

Abstract
Purpose
Kv4.2 subunits contribute to the pore-forming region of channels that express a transient, A-type K+ current (A-current) in hippocampal CA1 pyramidal cell dendrites. Here, the A-current plays an important role in signal processing and synaptic integration. Kv4.2 knockout mice show a near elimination of the A-current in area CA1 dendrites producing increased excitability in this region. In these studies, we evaluated young adult Kv4.2 knockout mice for spontaneous seizures and the response to convulsant stimulation in the whole animal in vivo and in hippocampal slices in vitro.
Methods
Electroencephalogram electrode-implanted Kv4.2 knockout and wildtype mice were observed for spontaneous behavioral and electrographic seizures. The latency to seizure and status epilepticus onset in Kv4.2 knockout and wildtype mice was assessed following intraperitoneal injection of kainate. Extracellular field potential recordings were performed in hippocampal slices from Kv4.2 knockout and wildtype mice following the bath application of bicuculline.
Results
No spontaneous behavioral or electrographic seizures were observed in Kv4.2 knockout mice. Following kainate, Kv4.2 knockout mice demonstrated a decreased seizure and status epilepticus latency as well as increased mortality compared to wildtype littermates. The background strain modified the seizure susceptibility phenotype in Kv4.2 knockout mice. In response to bicuculline, slices from Kv4.2 knockout mice exhibited an increase in epileptiform bursting in area CA1 as compared to wildtype littermates.
Discussion
These studies show that loss of Kv4.2 channels is associated with enhanced susceptibility to convulsant stimulation, supporting the concept that Kv4.2 deficiency may contribute to aberrant network excitability and regulate seizure threshold.
Keywords: A-type K channel, channelopathies, seizures, status epilepticus, mouse
Introduction
Kv4.2 subunits compose the pore-forming channel that contributes to the transient, rapidly activating and inactivating outward K+ current (A-current) in CA1 pyramidal cell dendrites (Kim et al., 2005; Chen et al., 2006). The A-current in this region regulates the back-propagating action potential and synaptic integration (Hoffman et al., 1997). Thus, Kv4.2 channels are critical regulators of postsynaptic excitability and aberrant function or loss of Kv4.2 channels is likely to facilitate hyperexcitability and potentially seizure initiation and propagation in the hippocampus.
Alterations in the Kv4.2 channel have been demonstrated in animal models of epilepsy. A decrease in Kv4.2 mRNA levels was found in rat hippocampus following pentylenetetrazol-induced seizures (Tsaur et al., 1992). In a model of cortical malformations that results in increased hippocampal excitability and a decrease in seizure threshold, there was a marked decrease in the expression of Kv4.2 channel subunits (Castro et al., 2001). Furthermore, in a rodent model of limbic seizures induced by pilocarpine, there was a significant decrease in Kv4.2 subunit expression in the hippocampus of chronically epileptic rats (Bernard et al., 2004). These studies revealed more prominent action potential backpropagation in CA1 dendrites from the epileptic compared to sham animals, indicative of increased dendritic excitability in the epileptic animals. The decrease in Kv4.2 expression in the epileptic animals was thought to underlie this effect.
Ion channelopathies have been implicated in several types of human epilepsy with complex partial seizures, including temporal lobe epilepsy (Biervert et al., 1998; Charlier et al., 1998; Singh et al., 1998; Eunson et al., 2000; Klein et al., 2004; Andermann et al., 2005; Du et al., 2005). Recently in a patient with temporal lobe epilepsy, a mutation was identified in KCND2, the gene encoding Kv4.2, which resulted in a 44-amino acid truncation in the Kv4.2 carboxyl terminal (Singh et al., 2006). Expression of the Kv4.2 truncation mutant in HEK cells resulted in attenuated Kv4.2 currents (Singh et al., 2006), indicating that the truncation was functionally significant. Together, these studies suggest that loss-of-function of Kv4.2 may contribute to acute and chronic seizures.
In this report, we used Kv4.2 knockout mice to investigate whether genetic loss of Kv4.2 increases susceptibility to convulsant stimulation. While loss of Kv4.2 at the genomic level increases hippocampal excitability and decreases the threshold for long-term potentiation induction in area CA1, our results suggest that knockout of Kv4.2 is not associated with an epilepsy phenotype in young adult animals. However, the findings of Kv4.2 downregulation in an acute seizure model (Tsaur et al., 1992) and in chronic epilepsy (Bernard et al., 2004), and the loss-of-function Kv4.2 mutation in a patient with temporal lobe epilepsy (Singh et al., 2006) suggest that Kv4.2 may function as a candidate seizure susceptibility gene. The findings in the current study provide further support for this possibility.
Materials and Methods
Animals
Littermates
The Kv4.2 knockout mice used in this study were generated in a 129S6/SvEv background (Guo et al., 2005). The knockout mice were backcrossed with wildtype 129S6/SvEvTac to produce (F6 or greater) littermates that were used in these studies except when specified otherwise. The experimental animals were between 12 to 30 weeks old. Kv4.2 knockout and wildtype animals were offspring from heterozygote parents. Genotyping of the littermates was performed by PCR using Kv4.2-specific primers (forward, GTG GAT GCC TGT TGC TTC; reverse, CCC ACA AGG CAG TTC TTT TA) and neo-specific primers (forward, AGG ATC TCC TGT CAT CTC ACC TTGCTC CTG; reverse, AAG AAC TCG TCA AGA AGG CGA TAG AAG GCG).
Non-littermates
Wildtype 129S6/SvEv, the background strain of Kv4.2 knockout mice were purchased from Taconic Farm USA. Inbred Kv4.2 knockout animals were generated by crossing homozygote knockout mice obtained from our colony of littermates, for at least 6 generations.
Mice were housed at the Baylor College of Medicine Center for Comparative Medicine at an ambient temperature of 22 °C, with a 14-h light and 10-h dark (20:00 to 06:00 h) diurnal cycle and were given ad libitum access to food and water. All procedures concerning animals were approved by the Institutional Animal Care and Use Committee of the Baylor College of Medicine.
Electrode implantation, electroencephalography and kainate-induced seizures
Kv4.2 littermate and non-littermate mice were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (16 mg/kg) (obtained from Baylor College of Medicine) delivered intraperitoneally and positioned in a stereotaxic frame. Three monopolar cortical surface recording electrodes were implanted: one in the left anterior cortex (2.0 mm posterior and 2.2 mm lateral to bregma), another in the right anterior cortex (2.0 mm posterior and 2.2 mm lateral to bregma) and a third in the left posterior cortex (5.0 mm posterior and 2.2 mm lateral to bregma). A fourth recording electrode was placed in the right hippocampus (2.2 mm posterior and 1.6 mm lateral to bregma and 1.6 mm deep). A reference screw electrode was placed in the skull on the right side, anterior to the frontal suture. A midline ground electrode was placed posterior to the occipital suture.
Four to seven days after electrode implantation, baseline video-EEG recordings were made for up to 2 hours using a Stellate digital video-EEG machine. Following baseline recordings, seizures were induced by the administration of kainate (20 or 40 mg/kg) intraperitoneally. In the seizure susceptibility studies, 40 mg/kg kainate was used to induce seizures in littermates. In the genetic background studies comparing non-littermates and littermates, a lower dose of kainate, 20 mg/kg was applied to avoid a ceiling effect. The animals were monitored both behaviorally using the Racine scale (Racine, 1972) and electrographically by continuous video-EEG recording for up to 4 additional hours after seizure induction. Tracings were visually inspected for electrographic seizure activity as previously defined (Anderson et al., 1997). Control groups included mice that had been treated with kainate vehicle (saline).
Slice preparation
Thirty to sixty day old Kv4.2 wildtype and knockout littermate mice were rapidly decapitated and the brains dissected out into ice-cold (4 °C) cutting solution. Cutting solution contained (mM): 115 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 7 dextrose. Transverse cortical-hippocampal slices (400 μm thick) were acutely prepared from the middle section of both the left and right hippocampus using a Vibratome, Series 3000 plus (Ted Pella Inc., Redding, CA, USA). Slices were collected from each animal and one or more of these slices were randomly used for each recording. The slices were transferred to a submerged holding chamber containing oxygenated ACSF for at least one hour before transfer to a recording chamber. ACSF contained (mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2.0 CaCl2, 1 MgCl2, 10 dextrose (PH 7.4 when saturated with 95% O2 and 5% CO2).
Field potential recordings
To perform field potential recordings, slices from knockout and wildtype mice were transferred one at a time to a recording chamber on a fixed Axioscope stage. The chamber was maintained at room temperature and continuously perfused with oxygenated ACSF at a rate of 1.5–2 ml per minute (Sari and Kerr, 2001). An ACSF filled glass recording electrode with a resistance of 3–5 MΩ was placed in the pyramidal layer of hippocampal area CA1 to record extracellular field potentials and population spikes. Recordings were made with an HEKA EPC-10 amplifier with PatchMaster software. A bipolar stimulating electrode was placed in the stratum radiatum of the CA1 region to stimulate the Schaffer-collateral/commissural pathway. Stimulation was performed using square pulses of 100 μs duration, set at a strength which gave rise to approximately 1/3 of the maximal population spike amplitude. Recordings began 30 minutes after electrode placement to ensure stable responses. Five micromolar bicuculline was applied by bath perfusion for 20 minutes before evaluating the evoked and spontaneous responses. Spontaneous responses were collected 5 minutes after the last evoked response was collected.
Excitability of the slice was assessed by determining the field potential duration which was defined as the distance between the initial rise of the field potential and the intersection of the falling phase of the field potential with the baseline (Karnup and Stelzer, 2001). In cases where after-discharges were present, the end of the field potentials was defined as the end of the last population spike in the last after-discharge. For wildtype mice, recordings were made from 12 slices that were obtained from 6 animals. For knockout mice, recordings were made from 15 slices that were obtained from 7 animals. The change in excitability is reported as percent field potential duration in the presence of bicuculline divided by field potential duration in ACSF only.
Statistical analysis
The GraphPad Prism software package was used for statistical analysis of the data. Data are expressed as mean ± standard error of mean (S.E.M.). One-way analysis of variance with post-hoc analysis, unpaired two tailed student’s t test or chi-square test was used for comparison. Statistical significance was taken as p ≤ 0.05.
Results
Enhanced susceptibility to convulsant stimulation in Kv4.2 knockout compared to wildtype mice in vivo
We observed naïve Kv4.2 knockout mice for spontaneous seizures since a loss-of-function mutation in Kv4.2 had previously been identified in a patient with temporal lobe epilepsy (Singh et al., 2006). Video-EEG monitoring of 12–30 week-old Kv4.2 knockout (n=22) and wildtype (n=20) littermates was performed for up to 6 hours per day and 3 days per week for 3 months. An accumulated 180 hours of recording per genotype revealed no epileptiform activity or electrographic seizures in the littermates.
To evaluate whether Kv4.2 knockout alters seizure threshold we used the chemoconvulsant kainate (40 mg/kg), injected intraperitoneally (i.p.), to induce seizures in Kv4.2 knockout and wildtype littermate mice implanted with EEG electrodes. All of the wildtype mice that developed seizures following kainate had either focal hippocampal (4/8) or focal cortical (4/8) onset (representative cortical onset seizure shown in Figure 1). In contrast, 50% (4/8) of the knockout mice that developed electrographic seizures with kainate treatment showed simultaneous ipsilateral cortical and hippocampal onset (Figure 1), while 37.5% (3/8) of the knockout mice exhibited focal hippocampal and 12.5% (1/8) had focal cortical seizure onset. In both the wildtype and knockout mice regardless of the location of onset, the electrographic seizures began with a high frequency (30 Hz) recruiting rhythm lasting 15 to 25 seconds followed by high voltage, synchronous, 4–6 Hz spike-and-wave discharges ranging from 25 to 40 seconds in duration and ended with a period of generalized voltage attenuation (Figure 1). Status epilepticus in both genotypes consisted of continuous, synchronous, high voltage, 2.5–3 Hz generalized spike-and-wave activity (Figure 1).
Figure 1.

Electrographic seizures and status epilepticus in Kv4.2 wildtype and knockout littermates. Representative simultaneous cortical (Ctx) and hippocampal (Hip) EEG recordings are shown for (A) Kv4.2 wildtype (+/+) and (B) Kv4.2 knockout (−/−) littermates before and after intraperitoneal injection of kainate (40 mg/kg). (A and B) The top two traces shown for each genotype are samples of cortex and hippocampal baseline EEG recordings done thirty minutes prior to kainate injection in the animals. The 2nd, 3rd and 4th pairs of traces below the top pair for the Kv4.2 knockout and wildtype mice are continuous EEG recordings of electrographic seizures that began after kainate injection. The electrographic elements of the seizures are similar in both genotypes except for differences in the location and latency of seizure onset. In both wildtype and knockout mice, seizure onset is marked by a 30 Hz recruiting rhythm which is immediately followed by high voltage synchronous 4–6 Hz generalized spike and wave activity and a period of post-ictal depression. However, in the trace for the wildtype mouse there is seizure onset focally in the cortex, while in the trace for the knockout mouse there is seizure onset diffusely in the cortex and hippocampus. This latter finding was not seen in any of the wildtype mice, while 50% of the knockout mice showed this diffuse seizure onset (see results section). Electrographic status epilepticus is marked by continuous high voltage 2.5–3 Hz generalized spike and wave activity. The latency to first electrographic seizure for the wildtype mouse shown is 108 seconds and to status epilepticus is 30 minutes after kainate injection (A). The latency to electrographic seizure onset for the knockout mouse is 42 seconds and to status epilepticus is 15 minutes after kainate injection (B).
The latency of the electrographic seizure onset was significantly shorter in the Kv4.2 knockout compared to wildtype animals (59.6±4.0 versus 122±16 seconds, respectively, n=6–8, p<0.01) (Figure 2A). In addition, Kv4.2 knockout mice developed electrographic status epilepticus significantly faster than littermate wildtype mice (15.1±1.8 versus 38.9±6.2 minutes, respectively, n=6–8, p<0.01) (Figure 2B).
Figure 2.

Kv4.2 knockout littermates show increased susceptibility to kainate stimulation. Following kainate (40 mg/kg) injection intraperitoneally in Kv4.2 wildtype (+/+) (n = 8) and Kv4.2 knockout (−/−) (n = 6) littermates, the animals were monitored with video-EEG for electrographic and behavioral seizures according to the Racine scale. (A) Knockout animals developed electrographic seizures significantly faster than wildtype mice (n=6–8). One hundred percent of the knockout and 80% of the wildtype mice developed seizures in response to kainate (data not shown). (B) Electrographic status epilepticus developed significantly faster in the knockout mice compared to wildtype littermates (n=6–8). (C) The latency to onset of forelimb clonus was significantly shorter in the knockout compared to wildtype mice. (D) Behavioral status epilepticus also developed significantly faster in the knockout versus wildtype littermates (n=6–8). Thus, both electrographic and behavioral seizures occurred significantly earlier in the knockout compared to the littermate wildtype mice. *p < 0.05, **p < 0.01, Student’s t test.
Behaviorally, seizures in both littermate genotypes were indistinguishable. However, the Kv4.2 knockout mice developed forelimb clonus significantly faster than the wildtype animals (9.4±1.5 versus 17.0±1.9 minutes, respectively, n=6–8, p<0.05) (Figure 2C). Behavioral status epilepticus, indicated by rearing and falling, also developed significantly faster in the Kv4.2 knockout compared to the wildtype mice (28.0±3.6 versus 40.2±3.6 minutes, respectively, n=6–8, p<0.05) (Figure 2D).
Kv4.2 knockout mice exhibit increased mortality following seizure induction
One complication of neurotoxin-induced models of seizures and status epilepticus in rodents is a high mortality rate following the induction of seizures. For the 129S6/Sv background strain used in these studies, reports show an 8–10% mortality rate within 4–6 hours of kainate-induced seizures (Shannon and Yang, 2004);(McKhann et al., 2003). In our studies, 25% (2/8) of the wildtype littermates died during the first 6 hours following seizure induction with kainate (40 mg/kg) (Figure 3). In marked contrast, 100% (6/6) of the knockout littermates died within 4 hours of seizure induction (Figure 3). The high mortality rate in the Kv4.2 knockout mice following kainate-induced seizures and status epilepticus prevented us from performing chronic epilepsy studies in the littermates.
Figure 3.
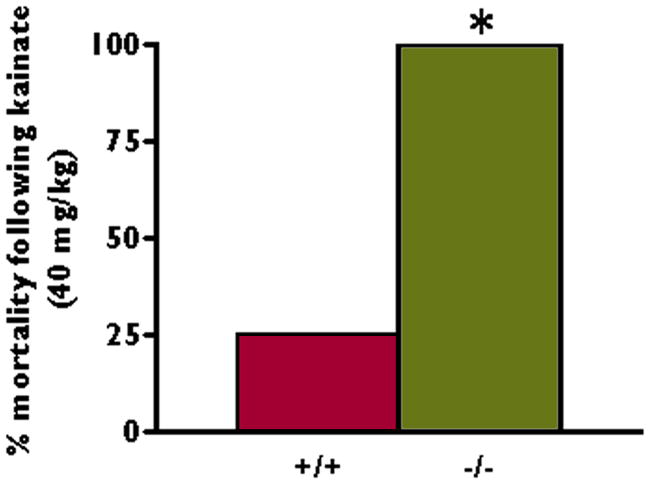
Kv4.2 deficiency is associated with high mortality after convulsant stimulus. Kv4.2 knockout (−/−) and Kv4.2 wildtype (+/+) littermate mice were given kainate (40 mg/kg) intraperitoneally. Acutely following the onset of status epilepticus, 100% (6/6) of the Kv4.2 knockout mice and 25% (2/8) of the littermate wildtype mice died. *p < 0.05, chi-square test.
Genetic background influences seizure susceptibility in Kv4.2 knockout mice
Given the variable expression of epilepsy in the human kindred with the loss-of-function mutation in Kv4.2 (Singh et al., 2006), we investigated whether the genetic background modulates the seizure phenotype in Kv4.2 knockout mice. For these studies, we used a reduced dosage of kainate (20 mg/kg) to avoid a ceiling effect and better isolate the influence of the genetic background on seizure susceptibility. Purchased non-littermate mice from the background strain were compared to inbred Kv4.2 knockout mice. Kainate (20 mg/kg) resulted in a significantly shorter latency to the onset of forelimb clonus in the inbred knockout mice compared to the purchased non-littermate 129S6/SvEvTac mice (17.1±1.2 versus 26.4±3.0 minutes respectively, n=8, p<0.05) (Figure 4). One hundred percent (8/8) of the inbred knockout mice compared to 80% (8/10) of the non-littermate wildtype mice developed seizures following kainate treatment. In contrast, there was no significant difference in the latency to the onset of forelimb clonus between the littermate knockout and wildtype mice at the 20 mg/kg dosage of kainate (16.8±2.0 versus 18.7±1.5 minutes respectively, n=8, p>0.05) (Figure 4). One hundred percent (8/8) of the littermate knockout compared to 72.7% (8/11) of the littermate wildtype mice developed seizures following the 20 mg/kg dosage of kainate.
Figure 4.

Enhanced seizure susceptibility phenotype in Kv4.2 knockout mice when compared to non-littermate wildtype mice. Following kainate (20 mg/kg) injection intraperitoneally into the 129S6/SvEvTac purchased wildtype (+/+) and inbred non-littermate Kv4.2 knockout (−/−) mice and littermate wildtype and Kv4.2 knockout mice, behavioral seizures were scored according to the Racine scale. (A) Non-littermate knockout mice developed forelimb clonus significantly faster than the wildtype 129S6/SvEvTac mice (n = 8, *p < 0.05, Student’s t test). (B) After backcrossing the Kv4.2 knockout mice with the 129S6/SvEvTac background strain to the F6 and higher generations, the seizure susceptibility studies were repeated with the same dosage of kainate using littermate knockout and wildtype mice. At this dose of kainate (20 mg/kg) there was no significant difference in seizure susceptibility between the littermate knockout (−/−) and wildtype (+/+) mice (n = 8).
Increased epileptiform activity in area CA1 of Kv4.2 knockout mice after convulsant stimulation in vitro
We performed extracellular field potential recordings in hippocampal slices from littermate knockout and wildtype mice in the presence of bicuculline (5 μM). This concentration of bicuculline was below the threshold to induce maximal bursting in slices from wildtype mice (Karnup and Stelzer, 2001). Orthodromic stimulation of the Schaffer collateral/commissural pathway was performed before and after the application of bicuculline to hippocampal slices from both genotypes and the field potentials were recorded in the stratum pyramidale of hippocampal area CA1. Prior to the application of bicuculline and in the absence of orthodromic stimulation, no spontaneous bursting activity was observed in area CA1 in slices from knockout or wildtype mice. In the presence of normal artificial cerebrospinal fluid, field potential recordings in area CA1 showed a single population spike in slices from both knockout and wildtype mice following orthodromic stimulation of the pathway (Figure 5A). There was no significant difference in the field potential duration recorded in slices from knockout and wildtype mice (37.2±2.1 versus 47.2±5.3 ms, n=12–15). Subsequently, bicuculline perfusion induced multiple population spikes in all of the hippocampal slices from knockout and wildtype mice. The evoked field potential duration was longer than baseline in 93% (14/15) of the slices from knockout and 75% (9/12) of the slices from wildtype mice (Figure 5A). Summary data show that the relative bicuculline-induced increase in field potential duration recorded in the slices from knockout was more than double that in the slices from wildtype mice (401±92 versus 163±24%, respectively, n=12–15, p<0.05) (Figure 5A). After-discharges were present in area CA1 in some of the slices from Kv4.2 knockout mice (33.3%, 5/15), but not in any of the slices from wildtype mice. This finding suggest increased reverberation of the hippocampal circuitry in the slices from knockout mice.
Figure 5.

Bicuculline-induced excitability is enhanced in hippocampal slices from Kv4.2 knockout mice. (A) Representative evoked field potential traces from transverse hippocampal slices prepared from Kv4.2 wildtype (+/+) and knockout (−/−) littermate mice before (Control) and after bicuculline (5 μM; Bicuculline) application are shown. Bicuculline application led to bursting in response to orthodromic stimulation in both genotypes; however, the response was more prolonged in the slices from the Kv4.2 knockout compared to littermate wildtype mice. A summary of the evoked response durations after bicuculline application is shown for both Kv4.2 knockout (n = 15) and wildtype (n = 12) littermates. *p < 0.05, Student’s t test. (B) Representative spontaneous field potential responses following bicuculline (5 μM) application to slices from the Kv4.2 wildtype (+/+) and knockout (−/−) littermate mice are shown. Bursting is more prolonged in the slices from the Kv4.2 knockout compared to wildtype mice. Summary data for bicuculline-induced spontaneous burst duration for slices from the Kv4.2 wildtype (n = 5) and knockout (n = 7) animals are shown. *p < 0.05, Student’s t test.
In the absence of Schaffer collateral/commissural stimulation, bicuculline induced similar proportions of spontaneous bursting in slices from knockout and wildtype mice [46.7% (7/15) and 41.7% (5/12), respectively]. However, the spontaneous field potential duration was significantly longer in slices from Kv4.2 knockout compared to wildtype mice (375±58 versus 163±57 ms, respectively, n=12–15, p<0.05) (Figure 5B). Furthermore, after-discharges occurred more frequently following the spontaneous bursts in slices from knockout compared to wildtype mice [85.6% (6/7) versus 20% (1/5) respectively].
Discussion
The results presented here add to the previous studies which have shown Kv4.2 downregulation in acute seizure (Tsaur et al., 1992) and limbic epilepsy (Bernard et al., 2004; Birnbaum et al., 2004 for review) models. Here, we have demonstrated that Kv4.2 knockout compared to wildtype littermate mice have increased sensitivity to convulsant stimulation at the whole animal level in vivo and at the network level in vitro. Systemic application of a chemoconvulsant resulted in a decreased latency to seizures and status epilepticus in Kv4.2 knockout versus wildtype littermates. The magnitude of this decrease is genetic background dependent. In concordance with the studies performed in vivo, hippocampal slice preparations from Kv4.2 knockout mice exhibited a greater increase in epileptiform activity compared to slices from wildtype mice following convulsant stimulation.
Loss of Kv4.2 channels in knockout mice was predicted to augment convulsant-induced hippocampal seizures. Indeed, convulsant stimulation with systemic kainate resulted in a decreased latency to seizures and status epilepticus in the Kv4.2 knockout compared to wildtype mice. This is consistent with the recent report showing that a decreased stimulation threshold is required to induce maximal hippocampal LTP in Kv4.2 knockout compared with wildtype mice (Chen et al., 2006). Furthermore, the simultaneous unilateral onset of electrographic seizures in the hippocampus and cortex in half of the knockout compared to none of the wildtype mice demonstrates that the loss of Kv4.2 channels leads to seizure onset with more diffuse hemispheric involvement in some animals. This finding is supported by work showing that Kv4.2 is involved in the modulation of excitability and synaptic responses in both the hippocampus and cortex (Nerbonne, 2008).
In this study, video-EEG monitoring of naive young adult Kv4.2 knockout mice did not reveal spontaneous seizures or epileptiform activity. However, we have observed that aged Kv4.2 knockout mice exhibit behavioral seizures in response to startle or acoustic stimuli, and show handling-induced seizures (unpublished observations). We cannot definitively exclude the possibility that the younger mice used in our studies have epileptiform activity with intermittent EEG monitoring. Continuous monitoring is currently unavailable in our animal facility; however, we used prolonged and frequent intermittent monitoring of the mice and were unable to detect any epileptiform activity in the wildtype or knockout mice under basal conditions. Thus, if the animals were having experiencing epileptiform activity and seizures, it is likely to be quite rare.
Another possible explanation for the lack of spontaneous seizures in the Kv4.2 knockout mice may be related to compensatory mechanisms in these animals (Chen et al., 2006). Our findings indicate that the threshold for forebrain excitability in the Kv4.2 knockouts is lower compared to the wildtype mice, but this may not be enough to generate spontaneous epileptiform activity or seizures in the presence of compensatory mechanisms. Indeed, compensatory mechanisms have recently been reported in the Kv4.2 knockout animals (Chen et al., 2006; Andrasfalvy et al., 2008; Nerbonne et al., 2008). In the hippocampal CA1 region of Kv4.2 knockout mice, non-Kv4.2 A-type currents and GABAergic responses are elevated (Chen et al., 2006) (Andrasfalvy et al., 2008). When the increase in GABAergic compensation was blocked, action potentials were more likely to be followed by burst firing in hippocampal area CA1 of the Kv4.2 knockout mice. In the cortex of Kv4.2 knockout mice, inhibitory currents IK (delayed rectifier current), and Iss (late component of the outward potassium current) are increased (Nerbonne et al., 2008). By robustly increasing excitability, such as with convulsant stimulation, the compensatory mechanisms are likely overcome, and the increase in excitability due to the loss of Kv4.2 is expressed.
The results from experiments in vitro in the hippocampal slices parallel that of the in vivo studies. There was no observable spontaneous bursting in hippocampal slices from knockout mice and the baseline evoked responses from littermate wildtype and knockout mice showed no epileptiform activity. However, convulsant stimulation augments seizure-like activity in slices from knockouts. In these slices, a low concentration of bicuculline (Wong et al., 1986; Traynelis and Dingledine, 1988) induced prolonged bursts with after-discharges, resembling interictal bursting activity. This bursting activity is similar to that described with higher concentrations of bicuculline (Karnup and Stelzer, 2001) and is thought to be due to the reverbation of the hippocampal network between the CA1 and CA3 region (Hablitz, 1984). As discussed above, compensatory mechanisms in the knockout mice probably prevent hyperexcitability in the excitatory neuron and the local hippocampal network under basal conditions. However, when the local network is challenged with a convulsant agent, the compensatory mechanisms may not be adequate to prevent the hyperexcitability. Together, these measures indicate increased hippocampal network reverberation in the hippocampal slices from Kv4.2 knockout relative to those from wildtype animals. These findings provide further support for a role of Kv4.2 channels in regulating hippocampal excitability.
The reason for the decreased threshold in network excitability in the hippocampal slice of knockout animals is likely to be at the cellular level. In area CA1, the pyramidal dendrites exhibit a tendency to fire in burst mode when intra-dendritically stimulated (Wong and Stewart, 1992; Golding et al., 1999). However, this burst firing normally does not propagate into the somatic region. The dendritic A-current is a potential mechanism underlying this observation. Blocking A-current with 4-aminopyridine (4-AP) causes seizure-like activity both in vivo and in vitro (Glover, 1982; Perreault and Avoli, 1991; Traub et al., 1995). However, 4-AP is a non-specific K+ channel blocker, which in addition to A-current, affects other presynaptic and postsynaptic K+ channel currents. (Sheng et al., 1992; Klee et al., 1995; Pedarzani et al., 1998). In the Kv4.2 knockout mice used in our studies, the A-current is selectively eliminated in the CA1 hippocampal dendrites with the preservation of non-Kv4.2 somatic A-currents (Chen et al., 2006). Indeed, when the compensatory increase in GABAergic responses were eliminated in the CA1 region of the knockout animals, action potentials were followed by burst firing (Andrasfalvy et al., 2008). Apart from the CA1 region, Kv4.2 is well-expressed in the principal neurons in other subfields of the hippocampus (Menegola and Trimmer, 2006), and a 4-aminopyridine sensitive A-type current is present in these regions (Beck et al., 1992; Mitterdorfer and Bean, 2002). Thus Kv4.2 may underlie the A-type current in these regions of the hippocampus and thereby affect the excitability of these neurons. However, the relationship between Kv4.2 and the A-type current in these regions is not yet characterized. The participation of these neurons in the seizure circuit of the knockout animals could contribute to the increase in seizure susceptibility in the hippocampal slice.
Genetic background is known to affect seizure susceptibility in both animals and humans. For instance, 129S6/SvEv mice require twice as much kainate (75 mg/kg) as other mouse strains such as C57 to maintain high grade seizures including status epilepticus (McKhann et al., 2003). Human, studies have shown that the phenotype of a channelopathy can be modulated by the genetic background (Schulze-Bahr et al., 1999; Mulley et al., 2003). Furthermore, the discordant expression of temporal lobe epilepsy in the patient with the Kv4.2 loss-of-function mutation, while the patient’s father who has the identical mutation does not have epilepsy (Singh et al., 2006), suggests that other factors influence the expression of seizures in this genotype. The seizure resistance of the 129S6/SvEvTac background strain (McKhann et al., 2003) of the Kv4.2 knockout mice may contribute to the phenotype of the Kv4.2 knockout mice. Perhaps in a less seizure resistant genetic background, knockout of Kv4.2 might result in spontaneous seizures. The findings from our studies comparing susceptibility to convulsant stimulation in non-littermate versus littermate Kv4.2 knockout and wildtype mice indicate that the susceptibility to convulsant stimulation with kainate in this genotype is modulated by the background mouse strain. These findings underscore the importance of using littermate wildtype controls for studies with knockout and transgenic mice, and highlight the importance of the genetic background as a modifying factor in the expression of seizures.
Kv4.2 knockout was associated with 100% mortality during status epilepticus. In comparison, 25% of the wildtype littermates with status epilepticus died. A 10–15 % mortality rate in the wildtype animals previously has been reported in a kainate model of seizures and status epilepticus (McKhann et al., 2003). While the mechanism underlying the high mortality rate in the Kv4.2 knockout mice is unknown, the prominent role of Kv4.2 in the early repolarization phase of the rodent myocyte action potential (Snyders, 1999), suggests the possibility of a cardiac mechanism for sudden death during status epilepticus in these mice. Under physiological conditions at rest, the Kv4.2 knockout mice do not express a cardiac phenotype compared to wildtype animals (Guo et al., 2005). However, during frequent seizures and status epilepticus, autonomic hyperactivity may result in a progressive cardiac dysrhythmia (Walton, 1993). Under these conditions, genetic absence of Kv4.2 in rodents may have lethal consequences.
The findings reported here strengthen the coupling of a channelopathy with seizures. Kv4.2 deficiency in the setting of an appropriate genetic background predisposes to increased forebrain excitability and a reduction in the seizure threshold. Therefore, enhancement of Kv4.2 function may represent a novel therapeutic strategy in the treatment of seizure disorders.
Acknowledgments
NIH/NINDS: NS049427 (AEA), NS039943 (AEA), K01-NS046328 (LFB), F32NS056664 (JNL), Epilepsy Foundation Postdoctoral Fellowship (JNL), Partnership for Pediatric Research in Epilepsy/Epilepsy Foundation (AEA), and Citizens United for Research in Epilepsy (AEA). We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- CA1
Cornu Ammonis 1
- EEG
electroencephalogram
- EPSP
excitatory postsynaptic potential
- Kv4.2
voltage-gated calcium-independent Shal-type potassium ion channel subunit 4.2
Footnotes
None of the authors has any conflict of interest to disclose.
References
- Andermann F, Kobayashi E, Andermann E. Genetic focal epilepsies: state of the art and paths to the future. Epilepsia. 2005;46(Suppl 10):61–67. doi: 10.1111/j.1528-1167.2005.00361.x. [DOI] [PubMed] [Google Scholar]
- Anderson AE, Hrachovy RA, Swann JW. Increased susceptibility to tetanus toxin-induced seizures in immature rats. Epilepsy Res. 1997;26:433–442. doi: 10.1016/s0920-1211(96)01010-8. [DOI] [PubMed] [Google Scholar]
- Andrasfalvy BK, Makara JK, Johnston D, Magee JC. Altered synaptic and non-synaptic properties of CA1 pyramidal neurons in Kv4.2 KO mice. J Physiol. 2008;586:3881–3992. doi: 10.1113/jphysiol.2008.154336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Ficker E, Heinemann U. Properties of two voltage-activated potassium currents in acutely isolated juvenile rat dentate gyrus granule cells. J Neurophysiol. 1992;68:2086–2099. doi: 10.1152/jn.1992.68.6.2086. [DOI] [PubMed] [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- Birnbaum SG, Varga AW, Yuan LL, Anderson AE, Sweatt JD, Schrader LA. Structure and function of Kv4-family transient potassium channels. Physiol Rev. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- Castro PA, Cooper EC, Lowenstein DH, Baraban SC. Hippocampal heterotopia lack functional Kv4.2 potassium channels in the methylazoxymethanol model of cortical malformations and epilepsy. J Neurosci. 2001;21:6626–6634. doi: 10.1523/JNEUROSCI.21-17-06626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, Kotagal P, Luders HO, Shi J, Cui J, Richerson GB, Wang QK. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733–738. doi: 10.1038/ng1585. [DOI] [PubMed] [Google Scholar]
- Eunson LH, Rea R, Zuberi SM, Youroukos S, Panayiotopoulos CP, Liguori R, Avoni P, McWilliam RC, Stephenson JB, Hanna MG, Kullmann DM, Spauschus A. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol. 2000;48:647–656. [PubMed] [Google Scholar]
- Glover WE. The aminopyridines. Gen Pharmacol. 1982;13:259–285. doi: 10.1016/0306-3623(82)90046-5. [DOI] [PubMed] [Google Scholar]
- Golding NL, Jung HY, Mickus T, Spruston N. Dendritic calcium spike initiation and repolarization are controlled by distinct potassium channel subtypes in CA1 pyramidal neurons. J Neurosci. 1999;19:8789–8798. doi: 10.1523/JNEUROSCI.19-20-08789.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Jung WE, Marionneau C, Aimond F, Xu H, Yamada KA, Schwarz TL, Demolombe S, Nerbonne JM. Targeted deletion of Kv4.2 eliminates I(to,f) and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ Res. 2005;97:1342–1350. doi: 10.1161/01.RES.0000196559.63223.aa. [DOI] [PubMed] [Google Scholar]
- Hablitz JJ. Picrotoxin-induced epileptiform activity in hippocampus: role of endogenous versus synaptic factors. J Neurophysiol. 1984;51:1011–1027. doi: 10.1152/jn.1984.51.5.1011. [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Karnup S, Stelzer A. Seizure-like activity in the disinhibited CA1 minislice of adult guinea-pigs. J Physiol. 2001;532:713–730. doi: 10.1111/j.1469-7793.2001.0713e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;569:41–57. doi: 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee R, Ficker E, Heinemann U. Comparison of voltage-dependent potassium currents in rat pyramidal neurons acutely isolated from hippocampal regions CA1 and CA3. J Neurophysiol. 1995;74:1982–1995. doi: 10.1152/jn.1995.74.5.1982. [DOI] [PubMed] [Google Scholar]
- Klein A, Boltshauser E, Jen J, Baloh RW. Episodic ataxia type 1 with distal weakness: a novel manifestation of a potassium channelopathy. Neuropediatrics. 2004;35:147–149. doi: 10.1055/s-2004-817921. [DOI] [PubMed] [Google Scholar]
- McKhann GM, 2nd, Wenzel HJ, Robbins CA, Sosunov AA, Schwartzkroin PA. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience. 2003;122:551–561. doi: 10.1016/s0306-4522(03)00562-1. [DOI] [PubMed] [Google Scholar]
- Menegola M, Trimmer JS. Unanticipated region- and cell-specific downregulation of individual KChIP auxiliary subunit isotypes in Kv4.2 knock-out mouse brain. J Neurosci. 2006;26:12137–12142. doi: 10.1523/JNEUROSCI.2783-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitterdorfer J, Bean BP. Potassium currents during the action potential of hippocampal CA3 neurons. J Neurosci. 2002;22:10106–10115. doi: 10.1523/JNEUROSCI.22-23-10106.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulley JC, Scheffer IE, Petrou S, Berkovic SF. Channelopathies as a genetic cause of epilepsy. Curr Opin Neurol. 2003;16:171–176. doi: 10.1097/01.wco.0000063767.15877.c7. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Gerber BR, Norris A, Burkhalter A. Electrical remodelling maintains firing properties in cortical pyramidal neurons lacking KCND2-encoded A-type K+ currents. J Physiol. 2008;586:1565–1579. doi: 10.1113/jphysiol.2007.146597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Krause M, Haug T, Storm JF, Stuhmer W. Modulation of the Ca2+-activated K+ current sIAHP by a phosphatase-kinase balance under basal conditions in rat CA1 pyramidal neurons. J Neurophysiol. 1998;79:3252–3256. doi: 10.1152/jn.1998.79.6.3252. [DOI] [PubMed] [Google Scholar]
- Perreault P, Avoli M. Physiology and pharmacology of epileptiform activity induced by 4-aminopyridine in rat hippocampal slices. J Neurophysiol. 1991;65:771–785. doi: 10.1152/jn.1991.65.4.771. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Sari P, Kerr DS. Domoic acid-induced hippocampal CA1 hyperexcitability independent of region CA3 activity. Epilepsy Res. 2001;47:65–76. doi: 10.1016/s0920-1211(01)00295-9. [DOI] [PubMed] [Google Scholar]
- Schulze-Bahr E, Wedekind H, Haverkamp W, Borggrefe M, Assmann G, Breithardt G, Funke H. The LQT syndromes--current status of molecular mechanisms. Z Kardiol. 1999;88:245–254. doi: 10.1007/s003920050283. [DOI] [PubMed] [Google Scholar]
- Shannon HE, Yang L. Seizure susceptibility of neuropeptide-Y null mutant mice in amygdala kindling and chemical-induced seizure models. Epilepsy Res. 2004;61:49–62. doi: 10.1016/j.eplepsyres.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Sheng M, Tsaur ML, Jan YN, Jan LY. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. 1992;9:271–284. doi: 10.1016/0896-6273(92)90166-b. [DOI] [PubMed] [Google Scholar]
- Singh B, Ogiwara I, Kaneda M, Tokonami N, Mazaki E, Baba K, Matsuda K, Inoue Y, Yamakawa K. A K(v)4.2 truncation mutation in a patient with temporal lobe epilepsy. Neurobiol Dis. 2006;24:245–253. doi: 10.1016/j.nbd.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales TO, Peiffer A, Anderson VE, Leppert M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- Snyders DJ. Structure and function of cardiac potassium channels. Cardiovasc Res. 1999;42:377–390. doi: 10.1016/s0008-6363(99)00071-1. [DOI] [PubMed] [Google Scholar]
- Traub RD, Colling SB, Jefferys JG. Cellular mechanisms of 4-aminopyridine-induced synchronized after-discharges in the rat hippocampal slice. J Physiol. 1995;489:127–140. doi: 10.1113/jphysiol.1995.sp021036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol. 1988;59:259–276. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- Tsaur ML, Sheng M, Lowenstein DH, Jan YN, Jan LY. Differential expression of K+ channel mRNAs in the rat brain and down- regulation in the hippocampus following seizures. Neuron. 1992;8:1055–1067. doi: 10.1016/0896-6273(92)90127-y. [DOI] [PubMed] [Google Scholar]
- Walton NY. Systemic effects of generalized convulsive status epilepticus. Epilepsia. 1993;34(Suppl 1):S54–58. doi: 10.1111/j.1528-1157.1993.tb05906.x. [DOI] [PubMed] [Google Scholar]
- Wong RK, Stewart M. Different firing patterns generated in dendrites and somata of CA1 pyramidal neurones in guinea-pig hippocampus. J Physiol. 1992;457:675–687. doi: 10.1113/jphysiol.1992.sp019401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RK, Traub RD, Miles R. Cellular basis of neuronal synchrony in epilepsy. Adv Neurol. 1986;44:583–592. [PubMed] [Google Scholar]