

Abstract
Major Depressive Disorder (MDD) has been conceptualized as a neural network-level disease. Few studies of the neural bases of depression, however, have used analytic techniques that are capable of testing network-level hypotheses of neural dysfunction in this disorder. Moreover, of those that have, fewer still have attempted to determine directionality of influence within functionally abnormal networks of structures. We used multivariate Granger causality analysis — a technique that estimates the extent to which preceding neural activity in one or more seed regions predicts subsequent activity in target brain regions — to analyze blood-oxygen-level dependent (BOLD) data collected during eyes-closed rest in depressed and never-depressed persons. We found that activation in the hippocampus predicted subsequent increases in ventral anterior cingulate cortex (vACC) activity in depression, and that activity in medial prefrontal cortex and vACC were mutually reinforcing in MDD. Hippocampal and vACC activation in depressed participants predicted subsequent decreases in dorsal cortical activity. This study shows that, on a moment-by-moment basis, there is increased excitatory activity among limbic and paralimbic structures, as well as increased inhibition in activity of dorsal cortical structures, by limbic structures in depression; these aberrant patterns of effective connectivity implicate disturbances in the mesostriatal dopamine system in depression. These findings advance neural theory of depression by detailing specific patterns of limbic excitation in MDD, by making explicit the primary role of limbic inhibition of dorsal cortex in the cortico-limbic relation posited to underlie depression, and by presenting an integrated neurofunctional account of altered dopamine function in this disorder.
Keywords: Depression, fMRI, Granger causality, multivariate, dopamine, neural network
Introduction
Over the past 15 years, research examining the neural substrates of Major Depressive Disorder (MDD) has documented a number of depression-associated anomalies in brain structure and function. With respect to brain structure, for example, meta-analytic syntheses have found reliably decreased hippocampal volume associated with depression, with volumetric decrease of the hippocampus correlated positively with chronicity of MDD (1, 2). Similarly, in the absence of pharmacological intervention, amygdala volume is reliably decreased in depression, more so with a longer history of depressive illness (3). Although found less consistently, investigators have also noted volumetric decreases in depressed individuals in anterior cingulate (4–6) and orbitofrontal (7, 8) cortices and striatum (9, 10).
Sufficient data have now accumulated from functional neuroimaging studies of MDD to permit their synthesis and systematic comparison. In the largest meta-analysis to date of functional neuroimaging data from samples of depressed individuals, Seminowicz and colleagues (11) compared regional brain metabolic data acquired during the initial assessment portion of three positron emission tomography (PET) studies of the neural bases of treatment effects in depression. Compared with never-depressed controls, depressed individuals were found to be characterized by abnormal activity in dorsolateral (12, 13), medial (14, 15) and orbital/ventrolateral (13, 16) prefrontal cortex (PFC), insular (17, 18) and parietal (18) cortices, rostral (19, 20) and ventral (6, 21) anterior cingulate cortex (ACC), as well as in the posterior cingulate cortex (15, 18), hippocampus (13, 22), thalamus (15, 23), and caudate (19, 23). Moreover, while activity in the amygdala has not been identified as abnormal in depression by such a systematic meta-analysis, a number of investigators have documented anomalous amygdala activity at baseline (16, 22) and in response to affective stimuli (24, 25) in depression.
Results from these investigations have been integrated into neural models of MDD (11, 26, 27). Among the most influential of these models is a formulation by Mayberg and her colleagues (27) that specifies a reciprocal relation between cortical structures that mediate cognition (e.g., dorsolateral PFC and inferior parietal cortex) and cortical and subcortical structures that support emotional functions (e.g., anterior insular cortex and hippocampus). According to this model, activity in limbic and paralimbic structures in depression dominates activity in dorsal cortical structures. Subsequent refinements to the original model specify that regions undergirding emotion-cognition integration (e.g., medial and orbital PFC) act as key mediators of this reciprocal cortico-limbic relation in depression (11).
Such neural models provide a useful theoretical framework for the clinical neuroscience of depression, and have been critical both in motivating ongoing empirical work examining the neural substrates of this disorder and in developing novel and effective neural-level interventions for MDD (15). It is important to recognize, however, that these models are not yet comprehensive in their scope and require important additional characterization. Perhaps most importantly, although depression is often characterized as a network-level neural disorder, we know little about the network-level properties of the brain in MDD beyond the constellation of structures that are consistently implicated in this disease. Specifically, our current formulations regarding which structures interact in an anomalous manner in MDD and the direction of influence among them require specification beyond that informed by studies of functional co-and counter-change among these structures in mood induction and treatment paradigms (27), which have relatively coarse temporal sampling, and from anterograde and retrograde tracing studies that map anatomical connectivity in non-disease model, non-human primates (26). This latter approach, while justified given the limited corpus of empirical work detailing structural and functional interrelations among neural ensembles in the human brain, nevertheless suffers from critical limitations. First, this approach often makes a “circuit diagram” assumption regarding cross-structure communication that implies that if Structure A sends axonal projections to Structure B, then Structure A, when active, will communicate with Structure B. This formulation ignores a large body of research indicating that active, anatomically connected structures do not communicate in an obligatory way by virtue of their anatomical connection but, instead, communicate dynamically in accord with task parameters and constraints (28). Second, this approach tends to ignore the potential for meaningful interactions between structures that are not connected mono-synaptically; indeed, recent evidence that bi-synaptically connected structures communicate in significant ways in depression (29) indicates that such interactions should be considered in neural models of this disorder. Finally, using anatomical connectivity data from non-disease model, non-human primates to characterize functional interactions among brain structures implicated in depression assumes that while activity in certain structures may be abnormal in depression, interaction among these structures is not; this assumption has been disconfirmed in preliminary work showing abnormal functional connectivity among structures showing abnormal function in depression (30, 31). The issues presented here with current multivariate, neurodynamic models of MDD suggest that the clinical neuroscience of depression will benefit from complementary model-free, multivariate analytic approaches examining functional neural network dynamics in real time in samples of depressed and nondepressed individuals.
A small handful of functional neuroimaging studies offer information about cross-structural communication and influence in depression. Lozano and colleagues (32), for example, implanted and activated stimulating electrodes in white matter adjacent to the ventral ACC (vACC; Brodmann Area 25) in individuals suffering from chronic, treatment-resistant depression. In addition to significantly reducing depressive symptomatology in the majority of participants in their study, this intervention also reduced vACC activity as well as activity in insular cortex, and medial and orbital/ventrolateral PFC; activity in dorsolateral PFC and dorsal ACC increased with stimulation of the vACC. These results are consistent with the formulation that modulation of vACC activity, in ameliorating depressive pathology, affects a constellation of limbic, paralimbic, and dorsal cortical structures that have been consistently implicated in MDD.
Taking a different approach to examining neural interaction and influence in depression, Seminowicz and others (11) applied structural equation modeling to PET data from a large sample of depressed persons, assessing the stability as connections were added and removed of a connectivity model derived from anatomical connectivity data from animals. The most stable connectivity model showed that cognitive-affective integration regions — medial and orbital PFC and rostral and ventral ACC — were key mediators of relations among other structures that have been associated with depression.
The present study was designed to specify more thoroughly aberrant functional interrelations in the brain in MDD by identifying and estimating anomalous cross-structure influence in depression. To do this, we used a multivariate implementation of Granger causality analysis (33), a model-free technique that has been used for estimating prior and posterior prediction between bivariate (34) and among multivariate (35) structure-specific blood-oxygen-level dependent (BOLD) time-series data.
Methods
Participants
Sixteen individuals diagnosed with Major Depressive Disorder (MDD) and 14 control subjects with no history of any DSM-IV diagnosed psychiatric disorder participated in this study. Participants were recruited from local psychiatric outpatient clinics as well as through website postings. All participants: 1) were between the ages of 18 and 50; 2) had no reported history of brain injury, lifetime history of primary psychotic ideation, or mania; 3) had no reported substance abuse within the past six months; and 4) had no physical limitations that prohibited them from undergoing an MRI examination. None of the participants in either group was taking antidepressant medication at the time of the study.
All depressed participants met criteria for a DSM-IV diagnosis of MDD based on their responses to the Structured Clinical Interview for DSM (SCID; 36); none of the control participants met diagnostic criteria for any current or past Axis-I disorder. In addition, all participants completed the Beck Depression Inventory-II (BDI-II; 37). Depressed individuals with a current comorbid diagnosis of panic or generalized anxiety disorder were excluded from the study. Informed consent was obtained from all participants, who were paid $25 per hour for their participation in the study. All aspects of this study complied with the ethical standards for treatment of human participants from the American Psychiatric Association.
Procedure
FMRI data acquisition
BOLD data were acquired with a 3.0T General Electric Signa MR scanner. Following scout scanning, two iterations of high order shimming were performed over the whole brain to reduce signal loss arising from field inhomogeneities. For the five-minute resting state scan, participants were instructed to lay still with their eyes closed and to avoid falling asleep. BOLD data were acquired with a single channel, whole-head imaging coil using a spiral pulse sequence (38) [repetition time (TR) = 1200 ms/frame, echo time (TE) 30 ms, flip angle = 77°, field of view (FOV) = 220 mm, number of frames = 250] that has been demonstrated to have superior recovery of BOLD signal in ventral prefrontal regions (39) of special interest to this investigation. A relatively rapid per-frame acquisition time of 1200 ms was used to increase the sensitivity of GC analysis to detect time-directed associations across BOLD time-series(34). We collected 18 axial slices of BOLD data per acquisition with this short TR sequence; by using 5 mm axial slices, we were able to acquire data from the most inferior aspect of the temporal lobes to the top of the brain, excluding the most superior aspect of somatomotor cortex (see Figure 1). Axial slices had 3.44 mm2 in-plane and 5 mm through-plane resolution. A high-resolution structural scan (124 axial slices, .859 mm2 in-plane and 1.2 mm through-plane resolution, TE = min, flip angle = 15°, FOV = 220 mm) was performed following BOLD scanning. We minimized participant head movement by inserting pads between the participant’s head and the fMRI headcoil.
Figure 1.
Depiction of imaging volume used to collect BOLD data.
FMRI data preprocessing
Preprocessing of fMRI data was conducted using Analysis of Functional Neural Images (AFNI) software (40). Time-series data were slice-time corrected relative to the ninth axial slice, and volume registered to correct for head translation and rotation during the scan (Fourier interpolation, two-pass). Data were spatially smoothed with a 4 mm Gaussian smoothing kernel. Functional images were then co-registered to high-resolution anatomical images and affine transformed into Talairach space (41). Because Granger causality analyses using relatively low lag orders, like those appropriate for the current study, operate on high frequency deflections in time-course data, BOLD time-series were not low-pass filtered. Low frequency fluctuations were removed from time-series data via regression (see Analysis section below).
Analysis
Description of approach
The primary objective of the analysis was to conduct multivariate Granger causality analysis on a network of structures found to exhibit anomalous function in depression. Identification of this network assumed a model in which the vACC is a primary functional convergence zone of these structures, an assumption that has been confirmed in prior research (11, 15). This network was identified in three steps: 1) localizing a vACC region of interest (ROI) that was over-recruited in the default-mode, or task-negative, network (42) in depression, as has been found in previous research (43); 2) with the vACC ROI so defined, conducting both vACC-to-whole-brain and whole-brain-to-vACC bivariate GC analysis on data from each participant; and 3) conducting voxel-wise comparisons of the resulting statistical maps from MDD and control groups to identify both regions with abnormal advance prediction of vACC activity in depression, and regions in which activity was predicted in advance by vACC activity more strongly in depressed than in nondepressed persons. We describe each of these steps more thoroughly below.
Identifying vACC ROI
Greicius et al. (43) demonstrated that the vACC is over-recruited in the default mode network in MDD. We sought to replicate this finding in order to identify the vACC ROI for this study. First, we identified the default mode network (DMN) in each participant by conducting a seed region-to-whole brain time-series correlation analysis similar to that used by Fox and colleagues (42). We then compared vACC involvement in this network in depressed and never-disordered persons. To identify the DMN in each participant, we regressed averaged BOLD time-series data from medial prefrontal and posterior cingulate seed regions (12 mm diameter, centered at −1 47 −4 and −5 −49 41, respectively; 42) against preprocessed voxel time-series data from the rest of the brain. Nuisance covariates included in the regression model were three translational and three rotational head-motion estimates and four regressors modeling zeroth- through third-order polynomial trends in the BOLD time series. To remove signal artifacts in voxel time series induced by cardiac pulsitility and respiratory fluctuations, we also included as a nuisance covariate average time-series data from a grey matter mask drawn on the Montreal Neurological Institute N27 brain. The correlation maps resulting from this regression procedure were then Fisher Z transformed to ensure that their statistics met the assumption of normality for conducting subsequent t-tests. Finally, transformed correlation maps from the MDD and control groups were compared using voxel-wise t-tests over the full extent of the vACC. The vACC was defined as the portion of the Talairach-defined (41) cingulate gyrus inferior to the most anterior aspect of the genu of the corpus callosum. The statistical threshold was set at p = .05, corrected for multiple comparisons across vACC voxels; appropriate per-voxel significance and cluster thresholds were calculated with AFNI’s AlphaSim.
Bivariate GC analysis
Consider the bivariate linear autoregressive model of two time-variant processes, x and y:
According to a definition from Granger (33), one time-variant process, x, “Granger causes” another time-variant process, y, across up to p temporal lags if preceding information from x predicts behavior in y, uniquely, relative to what preceding information about y can predict about behavior in y itself. In this model, zj(t) represents up to q exogenous processes (covariates, e.g., head motion parameters and physiological noise) independent of the path network (j=1,…,q). Contributions of each lagged variable to the prediction of its respective target are denoted by α; β corresponds to the covariate effect; and prediction errors of individual models are denoted by ε. If αyx is significantly different from zero, then it is said that x Granger causes y. One of the authors (GC) has implemented this definition in a linear autoregressive procedure (44) for estimating unique, time-directed prediction between the fMRI time-series of a seed region and the rest of the brain. We used this procedure to identify both regions whose time courses predict subsequent vACC activity and regions whose activity is predicted by preceding vACC activity abnormally in depression. Nuisance covariates in the regression model were the same as those used to identify the vACC ROI: six orthogonal motion estimates, polynomial functions modeling the BOLD drifting effect, and grey-matter signal. The seed region time-series in the model was preprocessed BOLD data extracted from a sphere centered on the peak of the vACC ROI (5 mm diameter, centered at −2, 6, −7) identified above. We estimated time-directed prediction between BOLD time-series across a lag of one TR (1200 ms) in order to maximize the temporal resolution of our estimates of neural influence. Finally, voxel-wise comparisons of resulting GC fit coefficients across diagnostic groups were performed with t-tests (p = .05, corrected) across the whole imaging volume.
Multivariate GC analysis
The bivariate GC definition presented above can be extended to multivariate conditions per the following generic multivariate autoregressive model:
We implemented this definition in a multivariate autoregressive procedure for determining unique time-directed prediction across multiple time-variant processes. We subjected to this procedure voxel time-courses from regions showing either an abnormal leading or lagging temporal relation with vACC activity in depression.1 For each participant, preprocessed time-series data were extracted from peak voxel locations in the regions that showed differential temporal relation with the vACC between depressed and control participants. These time-series data for each participant were then entered into multivariate GC analysis. The resulting GC path coefficients characterizing the strength and direction of the temporal relation among the structures, and the corresponding t-statistics, entered into the model were then compared between depressed and control groups (p = .05, uncorrected) using a linear mixed-effects multilevel model.
Results
Demographic and clinical variables
Demographic and clinical characteristics of the two groups of participants are presented in Table 1. The depressed and control groups did not differ significantly in age, t(28) = 1.50, or gender composition, χ2(28) = .50, ps < .05. The groups did differ significantly in years of education: the control participants had 1.5 years more education, on average, than did the depressed participants, t(28) = 2.16, p < .05. As expected, the depressed participants had higher BDI-II scores than did participants in the control group, t(28) = 13.12, p < .05 (see Table 1).
Table 1.
Participant demographic and clinical characteristics
| Control | Depressed | |
|---|---|---|
| Age, mean(SE) | 30.43 (2.35) | 34.63 (1.60) |
| Education, mean (SE)* | 17.14 (.8) | 15.44 (.60) |
| % Female | 43% | 62% |
| BDI-II, mean (SE)* | 3.79 (1.21) | 37.94 (2.19) |
Note:
p < .05; SE = Standard error of the mean; BDI-II = Beck Depression Inventory-II
Default-mode network comparison
The between-groups, within-vACC comparison of default-mode network statistical maps showed a region of significantly increased contribution (peak: −2, 6, −7, Brodmann Area 25) to the default-mode network in MDD; see Figure 2. Incidentally, the independent samples t-statistic value at this vACC location was the largest considering voxels outside the vACC as well.
Figure 2.
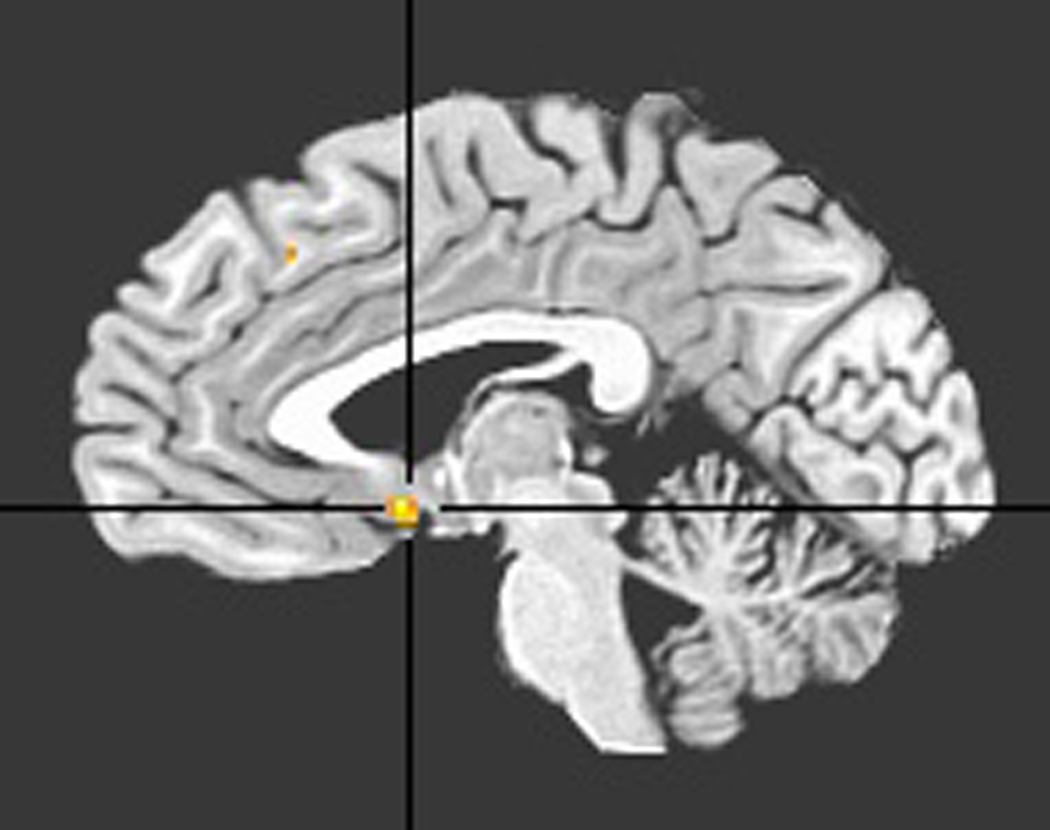
Ventral ACC ROI derived from comparison of default-mode networks of depressed with control groups.
Bivariate Granger causality analysis
The bivariate whole-brain-to-vACC GC analysis showed that activity in the medial prefrontal cortex (MPFC) and hippocampus predicted subsequent increases in vACC activity to a significantly greater extent in the depressed than in the control participants. This analysis also showed that increasing activity in dorsolateral prefrontal cortex (DLPFC) predicted subsequent decreasing vACC activity in depression (see Figure 3). Finally, the vACC-to-whole-brain GC analysis showed that vACC activity predicted subsequent decreases in activation in the posterior cingulate/cuneus (PCC), dorsomedial prefrontal cortex (DMPFC), and ventral striatum, bilaterally, in depression (see Figure 4).
Figure 3.

T-statistic map of MDD-versus-control comparison of vACC-to-whole brain GC analysis fit coefficients. Increased activity in the hippocampus and MPFC predicts subsequent increases in vACC activity more in MDD than in control participants. Activity increases in DLPFC predict activity decreases in the vACC more in MDD. Group mean coefficients are averaged from suprathreshold cluster. Group means in bold are significantly different from zero (two-tailed test, all ps < .01, uncorrected).
Figure 4.

T-statistic map of MDD-versus-control comparison of whole brain-to-vACC GC analysis fit coefficients. Increased activity in the vACC predicts subsequent decreases in activity in the PCC, DMPFC and ventral striatum, bilaterally more in MDD than in control participants. Group mean coefficients are averaged from suprathreshold cluster, ventral striatum clusters averaged together. Group means in bold are significantly different from zero (two-tailed test, all ps < .01, uncorrected).
Multivariate Granger causality analysis
The multivariate GC analysis incorporating regions identified in the bivariate GC analyses showed unique results not noted in the bivariate analysis. We observed reciprocal augmenting of activity between vACC and MPFC to a significantly greater extent in the depressed than in the control participants. Further, we found evidence of dampening of DLPFC by hippocampal activity in depression. We also noted significant dampening of ventral striatum by DMPFC activity in MDD more than in control participants. Several of the functional relations identified in the bivariate analyses were maintained in the multivariate analysis: in the depressed participants, increasing hippocampal activity predicted subsequent increases in activation in the vACC, and DLPFC activity predicted decreasing activity in the vACC. Finally, activity in the vACC predicted subsequent deactivation of DMPFC, PCC, and ventral striatum in depressed more than in control participants (see Figure 5).
Figure 5.

Statistical map of between-groups comparison of path coefficients from multivariate GC analysis including vACC, ventral striatum (vSTR), hippocampus (HPC), MPFC, DLPFC, DMPFC, and PCC. Blue/red arrows indicate significantly greater inhibition/activation of subsequent target activity in MDD versus control where a within-group effect was also present in the MDD group (two-tailed tests, all ps < .05, uncorrected). Table values are group mean path coefficients with prediction going from row to column. Group means in bold are significantly different from zero.
Discussion
In this study, we used bivariate Granger causality analysis to identify a set of structures for subsequent multivariate GC analysis in which activity precedes or follows activation in the vACC differently in depressed than in nondepressed individuals. These structures, implicated in depression in a number of functional neuroimaging studies, include the hippocampus (13, 19), DLPFC (13, 19), MPFC (45, 46), PCC (18, 47) and ventral striatum (48, 49). Using multivariate GC analysis, we found evidence of a mutually excitatory relation between vACC and MPFC and of an inhibitory relation between the hippocampus and DLPFC in MDD. Replicating results from the bivariate analysis, we also found in the multivariate analysis subsequent dampening by the vACC of activity in the PCC, DMPFC, and ventral striatum, and bolstering of activity in the vACC by the hippocampus in depressed, but not in control, participants.
The multivariate GC findings reported here represent a significant advance for current neurodynamic models of major depression. Cortico-limbic inhibition models of depression are equivocal concerning the mechanisms by which limbic and paralimbic activity come to dominate over cortical activity in MDD; this dominance could result from attenuation of cortical-to-limbic inhibition, from strengthening of limbic-to-cortical inhibition in depression, or from these two processes working in synchrony. In demonstrating that vACC activity predicts subsequent decreases in activity in dorsal cortical regions (PCC and DMPFC) in depression, and that hippocampal activity predicts subsequent reduction of DLPFC activity, our data support the formulation that limbic-to-cortical inhibition is strengthened in MDD. The pattern of results that would support the formulation of decreased cortical-to-limbic inhibition in depression, i.e., subsequently decreased limbic activity following dorsal cortical activation in control more than depressed persons, was not obtained. Further, in documenting excitation of the vACC by the hippocampus and mutual excitation between the vACC and MPFC in depression, the current findings suggest that dampening of dorsal cortical activity by limbic activity is a consequence of spreading excitation in depression among structures comprising limbic-paralimbic networks.
The current results that hippocampal activation precedes both increased vACC activity and decreased DLPFC activity in depression, considered together with our finding that the hippocampus itself is not differentially affected in depression by preceding activity in other structures identified in our analysis, are consistent with the formulation that the hippocampus plays a key role in affecting depressotypic neural responses. The finding that hippocampal activity predicts subsequently increased vACC activity in depression fills a gap in the body of data supporting the formulation that an overactive hippocampus-to-vACC connection plays an important role in depressive pathology (50, 51). Research examining white matter tractography in humans has documented direct connectivity between the hippocampus and vACC (51) that is augmented in bipolar disorder (52). Moreover, hippocampal subfield stimulation studies in rodents (53) indicate that this hippocampus-to-vACC connection is excitatory. Further, using a rodent model of depression, Airan and colleagues documented increased neural transmission from the CA1 hippocampal subfield in depression, which is decreased with administration of imipramine (50). Considered collectively, these results predict and underscore the significance of the finding in the present study of increased effective connectivity of hippocampus-to-vACC in MDD.
The current data also demonstrate that hippocampal activity predicts subsequently decreased activation in the DLPFC in depression. That hippocampal dysfunction can have an adverse effect on DLPFC functioning is supported by animal model studies showing that lesions to the hippocampus promote subsequent DLPFC dysfunction (54) and affective disturbance (55). An association between aberrant hippocampus-DLPFC coupling and emotional disturbance has also been documented in humans by Meyer-Lindenberg and colleagues, who reported that unmedicated schizophrenic individuals demonstrated abnormally persistent functional connectivity between the hippocampus and DLPFC during a working memory task (56). A mechanism by which this aberrant coupling may underlie affective illness is underscored by research showing that the hippocampus plays a necessary mediating role in DLPFC regulation of the striatal dopamine system (57, 58). Thus, hippocampus-to-DLPFC inhibition, as found in depressed persons in the current study, may interfere with DLPFC regulation of the striatal dopamine system and play a role in the anhedonic responding that characterizes major depression.
The present study documents the novel neurodynamic finding of aberrant mutual excitation between MPFC and vACC in MDD. This co-facilitation of activity in depression between the MPFC — which is involved in representation and evaluation of the self (59) — and the vACC — which is postulated to subserve viscero-affective states (60) —may represent a neurofunctional account of maladaptive, self-directed rumination associated with MDD. Indeed, examining the correlation between the depressive rumination subscale of the Response Style Questionnaire (RSQ) and path coefficients of MPFC-to-vACC connectivity in the present study supports this hypothesis: the stronger the excitation of the vACC by the MPFC, the more depressive rumination reported by depressed participants [r(14) = .55; p < .05, two-tailed, controlling for the positive correlation between BDI-II and RSQ scores].
Importantly, a growing body of work is documenting other significant associations between the vACC and the MPFC in major depression. For example, reducing activity in the vACC through electrode stimulation of adjacent white matter tracts in depression also decreases MPFC activity (15). In addition, depressed persons who respond therapeutically to sleep deprivation show greater metabolism in the vACC and MPFC than do nonresponders (61). Finally, research demonstrating that dopamine function is enhanced by sleep deprivation (62) suggests that elevated metabolism in MPFC and vACC is a consequence of disturbed dopamine function that has been hypothesized to underlie depression (63). This postulation is corroborated by recent work showing that MPFC and vACC activity increases in response to catecholamine depletion in depressed but not in healthy individuals (46). A unifying account of these findings that should be examined explicitly in future research is that decreased inhibitory dopaminergic input from the ventral tegmental area (VTA) to the thalamus in depression disinhibits excitatory input from the thalamus to the MPFC and vACC (61).
While the present results are clearly relevant to formulations of neural influence in models of the pathophysiology of depression, we should note three important limitations to the methods described in this paper. First, the relatively sluggish hemodynamic response measured by fMRI in estimating brain activity does not allow us to differentiate temporally neural responses that occur less than 100 milliseconds apart. It will be important in future research to use functional neuroimaging techniques such as magnetoencephalography that measure brain activity more directly to examine aberrant neural influence in MDD. Second, we worked from a model in which abnormal neural communication in depression is mediated through the vACC; consequently, only structures that showed abnormal activation leading or lagging that of the vACC were included in the multivariate GC model. While we functionally localized several structures that have been implicated reliably in depression, other structures, including the amygdala, orbitofrontal cortex, and insula, were not localized with the current methods. Given the consistency with which these structures have been found to be characterized by anomalous patterns of activation in depression (11), it is possible either that the first-order lag examined in this study was not optimal for identifying aberrant communication between these structures and the vACC in depression, or that they are not part of an aberrantly functioning network mediated by the vACC in MDD. Finally, it is important to bear in mind that the current findings were obtained during resting state fMRI scanning. Thus, we can infer only that the aberrant neural functioning in depression reported here is associated with depressive pathology in general, and not with a particular type of depressotypic thought or affect. Recent work has begun to identify bivariate neural causal relations during active processing of affective stimuli in depression (64).
The present study is the first to examine time-directed dynamic relations at the multivariate, neural systems level in MDD. Our findings advance neural theory of depression in specifying the short-term functional interrelations among structures implicated in this disorder. In continuing to develop a comprehensive neural theory of MDD, future work should extend this approach to include broader temporal gradients of significance to the pathophysiology of MDD, potentially incorporating multiple neurofunctional assessments as depressed persons transition from the state of acute illness to remission.
Supplementary Material
Acknowledgements
We gratefully acknowledge the contributions of Becka Johnson, Emily Dennis, Sarah Victor, Melissa Henry, and Lindsey Sherdell in assisting with the collection, analysis and presentation of data for the present study. We thank Amit Etkin for his critique of an earlier version of this manuscript.
Preparation of this manuscript was supported by Grant MH59259 from the National Institute of Mental Health awarded to Ian H. Gotlib and Grant MH079651 from the National Institute of Mental Health awarded to J. Paul Hamilton.
All authors had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
We conducted quality assurance tests on these time-courses to ensure that they were stationary and that the residuals were non-skewed and non-heteroscedastic; see Supplementary Table 1.
Conflict of Interest Statement
All authors of this manuscript report no competing or financial interests.
Contributor Information
J. Paul Hamilton, Department of Psychology, Stanford University, Stanford, CA 94305, USA
Gang Chen, Scientific and Statistical Computing Core, National Institute of Mental Health, Bethesda, MD 20892, USA
Moriah E. Thomason, Department of Psychology, Stanford University, Stanford, CA 94305, USA
Mirra E. Schwartz, Department of Psychology, Stanford University, Stanford, CA 94305, USA
Ian H. Gotlib, Department of Psychology, Stanford University, Stanford, CA 94305, USA
References
- 1.Campbell S, Marriott M, Nahmias C, MacQueen GM. Lower hippocampal volume in patients suffering from depression: A meta-analysis. American Journal of Psychiatry. 2004 Apr;161(4):598–607. doi: 10.1176/appi.ajp.161.4.598. [DOI] [PubMed] [Google Scholar]
- 2.Videbech P, Ravnkilde B. Hippocampal volume and depression: A meta-analysis of MRI studies. American Journal of Psychiatry. 2004 Nov;161(11):1957–1966. doi: 10.1176/appi.ajp.161.11.1957. [DOI] [PubMed] [Google Scholar]
- 3.Hamilton JP, Siemer M, Gotlib IH. Amygdala volume in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Molecular Psychiatry. 2008 Nov;13(11):993–1000. doi: 10.1038/mp.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Botteron KN, Raichle ME, Drevets WC, Heath AC, Todd RD. Volumetric reduction in left subgenual prefrontal cortex in early onset depression. Biological Psychiatry. 2002 Feb;51(4):342–344. doi: 10.1016/s0006-3223(01)01280-x. [DOI] [PubMed] [Google Scholar]
- 5.Caetano SC, Kaur S, Brambilla P, Nicoletti M, Hatch JP, Sassi RB, et al. Smaller cingulate volumes in unipolar depressed patients. Biological Psychiatry. 2006 Apr;59(8):702–706. doi: 10.1016/j.biopsych.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 6.Drevets WC, Price JL, Simpson JR, Todd RD, Reich T, Vannier M, et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997 Apr;386(6627):824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- 7.Bremner JD, Vythilingam M, Vermetten E, Nazeer A, Adil J, Khan S, et al. Reduced volume of orbitofrontal cortex in major depression. Biological Psychiatry. 2002 Feb;51(4):273–279. doi: 10.1016/s0006-3223(01)01336-1. [DOI] [PubMed] [Google Scholar]
- 8.Lacerda ALT, Keshavan MS, Hardan AY, Yorbik O, Brambilla P, Sassi RB, et al. Anatomic evaluation of the orbitofrontal cortex in major depressive disorder. Biological Psychiatry. 2004 Feb;55(4):353–358. doi: 10.1016/j.biopsych.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 9.Krishnan KRR, McDonald WM, Escalona PR, Doraiswamy PM, Na C, Husain MM, et al. Magnetic-Resonance-Imaging of the Caudate Nuclei in Depression - Preliminary-Observations. Archives of General Psychiatry. 1992 Jul;49(7):553–557. doi: 10.1001/archpsyc.1992.01820070047007. [DOI] [PubMed] [Google Scholar]
- 10.Parashos IA, Tupler LA, Blitchington T, Krishnan KRR. Magnetic-resonance morphometry in patients with major depression. Psychiatry Research-Neuroimaging. 1998 Nov;84(1):7–15. doi: 10.1016/s0925-4927(98)00042-0. [DOI] [PubMed] [Google Scholar]
- 11.Seminowicz DA, Mayberg HS, McIntosh AR, Goldapple K, Kennedy S, Segal Z, et al. Limbic-frontal circuitry in major depression: a path modeling metanalysis. Neuroimage. 2004 May;22(1):409–418. doi: 10.1016/j.neuroimage.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 12.Bench CJ, Friston KJ, Brown RG, Scott LC, Frackowiak RSJ, Dolan RJ. The Anatomy of Melancholia - Focal Abnormalities of Cerebral Blood-Flow in Major Depression. Psychological Medicine. 1992 Aug;22(3):607–615. doi: 10.1017/s003329170003806x. [DOI] [PubMed] [Google Scholar]
- 13.Drevets WC, Videen TO, Price JL, Preskorn SH, Carmichael ST, Raichle ME. A Functional Anatomical Study of Unipolar Depression. Journal of Neuroscience. 1992 Sep;12(9):3628–3641. doi: 10.1523/JNEUROSCI.12-09-03628.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonul AS, Kula M, Bilgin AG, Tutus A, Oguz A. The regional cerebral blood flow changes in major depressive disorder with and without psychotic features. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 2004 Sep;28(6):1015–1021. doi: 10.1016/j.pnpbp.2004.05.036. [DOI] [PubMed] [Google Scholar]
- 15.Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005 Mar;45(5):651–660. doi: 10.1016/j.neuron.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 16.Drevets WC, Bogers W, Raichle ME. Functional anatomical correlates of antidepressant drug treatment assessed using PET measures of regional glucose metabolism. European Neuropsychopharmacology. 2002 Dec;12(6):527–544. doi: 10.1016/s0924-977x(02)00102-5. [DOI] [PubMed] [Google Scholar]
- 17.Drevets WC, Raichle ME. Neuroanatomical Circuits in Depression - Implications for Treatment Mechanisms. Psychopharmacology Bulletin. 1992;28(3):261–274. [PubMed] [Google Scholar]
- 18.Mayberg HS. Limbic-cortical dysregulation: A proposed model of depression. Journal of Neuropsychiatry and Clinical Neurosciences. 1997 Sum;9(3):471–481. doi: 10.1176/jnp.9.3.471. [DOI] [PubMed] [Google Scholar]
- 19.Kennedy SH, Evans KR, Kruger S, Mayberg HS, Meyer JH, McCann S, et al. Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. American Journal of Psychiatry. 2001 Jun;158(6):899–905. doi: 10.1176/appi.ajp.158.6.899. [DOI] [PubMed] [Google Scholar]
- 20.Mayberg HS, Brannan SK, Mahurin RK, Jerabek PA, Brickman JS, Tekell JL, et al. Cingulate function in depression: A potential predictor of treatment response. Neuroreport. 1997 Mar;8(4):1057–1061. doi: 10.1097/00001756-199703030-00048. [DOI] [PubMed] [Google Scholar]
- 21.Pizzagalli DA, Oakes TR, Fox AS, Chung MK, Larson CL, Abercrombie HC, et al. Functional but not structural subgenual prefrontal cortex abnormalities in melancholia. Molecular Psychiatry. 2004 Apr;9(4):393–405. doi: 10.1038/sj.mp.4001501. [DOI] [PubMed] [Google Scholar]
- 22.Hornig M, Mozley PD, Amsterdam JD. HMPAO SPECT brain imaging in treatment-resistant depression. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 1997 Oct;21(7):1097–1114. doi: 10.1016/s0278-5846(97)00100-0. [DOI] [PubMed] [Google Scholar]
- 23.Brody AL, Saxena S, Stoessel P, Gillies LA, Fairbanks LA, Alborzian S, et al. Regional brain metabolic changes in patients with major depression treated with either paroxetine or interpersonal therapy - Preliminary findings. Archives of General Psychiatry. 2001 Jul;58(7):631–640. doi: 10.1001/archpsyc.58.7.631. [DOI] [PubMed] [Google Scholar]
- 24.Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA. Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: An fMRI study. Biological Psychiatry. 2001 Nov;50(9):651–658. doi: 10.1016/s0006-3223(01)01263-x. [DOI] [PubMed] [Google Scholar]
- 25.Siegle GJ, Steinhauer SR, Thase ME, Stenger VA, Carter CS. Can't shake that feeling: Assessment of sustained event-related fMRI amygdala activity in response to emotional information in depressed individuals. Biological Psychiatry. 2002 May;51(9):693–707. doi: 10.1016/s0006-3223(02)01314-8. [DOI] [PubMed] [Google Scholar]
- 26.Drevets WC. Neuroimaging studies of mood disorders. Biological Psychiatry. 2000 Oct;48(8):813–829. doi: 10.1016/s0006-3223(00)01020-9. [DOI] [PubMed] [Google Scholar]
- 27.Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, et al. Reciprocal limbic-cortical function and negative mood: Converging PET findings in depression and normal sadness. American Journal of Psychiatry. 1999 May;156(5):675–682. doi: 10.1176/ajp.156.5.675. [DOI] [PubMed] [Google Scholar]
- 28.Engel AK, Fries P, Singer W. Dynamic predictions: Oscillations and synchrony in top-down processing. Nature Reviews Neuroscience. 2001 Oct;2(10):704–716. doi: 10.1038/35094565. [DOI] [PubMed] [Google Scholar]
- 29.Siegle GJ, Thompson W, Carter CS, Steinhauer SR, Thase ME. Increased amygdala and decreased dorsolateral prefrontal BOLD responses in unipolar depression: Related and independent features. Biological Psychiatry. 2007 Jan;61(2):198–209. doi: 10.1016/j.biopsych.2006.05.048. [DOI] [PubMed] [Google Scholar]
- 30.Anand A, Li Y, Wang Y, Wu JW, Gao SJ, Bukhari L, et al. Activity and connectivity of brain mood regulating circuit in depression: A functional magnetic resonance study. Biological Psychiatry. 2005 May;57(10):1079–1088. doi: 10.1016/j.biopsych.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 31.Hamilton JP, Gotlib IH. Neural substrates of increased memory sensitivity for negative stimuli in major depression. Biological Psychiatry. 2008 Jun;63(12):1155–1162. doi: 10.1016/j.biopsych.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lozano AM, Mayberg HS, Giacobbe P, Hamani C, Craddock RC, Kennedy SH. Subcallosal cingulate gyrus deep brain stimulation for treatment-resistant depression. Biological Psychiatry. 2008 Sep;64(6):461–467. doi: 10.1016/j.biopsych.2008.05.034. [DOI] [PubMed] [Google Scholar]
- 33.Granger CWJ. Investigating causal relations by econometric models and cross-spectral methods. Econometrica. 1969;37(3):424–438. [Google Scholar]
- 34.Goebel R, Roebroeck A, Kim DS, Formisano E. Investigating directed cortical interactions in time-resolved fMRI data using vector autoregressive modeling and Granger causality mapping. Magnetic Resonance Imaging. 2003 Dec;21(10):1251–1261. doi: 10.1016/j.mri.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 35.Deshpande G, LaConte S, James GA, Peltier S, Hu XP. Multivariate Granger Causality Analysis of fMRI Data. Human Brain Mapping. 2009 Apr;30(4):1361–1373. doi: 10.1002/hbm.20606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.First MB, Spitzer RL, Gibbon M, Williams JBW. The Structured Clinical Interview for DSM-III-R Personality-Disorders (SCID-I) Journal of Personality Disorders. 1995;9(2):83–91. [Google Scholar]
- 37.Beck AT, Rush AJ, Shaw BF, Emery G. Cognitive therapy of depression. 1979 [Google Scholar]
- 38.Glover GH, Law CS. Spiral-in/out BOLD fMRI for increased SNR and reduced susceptibility artifacts. Magnetic Resonance in Medicine. 2001 Sep;46(3):515–522. doi: 10.1002/mrm.1222. [DOI] [PubMed] [Google Scholar]
- 39.Preston AR, Thomason ME, Ochsner KN, Cooper JC, Glover GH. Comparison of spiral-in/out and spiral-out BOLD fMRI at 1.5 and 3T. Neuroimage. 2004 Jan;21(1):291–301. doi: 10.1016/j.neuroimage.2003.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cox RW. AFNI: Software for analysis and visualization of functional magnetic resonance neuroimages. Computers and Biomedical Research. 1996;29:162–173. doi: 10.1006/cbmr.1996.0014. [DOI] [PubMed] [Google Scholar]
- 41.Talairach J, Tournoux P. Co-Planar Stereotaxic Atlas of the Human Brain. Stuttgart, Germany: Thieme; 1988. [Google Scholar]
- 42.Fox MD, Snyder AZ, Vincent JL, Corbetta M, Van Essen DC, Raichle ME. The human brain is intrinsically organized into dynamic, anticorrelated functional networks; Proceedings of the National Academy of Sciences of the United States of America; 2005. Jul, pp. 9673–9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greicius MD, Flores BH, Menon V, Glover GH, Solvason HB, Kenna H, et al. Resting-state functional connectivity in major depression: Abnormally increased contributions from subgenual cingulate cortex and thalamus. Biological Psychiatry. 2007;62(5):429–437. doi: 10.1016/j.biopsych.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen G, Hamilton JP, Thomason ME, Gotlib IH, Saad ZS, Cox RW. Multi-region granger causality tuned for FMRI data analysis; Annual Meeting of the International Society for Magnetic Resonance in Medicine; 2009. [Google Scholar]
- 45.Holthoff VA, Beuthien-Baumann B, Zundorf G, Triemer A, Ludecke S, Winiecki P, et al. Changes in brain metabolism associated with remission in unipolar major depression. Acta Psychiatrica Scandinavica. 2004 Sep;110(3):184–194. doi: 10.1111/j.1600-0447.2004.00351.x. [DOI] [PubMed] [Google Scholar]
- 46.Hasler G, Fromm S, Carlson PJ, Luckenbaugh DA, Waldeciz T, Geraci M, et al. Neural response to catecholamine depletion in unmedicated subjects with major depressive disorder in remission and healthy subjects. Archives of General Psychiatry. 2008 May;65(5):521–531. doi: 10.1001/archpsyc.65.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Joe AY, Tielmann T, Bucerius J, Reinhardt MJ, Palmedo H, Maier W, et al. Response-dependent differences in regional cerebral blood flow changes with citalopram in treatment of major depression. Journal of Nuclear Medicine. 2006 Aug;47(8):1319–1325. [PubMed] [Google Scholar]
- 48.Fu CHY, Williams SCR, Cleare AJ, Brammer MJ, Walsh ND, Kim J, et al. Attenuation of the neural response to sad faces in major depression by antidepressant treatment - A prospective, event-related functional magnetic resonance imaging study. Archives of General Psychiatry. 2004 Sep;61(9):877–889. doi: 10.1001/archpsyc.61.9.877. [DOI] [PubMed] [Google Scholar]
- 49.Surguladze S, Brammer MJ, Keedwell P, Giampietro V, Young AW, Travis MJ, et al. A differential pattern of neural response toward sad versus happy facial expressions in major depressive disorder. Biological Psychiatry. 2005 Feb;57(3):201–209. doi: 10.1016/j.biopsych.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 50.Airan RD, Meltzer LA, Roy M, Gong YQ, Chen H, Deisseroth K. High-speed Imaging reveals neurophysiological links to behavior in an animal model of depression. Science. 2007 Aug;317(5839):819–823. doi: 10.1126/science.1144400. [DOI] [PubMed] [Google Scholar]
- 51.Johansen-Berg H, Gutman DA, Behrens TEJ, Matthews PM, Rushworth MFS, Katz E, et al. Anatomical connectivity of the subgenual cingulate region targeted with deep brain stimulation for treatment-resistant depression. Cerebral Cortex. 2008;18(6):1374–1383. doi: 10.1093/cercor/bhm167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Houenou J, Wessa M, Douaud G, Leboyer M, Chanraud S, Perrin M, et al. Increased white matter connectivity in euthymic bipolar patients: diffusion tensor tractography between the subgenual cingulate and the amygdalo-hippocampal complex. Molecular Psychiatry. 2007;12:1001–1010. doi: 10.1038/sj.mp.4002010. [DOI] [PubMed] [Google Scholar]
- 53.Vertes RP. Interactions among the medial prefrontal cortex, hippocampus and midline thalamus in emotional and cognitive processing in the rat. Neuroscience. 2006;142(1):1–20. doi: 10.1016/j.neuroscience.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 54.Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: Schizophrenia as a reality test. Neuropsychopharmacology. 2000 Sep;23(3):223–239. doi: 10.1016/S0893-133X(00)00137-8. [DOI] [PubMed] [Google Scholar]
- 55.Lipska BK, Weinberger DR. Delayed effects of neonatal hippocampal damage on haloperidol-induced catalepsy and apomorphine-induced stereotypic behaviors in the rat. Developmental Brain Research. 1993 Oct;75(2):213–222. doi: 10.1016/0165-3806(93)90026-7. [DOI] [PubMed] [Google Scholar]
- 56.Meyer-Lindenberg AS, Olsen RK, Kohn PD, Brown T, Egan MF, Weinberger DR, et al. Regionally specific disturbance of dorsolateral prefrontal-hippocampal functional connectivity in schizophrenia. Archives of General Psychiatry. 2005 Apr;62(4):379–386. doi: 10.1001/archpsyc.62.4.379. [DOI] [PubMed] [Google Scholar]
- 57.Odonnell P, Grace AA. Synaptic-interactions among excitatory afferents to nucleus-accumbens neurons - hippocampal gating of prefrontal cortical input. Journal of Neuroscience. 1995;15(5):3622–3639. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saunders RC, Kolachana BS, Bachevalier J, Weinberger DR. Neonatal lesions of the medial temporal lobe disrupt prefrontal cortical regulation of striatal dopamine. Nature. 1998;393(6681):169–171. doi: 10.1038/30245. [DOI] [PubMed] [Google Scholar]
- 59.Northoff G, Bermpohl F. Cortical midline structures and the self. Trends in Cognitive Sciences. 2004 Mar;8(3):102–107. doi: 10.1016/j.tics.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 60.Drevets WC, Savitz J, Trimble M. The subgenual anterior cingulate cortex in mood disorders. CNS Spectr. 2008;13(8):663–681. doi: 10.1017/s1092852900013754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu J, Buchsbaum MS, Gillin JC, Tang C, Cadwell S, Wiegand M, et al. Prediction of antidepressant effects of sleep deprivation by metabolic rates in the ventral anterior cingulate and medial prefrontal cortex. American Journal of Psychiatry. 1999;156(8):1149–1158. doi: 10.1176/ajp.156.8.1149. [DOI] [PubMed] [Google Scholar]
- 62.Ebert D, Feistel H, Kaschka W, Barocka A, Pirner A. Single-photon emission computerized-tomography assessment of cerebral dopamine D2 receptor blockade in depression before and after sleep-deprivation – preliminary results. Biological Psychiatry. 1994;35(11):880–885. doi: 10.1016/0006-3223(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 63.Swerdlow NR, Koob GF. Dopamine, schizophrenia, mania and depression - toward a unified hypothesis of cortico-striato-pallido-thalamic function. Behavioral and Brain Sciences. 1987;10(2):197–207. [Google Scholar]
- 64.Thompson WK, Siegle G. A stimulus-locked vector autoregressive model for slow event-related fMRI designs. Neuroimage. 2009 Jul;46(3):739–748. doi: 10.1016/j.neuroimage.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.