the effects of HDAC inhibition on synaptic plasticity. To do this, we studied the effects of TSA on hippocampal E-LTP. Because our single 100 Hz train E-LTP induction protocol is independent of transcription and translation (for review, see Nguyen and Woo, 2003), we were able to define the molecular nature of HDAC inhibitor-enhanced LTP. Using this protocol, we found that TSA treatment transformed transient E-LTP into a transcription-dependent, long-lasting form of LTP (Fig. 6A). Given these findings, we attempted to identify specific transcriptional mechanisms that underlie the effects of TSA on E-LTP. We found that TSA does not enhance LTP in CREBαΔ mutant mice and cbpKIX/KIX mice, suggesting that a key mechanism in the enhancement of LTP by HDAC inhibition is the formation of a complex between CREB and the CBP KIX domain. These results also demonstrate that HDAC inhibitors do not simply compensate for CREB or CBP histone acetyltransferase activity (Fig. 6). HDAC inhibitors can ameliorate memory and LTP deficits in certain CBP mutant mice (Alarcon et al., 2004; Korzus et al., 2004). However, the mice in these previous studies were heterozygous knock-outs (Alarcon et al., 2004) or transgenic mice expressing a transgene that contained a point mutation in the CBP HAT domain (Korzus et al., 2004). Importantly, both of those strains contain wild-type CBP that is still able to bind CREB, recruit basal transcription machinery, and perform histone acetylation (Fig. 6E). The observation that both of those previously studied cbp mutant strains (Alarcon et al., 2004; Korzus et al., 2004) were responsive to HDAC inhibitor treatment is consistent with our results using our previously described CBPΔ1 transgenic mice, which in addition to a truncated dominant-negative form of CBP also retain two wild-type alleles of cbp (Wood et al., 2005). We found that TSA was capable of enhancing hippocampal E-LTP in slices from CBPΔ1 transgenic mice, just as in wild-type littermates (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). This differential effect of HDAC inhibitors on distinct cbp mutant mice also serves as a caveat for future study of the usefulness of such drugs to treat disorders stemming from cbp disruption. HDAC inhibitors may be suitable for treating deficits attributable to some cbp mutations, but they may be ineffective at treating others.
Figure 6.
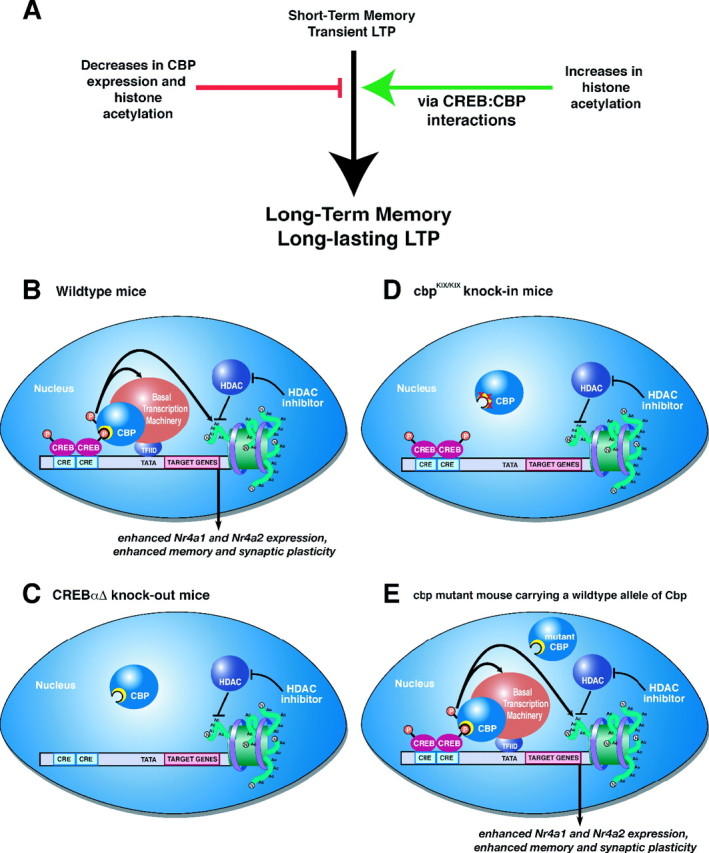
HDAC inhibition enhances memory and synaptic plasticity via a CREB:CBP interaction. A, Decreases in CBP activity and histone acetylation impair long-term memory and long-lasting forms of LTP. Conversely, increasing histone acetylation enhances long-term memory and LTP (as shown in this study; for other references, see Discussion). Our results demonstrate that these enhancements occur via a CREB:CBP complex and transcription of particular CREB-regulated genes such as Nr4a1 and Nr4a2. B, A schematic diagram of how activation of CREB by phosphorylation in wild-type mice recruits CBP via the CREB-binding domain (KIX; shown as yellow crescent). CBP in turn becomes activated (possibly by phosphorylation via CaMKIV) (Impey et al., 2002) and then regulates CREB-mediated transcription by interacting with the basal transcription machinery and associated cofactors and via its intrinsic histone acetyltransferase activity. HDAC inhibition requires CREB:CBP interaction to effectively enhance memory and synaptic plasticity. C, In the absence of CREB, as in the CREBαΔ mutant mice, increasing histone acetylation via HDAC inhibition has no effect because there is no transcription factor present to recruit the transcriptional coactivator CBP, which further recruits basal transcription machinery and acetylates histones to facilitate gene expression. D, Similarly, in cbpKIX/KIX mice that express a mutant CBP protein that blocks the main interaction with CREB, HDAC inhibition is also ineffective. This demonstrates that the interaction between CREB and CBP is necessary for the enhancement of memory and synaptic plasticity by HDAC inhibition, presumably because CBP function cannot be compensated for by simply increasing histone acetylation via HDAC inhibitors. Thus, it is likely that the role CBP plays in recruiting basal transcription machinery or other factors is crucial for gene expression required for the enhancement of memory and synaptic plasticity by HDAC inhibition. E, In other Cbp mutant mice, including Cbp heterozygous mice (Alarcon et al., 2004), transgenic mice expressing a mutant CBP protein with a mutation in the HAT domain (Korzus et al., 2004), and transgenic mice expressing a truncated inhibitory form of CBP (Wood et al., 2005), there is still at least one wild-type allele of Cbp present. This wild-type allele is sufficient to support the enhancement of memory and synaptic plasticity by HDAC inhibitors, because wild-type CBP can interact with CREB to recruit basal transcription machinery and acetylated histone proteins even in the presence of mutant CBP protein. This model is likely to apply to most partial CBP mutations in which wild-type CBP is still present.
Behaviorally, enhancement of memory consolidation for contextual fear conditioning induced by intrahippocampal injection of TSA was also dependent on CREB. Even treatment with twice the dose of TSA that produced memory enhancement in wild-type mice was incapable of enhancing memory in the CREBαΔ mutant mice. Importantly, histone acetylation is increased by the same amount in CREBαΔ mutant mice and wild-type littermates after TSA treatment (data not shown), demonstrating that TSA has similar overall effects on histone acetylation even in the presence of the CREBαΔ mutation. Much research suggests that CREB and CREB-mediated transcription (Bourtchuladze et al., 1994; Kogan et al., 1997; Gass et al., 1998; Josselyn et al., 2001, 2004; Barco et al., 2002; Graves et al., 2002; Kida et al., 2002; Pittenger et al., 2002; Balschun et al., 2003) is involved in both hippocampus and amygdala function during memory formation, although the studies are at times contradictory. For example, confounding effects of genetic background and gene dosage on behavioral phenotypes of CREB mutant mice exist and partly explain the disparity in fear conditioning results observed by different laboratories (for review, see Bucan and Abel, 2002; Lonze and Ginty, 2002; Kaplan and Abel, 2003). It is worth noting that these CREBαΔ mutant mice are not completely null for CREB family isoforms, because they still express the β isoform of CREB along with cAMP-responsive element modulator and ICER. Our current findings suggest that none of these CREB relatives are capable of mediating the effects of HDAC inhibitors on memory and plasticity.
Nr4a1 and Nr4a2 are CREB-regulated genes that may play a role in the enhancement in hippocampus-dependent memory after increased histone acetylation
We report here that of 12 CRE-containing genes shown previously to be involved in learning and memory, affected by histone acetylation, or both, only the expression of Nr4a1 was significantly increased after TSA treatment and fear conditioning. Because HDAC inhibitors are thought to act globally, we had expected that expression of most, if not all, of the examined genes would be affected by TSA treatment. The results contradict this assumption and are more in line with other studies (Van Lint et al., 1996; Sambucetti et al., 1999; Fass et al., 2003) showing that HDAC inhibitors can have selective and bidirectional effects on gene expression. Overall, these findings suggest that the enhancement of memory and synaptic plasticity by HDAC inhibition occurs through the transcriptional regulation of a particular subset of CREB genes.
We also found that the TSA-induced enhancement of Nr4a1 and Nr4a2 expression after fear conditioning is CREB dependent. Nr4a1 (also known as nur77 and NGFI-B) belongs to a subfamily of orphan nuclear receptors, whose other members are Nr4a2 [also known as Rnr-1 (ribonucleotide reductase-1), Not, and Nurr1] and Nr4a3 [also known as Nor-1 (neuron-derived orphan receptor-1) and Tec], all of which function as transcription factors (Ryseck et al., 1989; Steinmetz et al., 2001). Several studies have implicated Nr4a1 and Nr4a2 as CREB target genes (Fass et al., 2003; Darragh et al., 2005; von Hertzen and Giese, 2005a,b). Also, Fass et al. (2003) observed that forskolin-induced Nr4a1 expression was increased by TSA treatment, whereas Nr4a3 expression was not enhanced by TSA (Fass et al., 2003). These results are consistent with our findings that Nr4a1 and Nr4a2 expression is enhanced by TSA during memory consolidation, whereas Nr4a3 expression is not.
Importantly, Nr4a1 and Nr4a2 also seem to be involved in normal memory formation. Nr4a1 is expressed in the hippocampus after contextual fear conditioning (von Hertzen and Giese, 2005b; Keeley et al., 2006), and Nr4a2 is involved in learning of a spatial discrimination task (de Ortiz et al., 2000; Colon-Cesario et al., 2007). Nr4a1 and Nr4a2 may function in memory consolidation to activate secondary waves of transcription. Heterodimers composed of both Nr4a1 and Nr4a2 can enhance transcription from target promoters more than homodimers of each individual factor alone (Maira et al., 1999), suggesting that Nr4a1 and Nr4a2 expression may act as a functional unit to regulate gene expression during memory consolidation. It is important to note that, because we have not performed genome-wide analysis of transcription or assessed gene expression at all time points after training and TSA administration, there may be many other memory-related and CREB:CBP-regulated genes whose expression is altered by intrahippocampal TSA injection. Nevertheless, Nr4a1 and Nr4a2 may play a role in the enhancing effects of HDAC inhibition on hippocampus-dependent memory and synaptic plasticity. Future experiments will be required to determine the contribution of Nr4a1 and Nr4a2 to long-term memory as well as the enhancement of memory by HDAC inhibitors and to identify downstream targets of Nr4a1 and Nr4a2.
In our model, the enhancement of memory and synaptic plasticity by HDAC inhibition in the hippocampus (Fig. 6) is mediated by the cascade beginning with CREB:CBP interactions in which CBP regulates the expression of CREB-regulated genes involved in long-term memory formation and long-lasting LTP by acetylating histones at the promoters of key CREB target genes such as Nr4a1 and Nr4a2 and by recruiting basal transcription machinery and other transcriptional regulators to these specific promoters. The future investigation of particular HDACs using more specific HDAC inhibitors will undoubtedly be a fruitful area of investigation to understand the fundamental mechanisms of chromatin modification for the regulation of transcription underlying memory and synaptic plasticity. Overall, this work should help guide the use of HDAC inhibitors to treat cognitive disorders, especially those associated with CBP deficits, including Rubinstein-Taybi syndrome and neurodegenerative diseases.
Footnotes
This work was supported by Systems and Integrative Biology Training Grant GM07517 (to C.G.V.; M. Nusbaum, principle investigator), National Institutes of Health (NIH) Training Grant HL07953 (to C.G.V.; A. I. Pack, principle investigator), NIH Institutional National Research Service Award (NRSA) Training Grant AG00256 (to J.M.S.; A. Pack, principle investigator), a Nassau Fund undergraduate research award (to M.A.A.), a University of Pennsylvania postdoctoral training program fellowship in neurodegenerative diseases (to M.A.W.; V. M. Y. Lee, principle investigator), NRSA Grant MH069136 (to C.B.M.), and grants from the Merck Foundation, the Packard Foundation, the University of Pennsylvania Research Foundation, and the Whitehall Foundation and NIH Grant MH060244 (to T.A.). We thank Dr. Julie Blendy for initially providing the CREBαΔ mutant mice, Michele Kelly for her statistical assistance, and Dr. Rita Balice-Gordon for use of her microscope.
References
- Abel T, Martin KC, Bartsch D, Kandel ER. Memory suppressor genes: inhibitory constraints on the storage of long-term memory. Science. 1998;279:338–341. doi: 10.1126/science.279.5349.338. [DOI] [PubMed] [Google Scholar]
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Balschun D, Wolfer DP, Gass P, Mantamadiotis T, Welzl H, Schutz G, Frey JU, Lipp HP. Does cAMP response element-binding protein have a pivotal role in hippocampal synaptic plasticity and hippocampus-dependent memory? J Neurosci. 2003;23:6304–6314. doi: 10.1523/JNEUROSCI.23-15-06304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/s0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, Scott R, Tully T. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci USA. 2003;100:10518–10522. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucan M, Abel T. The mouse: genetics meets behaviour. Nat Rev Genet. 2002;3:114–123. doi: 10.1038/nrg728. [DOI] [PubMed] [Google Scholar]
- Chen X, Dai JC, Greenfield EM. Termination of immediate-early gene expression after stimulation by parathyroid hormone or isoproterenol. Am J Physiol Cell Physiol. 2002;283:C1432–C1440. doi: 10.1152/ajpcell.00221.2002. [DOI] [PubMed] [Google Scholar]
- Chwang WB, O'Riordan KJ, Levenson JM, Sweatt JD. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem. 2007;13:322–328. doi: 10.1101/lm.152906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colon-Cesario WI, Martinez-Montemayor MM, Morales S, Felix J, Cruz J, Adorno M, Pereira L, Colon N, Maldonado-Vlaar CS, Pena de Ortiz S. Knockdown of Nurr1 in the rat hippocampus: implications to spatial discrimination learning and memory. Learn Mem. 2007;13:734–744. doi: 10.1101/lm.407706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti AC, Kuo YC, Valentino RJ, Blendy JA. Inducible cAMP early repressor regulates corticosterone suppression after tricyclic antidepressant treatment. J Neurosci. 2004;24:1967–1975. doi: 10.1523/JNEUROSCI.4804-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darragh J, Soloaga A, Beardmore VA, Wingate AD, Wiggin GR, Peggie M, Arthur JS. MSKs are required for the transcription of the nuclear orphan receptors Nur77, Nurr1 and Nor1 downstream of MAPK signalling. Biochem J. 2005;390:749–759. doi: 10.1042/BJ20050196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol Bull. 1984;96:518–559. [PubMed] [Google Scholar]
- Davis S, Bozon B, Laroche S. How necessary is the activation of the immediate early gene zif268 in synaptic plasticity and learning? Behav Brain Res. 2003;142:17–30. doi: 10.1016/s0166-4328(02)00421-7. [DOI] [PubMed] [Google Scholar]
- de Ortiz SP, Maldonado-Vlaar CS, Carrasquillo Y. Hippocampal expression of orphan nuclear receptor gene hzf-3/nurr1 during spatial discrimination learning. Neurobiol Learn Mem. 2000;74:161–175. doi: 10.1006/nlme.1999.3952. [DOI] [PubMed] [Google Scholar]
- Fanselow MS. Conditioned and unconditional components of post-shock freezing. Pavlov J Biol Sci. 1980;15:177–182. doi: 10.1007/BF03001163. [DOI] [PubMed] [Google Scholar]
- Fass DM, Butler JE, Goodman RH. Deacetylase activity is required for cAMP activation of a subset of CREB target genes. J Biol Chem. 2003;278:43014–43019. doi: 10.1074/jbc.M305905200. [DOI] [PubMed] [Google Scholar]
- Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- Gass P, Wolfer DP, Balschun D, Rudolph D, Frey U, Lipp HP, Schutz G. Deficits in memory tasks of mice with CREB mutations depend on gene dosage. Learn Mem. 1998;5:274–288. [PMC free article] [PubMed] [Google Scholar]
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- Graves L, Dalvi A, Lucki I, Blendy JA, Abel T. Behavioral analysis of CREB alphadelta mutation on a B6/129 F1 hybrid background. Hippocampus. 2002;12:18–26. doi: 10.1002/hipo.10003. [DOI] [PubMed] [Google Scholar]
- Guzowski JF, Miyashita T, Chawla MK, Sanderson J, Maes LI, Houston FP, Lipa P, McNaughton BL, Worley PF, Barnes CA. Recent behavioral history modifies coupling between cell activity and Arc gene transcription in hippocampal CA1 neurons. Proc Natl Acad Sci USA. 2006;103:1077–1082. doi: 10.1073/pnas.0505519103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, Jennings EG, Zeitlinger J, Pokholok DK, Kellis M, Rolfe A, Takusagawa KT, Lander ES, Gifford DK, Fraenkel E, Young RA. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Modulation of both the early and the late phase of mossy fiber LTP by the activation of beta-adrenergic receptors. Neuron. 1996;16:611–617. doi: 10.1016/s0896-6273(00)80080-x. [DOI] [PubMed] [Google Scholar]
- Hummler E, Cole TJ, Blendy JA, Ganss R, Aguzzi A, Schmid W, Beermann F, Schutz G. Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc Natl Acad Sci USA. 1994;91:5647–5651. doi: 10.1073/pnas.91.12.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Alemi L, Harum KH. Learning, memory, and transcription factors. Pediatr Res. 2003;53:369–374. doi: 10.1203/01.PDR.0000049517.47493.E9. [DOI] [PubMed] [Google Scholar]
- Josselyn SA. What's right with my mouse model? New insights into the molecular and cellular basis of cognition from mouse models of Rubinstein-Taybi Syndrome. Learn Mem. 2005;12:80–83. doi: 10.1101/lm.93505. [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA, Jr, Neve RL, Nestler EJ, Davis M. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci. 2001;21:2404–2412. doi: 10.1523/JNEUROSCI.21-07-02404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Kida S, Silva AJ. Inducible repression of CREB function disrupts amygdala-dependent memory. Neurobiol Learn Mem. 2004;82:159–163. doi: 10.1016/j.nlm.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Kalkhoven E. CBP and p300: HATs for different occasions. Biochem Pharmacol. 2004;68:1145–1155. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessel TM, editors. Ed 4. New York: McGraw-Hill; 2000. Principles of neural science. [Google Scholar]
- Kaplan MP, Abel T. Genetic approaches to the study of synaptic plasticity and memory storage. CNS Spectr. 2003;8:597–610. doi: 10.1017/s1092852900018873. [DOI] [PubMed] [Google Scholar]
- Kasper LH, Boussouar F, Ney PA, Jackson CW, Rehg J, van Deursen JM, Brindle PK. A transcription-factor-binding surface of coactivator p300 is required for haematopoiesis. Nature. 2002;419:738–743. doi: 10.1038/nature01062. [DOI] [PubMed] [Google Scholar]
- Keeley BK, Wood MA, Isiegas C, Stein J, Hellman K, Hannenhalli S, Abel T. Differential transcriptional response to nonassociative and associative components of classical fear conditioning in the amygdala and hippocampus. Learn Mem. 2006;13:135–142. doi: 10.1101/lm.86906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, Silva AJ. CREB required for the stability of new and reactivated fear memories. Nat Neurosci. 2002;5:348–355. doi: 10.1038/nn819. [DOI] [PubMed] [Google Scholar]
- Klopotowska D, Matuszyk J, Rapak A, Gidzinska B, Cebrat M, Ziolo E, Strzadala L. Transactivation activity of Nur77 discriminates between Ca2+ and cAMP signals. Neurochem Int. 2005;46:305–312. doi: 10.1016/j.neuint.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Kogan JH, Frankland PW, Blendy JA, Coblentz J, Marowitz Z, Schutz G, Silva AJ. Spaced training induces normal long-term memory in CREB mutant mice. Curr Biol. 1997;7:1–11. doi: 10.1016/s0960-9822(06)00022-4. [DOI] [PubMed] [Google Scholar]
- Korzus E. The relation of transcription to memory formation. Acta Biochim Pol. 2003;50:775–782. [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalovsky D, Refojo D, Liberman AC, Hochbaum D, Pereda MP, Coso OA, Stalla GK, Holsboer F, Arzt E. Activation and induction of NUR77/NURR1 in corticotrophs by CRH/cAMP: involvement of calcium, protein kinase A, and MAPK pathways. Mol Endocrinol. 2002;16:1638–1651. doi: 10.1210/mend.16.7.0863. [DOI] [PubMed] [Google Scholar]
- Krug M, Lossner B, Ott T. Anisomycin blocks the late phase of long-term potentiation in the dentate gyrus of freely moving rats. Brain Res Bull. 1984;13:39–42. doi: 10.1016/0361-9230(84)90005-4. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Levine AA, Guan Z, Barco A, Xu S, Kandel ER, Schwartz JH. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci USA. 2005;102:19186–19191. doi: 10.1073/pnas.0509735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lund AH, van Lohuizen M. Epigenetics and cancer. Genes Dev. 2004;18:2315–2335. doi: 10.1101/gad.1232504. [DOI] [PubMed] [Google Scholar]
- Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Maira M, Martens C, Philips A, Drouin J. Heterodimerization between members of the Nur subfamily of orphan nuclear receptors as a novel mechanism for gene activation. Mol Cell Biol. 1999;19:7549–7557. doi: 10.1128/mcb.19.11.7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC. The long-term potential of LTP. Nat Rev Neurosci. 2003;4:923–926. doi: 10.1038/nrn1258. [DOI] [PubMed] [Google Scholar]
- Maren S. Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci. 2001;24:897–931. doi: 10.1146/annurev.neuro.24.1.897. [DOI] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Adv Cancer Res. 2004;91:137–168. doi: 10.1016/S0065-230X(04)91004-4. [DOI] [PubMed] [Google Scholar]
- Michael LF, Asahara H, Shulman AI, Kraus WL, Montminy M. The phosphorylation status of a cyclic AMP-responsive activator is modulated via a chromatin-dependent mechanism. Mol Cell Biol. 2000;20:1596–1603. doi: 10.1128/mcb.20.5.1596-1603.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedivi E, Fieldust S, Theill LE, Hevron D. A set of genes expressed in response to light in the adult cerebral cortex and regulated during development. Proc Natl Acad Sci USA. 1996;93:2048–2053. doi: 10.1073/pnas.93.5.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401–437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- Norton VG, Imai BS, Yau P, Bradbury EM. Histone acetylation reduces nucleosome core particle linking number change. Cell. 1989;57:449–457. doi: 10.1016/0092-8674(89)90920-3. [DOI] [PubMed] [Google Scholar]
- Oike Y, Hata A, Mamiya T, Kaname T, Noda Y, Suzuki M, Yasue H, Nabeshima T, Araki K, Yamamura K. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum Mol Genet. 1999;8:387–396. doi: 10.1093/hmg/8.3.387. [DOI] [PubMed] [Google Scholar]
- Parker D, Ferreri K, Nakajima T, LaMorte VJ, Evans R, Koerber SC, Hoeger C, Montminy MR. Phosphorylation of CREB at Ser-133 induces complex formation with CREB-binding protein via a direct mechanism. Mol Cell Biol. 1996;16:694–703. doi: 10.1128/mcb.16.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, Kandel ER. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron. 2002;34:447–462. doi: 10.1016/s0896-6273(02)00684-0. [DOI] [PubMed] [Google Scholar]
- Ryseck RP, Macdonald-Bravo H, Mattei MG, Ruppert S, Bravo R. Structure, mapping and expression of a growth factor inducible gene encoding a putative nuclear hormonal binding receptor. EMBO J. 1989;8:3327–3335. doi: 10.1002/j.1460-2075.1989.tb08494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambucetti LC, Fischer DD, Zabludoff S, Kwon PO, Chamberlin H, Trogani N, Xu H, Cohen D. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem. 1999;274:34940–34947. doi: 10.1074/jbc.274.49.34940. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Wetmore C, Olson L. Comparative study of brain-derived neurotrophic factor messenger RNA and protein at the cellular level suggests multiple roles in hippocampus, striatum and cortex. Neuroscience. 1996;74:161–183. doi: 10.1016/0306-4522(96)00093-0. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Zoghbi HY. Rett syndrome and MeCP2: linking epigenetics and neuronal function. Am J Hum Genet. 2002;71:1259–1272. doi: 10.1086/345360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Steinmetz AC, Renaud JP, Moras D. Binding of ligands and activation of transcription by nuclear receptors. Annu Rev Biophys Biomol Struct. 2001;30:329–359. doi: 10.1146/annurev.biophys.30.1.329. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Tischmeyer W, Grimm R. Activation of immediate early genes and memory formation. Cell Mol Life Sci. 1999;55:564–574. doi: 10.1007/s000180050315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat Struct Mol Biol. 2005;12:110–112. doi: 10.1038/nsmb0205-110. [DOI] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- Vettese-Dadey M, Grant PA, Hebbes TR, Crane- Robinson C, Allis CD, Workman JL. Acetylation of histone H4 plays a primary role in enhancing transcription factor binding to nucleosomal DNA in vitro. EMBO J. 1996;15:2508–2518. [PMC free article] [PubMed] [Google Scholar]
- von Hertzen LS, Giese KP. Alpha-isoform of Ca2+/calmodulin-dependent kinase II autophosphorylation is required for memory consolidation-specific transcription. NeuroReport. 2005a;16:1411–1414. doi: 10.1097/01.wnr.0000175244.51084.bb. [DOI] [PubMed] [Google Scholar]
- von Hertzen LS, Giese KP. Memory reconsolidation engages only a subset of immediate-early genes induced during consolidation. J Neurosci. 2005b;25:1935–1942. doi: 10.1523/JNEUROSCI.4707-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters CL, Blendy JA. Different requirements for cAMP response element binding protein in positive and negative reinforcing properties of drugs of abuse. J Neurosci. 2001;21:9438–9444. doi: 10.1523/JNEUROSCI.21-23-09438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Attner MA, Oliveira AMM, Brindle PK, Abel T. A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn Mem. 2006;13:609–617. doi: 10.1101/lm.213906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh SH, Lin CH, Gean PW. Acetylation of nuclear factor-kappaB in rat amygdala improves long-term but not short-term retention of fear memory. Mol Pharmacol. 2004;65:1286–1292. doi: 10.1124/mol.65.5.1286. [DOI] [PubMed] [Google Scholar]