

Abstract
Nonhost environmental reservoirs of pathogens play key roles in their evolutionary ecology and in particular in the evolution of pathogenicity. In light of recent reports of the plant pathogen Pseudomonas syringae in pristine waters outside agricultural regions and its dissemination via the water cycle, we have examined the genetic and phenotypic diversity, population structure, and biogeography of P. syringae from headwaters of rivers on three continents and their phylogenetic relationship to strains from crops. A collection of 236 strains from 11 sites in the United States, in France, and in New Zealand was characterized for genetic diversity based on housekeeping gene sequences and for phenotypic diversity based on measures of pathogenicity and ice nucleation activity. Phylogenetic analyses revealed several new genetic clades from water. The genetic structure of P. syringae populations was not influenced by geographic location or water chemistry, whereas the phenotypic structure was affected by these parameters. Comparison with strains from crops revealed that the metapopulation of P. syringae is structured into three genetic ecotypes: a crop-specific type, a water-specific type, and an abundant ecotype found in both habitats. Aggressiveness of strains was significantly and positively correlated with ice nucleation activity. Furthermore, the ubiquitous genotypes were the most aggressive, on average. The abundance and diversity in water relative to crops suggest that adaptation to the freshwater habitat has played a nonnegligible role in the evolutionary history of P. syringae. We discuss how adaptation to the water cycle is linked to the epidemiological success of this plant pathogen.
IMPORTANCE
Many pathogens have life cycles that involve survival and multiplication in nonhost environmental habitats. For human pathogens, numerous studies have revealed how adaptation to environmental habitats is linked to the evolution of their pathogenicity and emergence of pathogens. For plant pathogens, the link between adaptation to nonhost habitats and pathogenicity has not been explored. Here we have examined the genetic and phenotypic diversity of the plant pathogen Pseudomonas syringae in headwaters of rivers on three continents and compared it to that of strains from crops. This model pathogen was chosen because it is widely abundant in habitats associated with the water cycle and in particular in pristine waters outside agricultural regions. This work reveals that there is considerable exchange of populations between freshwater and agricultural habitats and that those in the former contribute considerably to the diversification of P. syringae.
INTRODUCTION
There is mounting evidence that the ecology of pathogens in their nonhost environmental reservoirs can have a significant impact on their ability to cause disease in their hosts. This has been demonstrated for human pathogens, such as Vibrio cholerae, with a marked capacity to survive and proliferate in environmental reservoirs (1, 2). Other examples include Legionella pneumophila (3), Pseudomonas aeruginosa (4), the Burkholderia cepacia complex (5), and Cryptococcus neoformans (6). Environmental reservoirs play three key roles in the evolutionary ecology of these pathogens (7). First, survival and proliferation in environmental reservoirs ensure a competitive advantage in the absence of the host. Second, genes for virulence factors can be acquired in these reservoirs. Third, some virulence factors also have roles in adaptation to the conditions of the environmental reservoirs and are thereby positively selected in the reservoirs in the absence of interaction with the primary host. In the quest to elucidate the foundation of parasitic fitness, this emerging insight has oriented research toward questions concerning the basis of adaptability to the environment—in complement to questions about specialization to the host (5).
For plant pathogens, there has been relatively little investigation of the impact of their ecology in environmental (nonagricultural) reservoirs on parasitic fitness. Although environmental reservoirs of many plant pathogens are known, they have been ignored in studies of the evolution of parasitic fitness—which have been almost exclusively agrocentric (7). For example, we have very little perspective on the relative genetic diversity of plant pathogens in environmental versus agricultural habitats, whereas for human pathogens such as V. cholerae, autochthonous environmental isolates are known to have more extensive genetic diversity than strains from clinical contexts, which tend to be primarily clonal (8). Similarly, the genotypes of strains of P. aeruginosa in open oceans are, surprisingly, distinct from those from all other habitats of this bacterium (9). Furthermore, we have no clear sense of the biogeographic distribution of plant pathogens in environmental habitats. For V. cholerae, it is known that the distribution of genotypes in water is markedly influenced by temperature and the extent of eutrophication (8). For both V. cholerae and P. aeruginosa, geographic location in ocean waters relative to the coast has a notable influence on the genetic structure of their populations (10, 11). In contrast, environmental and clinical strains of Legionella spp. manifest similar degrees of genetic diversity and the distribution of these genotypes does not have an obvious geographic pattern (12).
The plant pathogen Pseudomonas syringae is emerging as an opportune model for exploring ecology in environmental habitats. Our recent discovery of its abundance in substrates associated with the water cycle, and in particular its frequent presence in alpine rivers and lakes (13), provides a unique occasion to characterize this unknown diversity. The objectives of this work were to determine the genetic diversity and major phenotypic properties (pathogenicity and ice nucleation activity) of populations of P. syringae in river headwaters—upstream from agricultural regions on three continents—and to evaluate the biogeographical relationships among these populations and their relatedness to strains collected from disease epidemics. Via this approach, we attempted to gain novel insight into the evolutionary history of this bacterium that might not be perceived from studies of its direct relationship with crops.
RESULTS
P. syringae is ubiquitous in river source waters.
P. syringae was detected at concentrations of ca. 50 to 104 CFU/liter in rivers at all 11 sites sampled and constituted generally less than 10−3 of the total cultural bacteria in these water samples (Table 1). All of the strains characterized here had phenotypic traits compatible with P. syringae (production of a fluorescent pigment and absence of cytochrome c oxidase and arginine dihydrolase). Genotyping confirmed this identification as indicated below. Ninety-six percent of the strains also induced a hypersensitive response (HR) in tobacco. The rivers in this study represent sources of irrigation water in southeastern and southwestern France, in the northwestern United States, and in southern New Zealand. These data complement previous observations of P. syringae in river source waters in other geographic regions, viz, the Po River in Italy (13). These population density values are probably underestimates in that they do not account for variants of P. syringae that cannot grow on the culture medium used here.
TABLE 1 .
Origins of strains of P. syringae from water and characteristics of the water sampled
| Population density (log CFU/liter) | ||||||
|---|---|---|---|---|---|---|
| Country, region, date, and stream | Latitude, longitude(altitude [m]) | Site code | Conductivity (µS) | P. syringae | Total mesophilicbacteria | Strain codes |
| France, Alpes-de-Haute-Provence County | ||||||
| 3 April 2007 | ||||||
| Sauze, east branch | 44°20′40″N,6°40′54″E (2,100) | 10.1 | 522 | 4.30 | 7.25 | SZ002—SZ030 |
| Sauze, west branch | 44°20′40″N,6°40′48″E (2,100) | 11.1 | 1,105 | 3.00 | 7.30 | SZ031—SZ054 |
| 31 May 2007 | ||||||
| Sauze, east branch | 44°20′40″N,6°40′54″E (2,100) | 10.2 | 339 | 2.16 | 5.55 | SZ085—SZ113 |
| Sauze, west branch | 44°20′40″N,6°40′48″E (2,100) | 11.2 | 352 | 1.56 | 5.62 | SZ115—SZ125 |
| Sauze, source | 44°20′11″N,6°40′15″E (2,500) | 12 | 232 | 1.85 | 5.51 | SZ127—SZ145 |
| France, Hautes-Alpes County, 31 May 2007, Rioul Mounal, source | 44°32′ 09" N,6°42′12″E (2,100) | 0 | 102 | 1.83 | 6.79 | UB410—UB429 |
| France, Lozère County, 23 April 2007, Tarn, near source | 44°22′44″N,3°47′41″E (1,300) | 21 | 20 | 2.65 | 7.24 | TA001—TA022 |
| France, Aveyron County, 24 April 2007, Viaur, near source | 44°15′35″N,2°59′10″E (900) | 22 | 87 | 3.03 | 6.82 | Vi003—Vi024 |
| United States, Park County, MT, 12 August 2007, Pine Creek | 45°29′15″N,110°30′ 00" W (1,865) | 33 | 68 | 2.75 | 5.84 | USA001—USA019 |
| United States, Grand Teton National Park, WY, | ||||||
| 14 August 2007 | ||||||
| Pilgrim Creek | 43°55′42″N,110°33′42″W (2,140) | 37 | 200 | 2.69 | 6.82 | USA087—USA108 |
| Cascade Creek | 43°45′54″N,110°45′ 00" W (2,200) | 40 | 43 | 2.84 | 6.32 | USA031—USA053 |
| New Zealand, Central Otago, South Island, | ||||||
| 16 November 2007, Schoolhouse Creek | 45°12′05.14"S,168°59′ 09"E (677) | 42 | 1.42 | 3.45 | 5.81 | AI001—AI134 |
| 4 March 2009, Commissioners Creek | 45°14′50.41"S,168°55′ 53"E (800) | 43 | 1.49 | 4.03 | 5.92 | AL005—AL039 |
Strains of P. syringae from rivers represent clades that have not been detected previously.
The phylogenetic trees resulting from the neighbor-joining approach and from the maximum-likelihood approach were strictly identical in terms of the grouping of strains and their placement in the trees. Results here refer to the phylogeny based on the neighbor-joining approach.
Clades of P. syringae were differentiated based on phylogenetic analysis of 1,885 bp of concatenated cts, gapA, gyrB, and rpoD gene sequences (Fig. 1) such that the maximum genetic difference between strains in a single clade was not greater than 2.3%. This criterion is consistent with the genetic variability in previously described clades of P. syringae (groups 1 to 5) (47, 63) and places the reference strains in the expected clades. Based on this criterion, strains in group 2 could be differentiated into two clades, with each clade having less than 1% genetic variation within it and with a mean genetic distance of 2.8% between clades (Fig. 1). Otherwise, each clade was more than 3.5% different from the other clades (Table 2). In water, nine clades that have not been previously described were identified (the USA102 clade, the TA003 clade, the TA002 clade, the CC1524 clade, the UB246 clade, and four clades that were represented by only one strain each, i.e., UB370, USA032, TA020, and TA006). Twenty-three strains used for phylogenetic analyses fell into these nine clades. Among these, 74% induced an HR in tobacco. Strains that did not induce an HR in tobacco included three of four strains in the TA002 clade, one strain in the CC1524 clade, and both strains in the UB246 clade.
FIG 1 .

Neighbor-joining phylogenetic tree constructed on the basis of the concatenated sequences of the four housekeeping gene fragments gapA, cts, gyrB, and rpoD (1,885 bases) for 120 strains of P. syringae and 4 strains of similar species (P. viridiflava, P. fluorescens, and P. aeruginosa). The tree was rooted on P. aeruginosa PAO1. Bootstrap scores are indicated at each node, and a ruler for 5% dissimilarity of the concatenated sequences is presented below the tree. Names of branches indicated as groups correspond to the genomic groups reported previously (47, 63). Names of branches indicated as clades correspond to genomic groups not reported previously. For the part of the tree containing P. syringae strains, the substrates from which the strains on each branch were isolated are indicated. Substrates collected in nonagricultural environments are indicated by black rectangles, and those from agricultural environments are indicated by gray rectangles.
TABLE 2 .
Genetic variability within and between clades of P. syringaea
| Mean variability between clades | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cladeb | No. of strains | Mean variability within clade | Group 1 | Group 2a | Group 2b | Group 3 | Group 4 | SZ030 | USA102 | TA003 | TA002 | CC1524 |
| Group 1 | 16 | 1.45 | ||||||||||
| Group 2a | 23 | 0.79 | 8.71 | |||||||||
| Group 2b | 38 | 0.92 | 9.13 | 2.81 | ||||||||
| Group 3 | 6 | 1.20 | 7.85 | 5.96 | 6.62 | |||||||
| Group 4 | 3 | 0.18 | 8.45 | 8.44 | 8.69 | 8.16 | ||||||
| SZ030 | 7 | 1.03 | 8.98 | 3.53 | 3.71 | 6.46 | 8.63 | |||||
| USA102 | 3 | 1.61 | 9.75 | 9.46 | 9.61 | 9.22 | 8.16 | 9.80 | ||||
| TA003 | 4 | 1.25 | 10.08 | 9.78 | 9.78 | 9.72 | 9.89 | 9.69 | 2.99 | |||
| TA002 | 4 | 2.3 | 10.00 | 10.52 | 10.96 | 9.85 | 9.82 | 10.51 | 10.53 | 10.67 | ||
| CC1524 | 6 | 1.01 | 10.74 | 11.20 | 11.67 | 10.73 | 11.68 | 11.46 | 11.19 | 11.53 | 7.37 | |
| UB246 | 2 | 0.60 | 13.74 | 13.89 | 14.17 | 14.13 | 14.88 | 14.07 | 13.02 | 13.78 | 14.28 | 14.21 |
Variability is expressed as the percent difference in the 1,885-base-long concatenated partial sequences of the cts, gapA, gyrB, and rpoD genes.
Clades refer to branches on the phylogenetic tree in Fig. 1.
Interestingly, several identical concatenated sequence types were identified among the strains for which the four genes were sequenced. Strains CC0457 (isolated from diseased cantaloupe during an epidemic of bacterial blight near Avignon, France, in 2003) and AI116 (from water at site 42 in New Zealand) had identical concatenated sequences. Strains USA001 (from water at site 33 in the United States), USA109 (from water at site 37 in the United States), and CC1429 (isolated from an epilithic biofilm at site 33 in the United States in 2004) (15) also had identical concatenated sequences.
The P. syringae metapopulation is composed of three ecotypes: water strains, crop strains, and ubiquitous strains.
Further analyses of the structure of populations from water and from crops were based on the sequences of the cts gene only. When the phylogeny based on four housekeeping genes described above was compared to a tree constructed based solely on the sequence of the cts gene, strains were clustered identically. The use of the gyrB, rpoD, and gapA sequences in addition to that of cts led to the placement of strains UB370, TA006, and TA020 into clades that were distinct from their neighbors. Otherwise, all of the clades in both trees were identical. Hence, we used the cts sequences only, thereby allowing us to incorporate more strains into the subsequent analyses.
Haplotypes of the cts sequences of 236 strains from water were compared to those of 87 strains from epidemics by exploiting, for the latter, sequences available in public databases and those for some crop strains in our collection. The 323 strains split into 88 unique haplotypes (G/N [genetic richness, i.e., the total number of multilocus genotypes, G, divided by the total number of samples, N] = 0.37), 44 of which were represented by a single strain, and the dominant haplotype was represented by 54 strains. Of the 88 haplotypes analyzed, 21 represented strains from crops only, 59 represented strains from water only, and 8 represented strains from water and crops. To separate haplotypes into homogeneous groups, clustering was performed with STRUCTURE for 1 to 11 clusters (K). Two optimal numbers of clusters were identified, k = 2 and k = 4, based on the ΔK statistic of Evanno et al. (16). A subsequent principal-component analysis (PCA), followed by clustering based on Ward’s method, revealed four clusters of P. syringae strains (Fig. 2). Axes 1 and 2 of the PCA accounted, respectively, for 24.8% and 17.2% of the total genetic variability. The clusters discriminated by multivariate analysis were in perfect agreement with the clusters inferred by using the Bayesian clustering algorithm with k = 4 (Fig. 2). Assuming a threshold of Q > 0.8 for assignment to a cluster, about 88% of the distinct haplotypes clearly belonged to only one cluster, indicating that the four clusters were highly divergent.
FIG 2 .
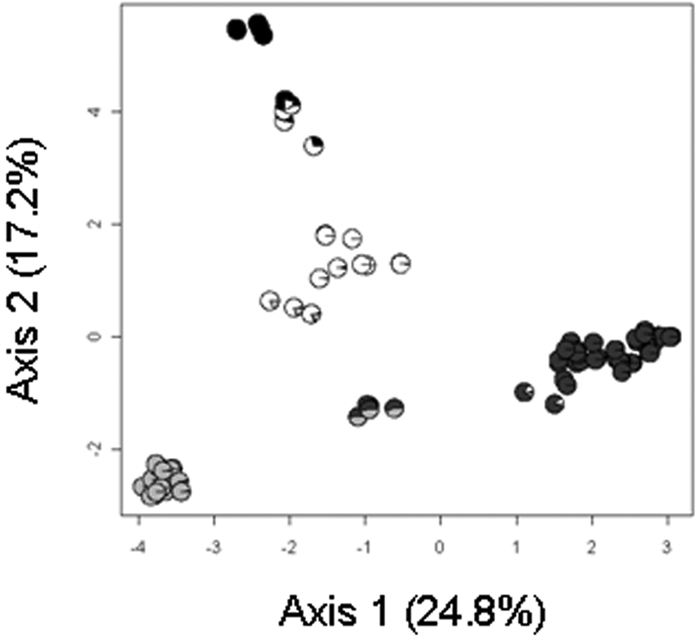
PCA performed on 88 haplotypes of P. syringae, followed by a nonhierarchical classification with clustering based on Ward’s method. For each strain, the probability of assignment to each of the four clusters obtained with the STRUCTURE software is reported on the PCA using a pie chart legend as follows: cluster 1, black; cluster 2, white; cluster 3, light gray; cluster 4, dark gray.
Clusters 2, 3, and 4 were composed of haplotypes representing strains from water and from crops (Fig. 3). Seven haplotypes that had equal probabilities of belonging to cluster 3 and cluster 4 represented strains from crops. Haplotypes in cluster 1, or that were equally likely to belong to clusters 1 and 2, represented strains from water only. Three of the strains in cluster 1 were also included in the phylogenetic analysis presented above. These strains all belonged to the CC1524 clade (Fig. 1). Likewise, the relationship between clades and the other clusters could be determined based on strains used in both the phylogenetic and cluster analyses. Cluster 2 was composed of strain USA032 and strains in the USA102 and TA003 clades but also included strain SZ131 in the UB246 clade. Cluster 3 was composed of strains in groups 2a and 2b and the SZ030 clade. Cluster 4 was composed of strains in group 1. Strains that had equal probabilities of being in cluster 3 and cluster 4 were in group 3. Strain TA020 and strains in the TA002 clade had equal probabilities of being in cluster 1 and cluster 2.
FIG 3 .
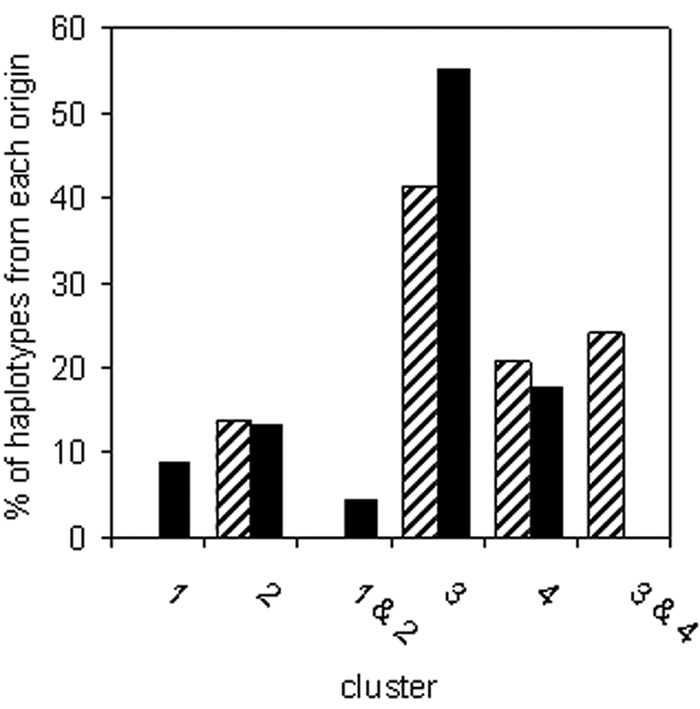
Distribution of cts haplotypes of P. syringae into four genetic clusters inferred with the Bayesian clustering algorithm of the STRUCTURE software. The frequency of haplotypes assigned to each of the clusters is indicated for haplotypes of strains from crops (hatched bars) and of strains from water (solid black bars). Of the 88 haplotypes analyzed, 78 were assigned to clusters with probabilities of inferred ancestry between 0.70 and 0.99. Three haplotypes were assigned at equal probabilities (ca. 0.50) to clusters 1 and 2, and seven haplotypes were assigned at equal probabilities to clusters 3 and 4.
Results of both clustering and phylogenetic analyses were coherent in terms of the co-occurrence and separation of genetic groups of P. syringae between water and crops. Water harbored genotypes not found among the strains from crops analyzed here. These genotypes corresponded to cluster 1 haplotypes: the CC1524 and TA002 clades and strain TA020 (Fig. 1). Furthermore, phylogenetic analysis based on all four housekeeping gene sequences indicated that crop strains in cluster 2 were not part of the USA102, TA003, and UB246 clades, even though they had similar cts sequences. Hence, the latter three clades contained no representatives from crops. Among these five clades, the CC1524 and TA002 clades harbored strains from other nonagricultural substrates (Fig. 1). Likewise, crops harbored a genotype not detected among the water strains: group 3, which comprises reference strain 1448A. Strains in groups 1, 2a, and 2b were found in water and in crops. These clades were widespread among numerous substrates, and groups 2a and 2b, in particular, were detected in all of the substrates from which isolations have been made in this and previous studies (13, 15).
The genetic structure of P. syringae populations in water is not markedly influenced by geographic location.
The partial cts sequences of the 236 strains from water analyzed here revealed 67 haplotypes. For each of the three major regions where water was sampled (France, the United States, and New Zealand), 40 to 70% of the combined samples from that region constituted haplotypes specific to the region and 13 to 27% was composed of haplotypes shared by all three major regions (Fig. 4). Overall, not more than 50% of the haplotypes collected in each region were also found elsewhere. Nevertheless, FST analyses of the interaction of genetic population structure and geography revealed few significant differences among sites (Fig. 5). With the exception of the populations at site 43 and at the second sampling date for site 10 (31 May 2007), all populations were similar to at least one other population (within or across regions), in terms of the relative proportions of the different haplotypes and their overall genetic relatedness. The significant differences observed between the two samples at site 10 were due to loss of diversity in the sample obtained on the second date relative to that obtained on the first date: the samples obtained on both dates had two haplotypes in common, but the sample obtained on the first date had four additional haplotypes not detected in that obtained on the first date, whereas the sample from the second date had an additional haplotype not detected in that from the first date. It is interesting that this corresponded to a 100-fold reduction in the density of the P. syringae population at this site between the two sampling dates. At adjacent sampling site 11, there was a 25-fold reduction in population density between the two dates but this did not lead to a significant change in population structure. The differences in population structure at New Zealand sites 42 and 43 were more striking than the temporal differences observed at French site 10. At site 42, 19 haplotypes were detected; at site 43, 11 haplotypes were detected, of which only 4 were also detected at site 42. Furthermore, about 30% of the population at site 43 was composed of strains with haplotypes unique to that site.
FIG 4 .
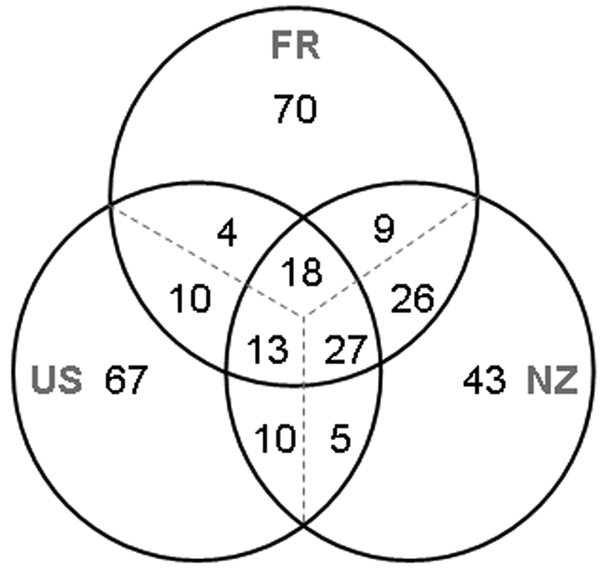
Frequency of haplotypes of P. syringae from water unique to the region sampled (France [FR], New Zealand [NZ], or the United States [US]) and in common between and among these regions. Frequency is expressed as the percentage of the total number of strains in the pooled samples from each region. Haplotypes were identified in terms of partial sequences of the cts gene.
FIG 5 .

Genetic similarity of populations of P. syringae, based on partial sequences of the cts gene, in terms of the sites and dates of isolation from water. Populations are represented by circles whose size is proportional to the relative concentration of the P. syringae population (number of bacteria per liter) in the waters that were sampled. The numbers in the circles correspond to the sampling sites listed in Table 1. The regional origins of samples are indicated by the gray ovals (US, United States; FR, France; NZ, New Zealand). Within the U.S. and France regions, the physical distance between populations shown is roughly proportional to their real geographic distance. The genetic structures of populations linked by black lines are not significantly different (P > 0.05) based on an FST test. The structures of those not linked by black lines are significantly different (P ≤ 0.05). The structures of the populations circled in black are significantly different from those of all of the other populations in this study.
The phenotypic structure of P. syringae populations in water is influenced by geographic location.
The origin of water samples had a significant effect on the aggressiveness of P. syringae populations on cantaloupe seedlings. This significant influence was observed for aggressiveness expressed as the mean absolute disease severity on cotyledons, the mean disease severity relative to that induced by control strain CC0094, and the mean frequency of cotyledons with a score of ≥1. A one-way analysis of variance (ANOVA) revealed a significant effect of sample origin on mean disease severity for the samples in Table 1 (P = 3.38 × 10−8). The sites with the lowest average aggressiveness of strains were all French sites (10.1, 11.1, and 21) (Table 3). However, the populations at sites 10 and 11 experienced marked increases in average aggressiveness between the first and second sampling dates, and for site 10, this increase was significant. Likewise, one-way ANOVA indicated that sample origin had a significant effect on the average temperature at which strains catalyzed freezing, as measured for suspensions of 104 (P = 0.023) and 106 (P = 0.036) cells. Here we present mean freezing temperatures for suspensions of 106 cells. These temperatures were the coldest for French samples 10.1 and 11.1 (Table 3). As for disease severity, the mean freezing temperature increased significantly—by 2°C or more—by the second sampling date at both of these sites. It is interesting that the change in phenotypic profile at site 10 corresponded to a significant change in genetic population structure but for site 11 it did not.
TABLE 3 .
Average aggressiveness and ice nucleation activity phenotypes of strains of P. syringae in waters from different origins
| Sourcea | No. of strains | Aggressivenessb | Ice nucleation activityd |
|---|---|---|---|
| 0 | 13 | 1.64 (A)c | −6.2 (AB)e |
| 10.1 | 15 | 0.08 (C) | −7.2 (B) |
| 10.2 | 14 | 1.68 (A) | −4.7 (A) |
| 11.1 | 14 | 0.23 (C) | −7.1 (B) |
| 11.2 | 10 | 0.72 (ABC) | −5.2 (A) |
| 12 | 11 | 0.29 (BC) | −6.3 (AB) |
| 21 | 13 | 0.13 (C) | −6.1 (AB) |
| 22 | 15 | 1.10 (ABC) | −5.3 (A) |
| 33 | 10 | 1.56 (AB) | −5.8 (AB) |
| 37 | 10 | 0.48 (ABC) | −4.9 (A) |
| 40 | 10 | 0.64 (ABC) | −5.8 (AB) |
| 42 | 70 | 1.02 (ABC) | −5.4 (A) |
Sources are described in Table 1. Phenotypes of strains from site 43 were not determined.
Shown is the overall mean disease severity (on a scale of 0 to 4), for all strains tested per source, as indexed on 12 cantaloupe cotyledons per strain.
Mean severity values followed by the same letters were not significantly different (P ≤ 0.05) based on Tukey’s honest significant difference test.
Shown is the overall mean temperature, for all strains tested per source, at which ice nucleation was induced by the suspensions of 106 cells tested per strain.
Mean temperatures followed by the same letter are not significantly different (P ≤ 0.05) based on the least significant difference test. (No significant differences were revealed with Tukey’s honest significant difference test.)
Water conductivity has a significant impact on the aggressiveness and ice nucleation activity, but not on the genetic structure, of populations.
To evaluate the impact of overall water chemistry on the structure of P. syringae populations, water samples were categorized based on low conductivity (<100 µS), medium conductivity (100 to 500 µS), and high conductivity (>500 µS). For genotypic characterization, the analysis was based on 134, 73, and 29 strains in each category, respectively, and for phenotypic characterization, analyses were based on 102, 73, and 29 strains in each category, respectively; FST analysis indicated that there was no significant effect of water conductivity on the genetic structure of P. syringae populations (AMOVA, among-group variation Va, P = 0.421). However, one-way analyses of variance showed that there was a significant effect of conductivity for all phenotypic variables (all measures of aggressiveness and ice nucleation activity) (P < 0.05). Tukey’s honest significant difference test showed that the effect of conductivity was due to a significantly reduced aggressiveness on cantaloupe and reduced ice nucleation activity of strains from waters with high conductivity. The mean severity of disease caused by strains from water with low and medium conductivity was 0.93 and 1.05, respectively, but for strains from water with high conductivity, the mean disease severity was 0.15. Likewise, the mean temperature of ice nucleation activity for suspensions of 106 cells of strains from low- and high-conductivity waters was −5.5°C and −5.3°C, respectively, whereas for strains from high-conductivity waters it was −7.1°C.
There is a significant interaction among phylogenetic relatedness, aggressiveness, and ice nucleation activity for P. syringae strains from water.
Genetic clusters of P. syringae, identified with Bayesian clustering as described above, had a significant effect on aggressiveness on cantaloupe and on ice nucleation activity (one-way ANOVA, P < 0.001 for all measures of these phenotypes). Strains in cluster 3 induced the overall highest mean severity. In particular, 30% of the strains in this cluster induced disease on cantaloupe with a mean severity score of 2 or greater. This frequency was significantly greater (P < 0.05) than that of any of the other clusters (Table 4). Likewise, on average, cluster 3 strains induced freezing at warmer temperatures than strains in the other clusters (Table 4). Clusters 2 and 3 had significantly higher frequencies of strains that could induce freezing at −5°C or at warmer temperatures (P < 0.05).
TABLE 4 .
Aggressiveness on cantaloupe seedlings and ice nucleation activity of strains in each of the cts haplotype clusters of P. syringae
| Clustera | No. of strains | Mean disease severity scoreb | % of strains with disease severity score of >2c | Mean freezing temp (°C)d | % of strains freezing at ≥−5°Ce |
|---|---|---|---|---|---|
| 1 | 20 | 0.23 | 5.0 B | −6.90 | 20.0 B |
| 2 | 19 | 0.35 | 5.3 B | −5.26 | 73.7 A |
| 3 | 121 | 1.20 | 30.0 A | −4.97 | 80.2 A |
| 4 | 43 | 0.46 | 0.0 B | −7.47 | 27.9 B |
Clusters of cts haplotypes were inferred with the Bayesian clustering algorithm of the STRUCTURE software.
Shown is the mean disease severity after 7 days of incubation of 12 inoculated cotyledons per strain. ANOVA indicated a significant effect of cluster on mean disease severity (P < 0.001).
The frequency of highly aggressive strains is expressed as the percentage of strains inducing a mean disease severity of 2 (on a scale of 0 to 4) on cantaloupe seedlings. Percentages followed by the same letter are not significantly different (P > 0.05) based on a pairwise Fisher exact test of the total number of strains in each phenotype category.
Shown is the mean warmest freezing temperature of 106 cells of each strain tested in each cluster. ANOVA indicated a significant effect of cluster on mean freezing temperature (P < 0.001).
The frequency of highly ice nucleation active strains is expressed as the percentage of strains freezing at −5°C or at warmer temperatures. Percentages followed by the same letter are not significantly different (P > 0.05) based on a pairwise Fisher exact test of the total number of strains in each phenotype category.
There was also a significant positive correlation between the temperature at which strains induced ice nucleation and their aggressiveness on cantaloupe. Based on Pearson’s product-moment correlation, the warmest temperature of ice nucleation for suspensions of 106 cells increased significantly with aggressiveness in terms of the mean disease score per strain (R = 0.274, P = 7.21 × 10−5), the mean disease score per strain as a fraction of the score for the control strain (CC0094) used in each series of inoculation (R = 0.296, P = 1.67 × 10−5), and the frequency of diseased plants per strain (R = 0.292, P = 2.14 × 10−5). Spearman’s rank correlation also indicated that these correlations were significant (P < 3 × 10−5 for all correlations). Correlations with ice nucleation activity in suspensions of 104 cells indicated identical significant trends.
To further evaluate the relationship between ice nucleation activity and aggressiveness on cantaloupe, strains were attributed to classes based in the intensity of these traits. Strains in aggressiveness class 0 (nonvirulent strains) did not cause symptoms on more than 1 of the 12 seedlings inoculated per strain. Strains in aggressiveness class 1 caused disease on two to eight seedlings. Strains in aggressiveness class 2 caused disease on 9 or more of the 12 seedlings. Likewise, strains in ice nucleation activity class 0 had no detectable capacity to induce freezing at temperatures warmer than −9°C in a population of 106 cells; those in ice nucleation activity class 1 had at least 1 cell among 106 able to catalyze ice formation at −6 to −9°C, and those in class 2 had at least 1 cell among 106 able to catalyze ice formation at −2 to −5°C. A contingency table of the number of strains in each of the paired classes further illustrates the significant relationship between ice nucleation activity and aggressiveness (Table 5). Nonvirulent strains had equal probabilities of being in any of the three classes of ice nucleation activity, whereas strains that were not ice nucleation active were rarely virulent on cantaloupe.
TABLE 5 .
Contingency table of the relationship of aggressiveness on cantaloupe to the ice nucleation activity of 203 water strains of P. syringae
| No. of strains in ice nucleation activity classb: | |||
|---|---|---|---|
| Aggressiveness classa | 0 | 1 | 2 |
| 0 | 24 | 22 | 37 |
| 1 | 3 | 10 | 35 |
| 2 | 6 | 12 | 54 |
Strains in aggressiveness class 0 (nonvirulent strains) did not cause symptoms on more than 1 of the 12 seedlings inoculated per strain, strains in aggressiveness class 1 caused disease on 2 to 8 seedlings, and strains in aggressiveness class 2 caused disease on 9 or more of the 12 seedlings.
Strains in ice nucleation activity class 0 had no detectable capacity to induce freezing at temperatures warmer than −9°C in a population of 106 cells. Strains in ice nucleation activity class 1 had at least 1 cell among 106 able to catalyze ice formation at −6 to −9°C. Strains in class 2 had at least 1 cell among 106 able to catalyze ice formation at −2 to −5°C. P = 0.0000, Fisher’s exact test.
DISCUSSION
The whole of the population biology of P. syringae reported to date concerns populations associated with plants, and in particular with plant canopies. Natural populations of P. syringae on plant canopies are generally dominated by strains that are pathogenic to the cropped plants in the locality studied (17–20), illustrating the selective pressure of plants in the apparent diversity and structure of populations of plant pathogens. By exploring the diversity of P. syringae populations in freshwater, a habitat where this bacterium is ubiquitous and likely to be subjected to selective forces quite different from those engendered by plants, we have revealed four major insights into its population biology that also give clues to its evolutionary history and strategies for adaptation.
First, populations of P. syringae in water contain genetic clades not previously reported for strains from crops. These clades, for the most part, are closer to the root of the phylogenetic tree than the previously reported clades. Most of the strains in these clades induce hypersensitivity in tobacco, suggesting that they have type III secretion systems. In other words, they have the cellular structures that effectively inject some effectors into plants, signaling that they are pathogens, thereby inducing a resistance response in the plants (14). However, their low aggressiveness on cantaloupe suggests that they do not have sufficient molecular arms to cause much disease in this plant species. It is possible that these new clades are, in fact, associated with crops and have not yet been reported or that they are below current detection levels. Alternatively, they could be pathogens of plants not grown as crops. The focus of plant pathology has been mostly limited to cultivated plants. As cultivated plants are a minority in the plant kingdom, pathogens of noncrop plants could have easily been overlooked.
Second, the overall genetic structure of P. syringae populations in water is not significantly affected by geographic location. Sequencing of more genes of the strains studied here might contribute nuances to this initial observation. But based on the data as presented, there seems to be a considerable force contributing to the pandemicity of lines of this bacterium, most likely via transport with the water cycle (13). This is well illustrated by the numerous haplotypes found both in water and on crops and in particular by the presence of apparent clones such as AI116 and CC0457 found in both water in New Zealand and crops in France. Nevertheless, 43% to 70% of the combined populations from each continent contained haplotypes of the cts gene that were unique to that continent. These haplotypes were not dominant and thereby did not have a significant impact on overall population structure. However, we cannot rule out the possibility that the haplotypes labeled as unique to a location are indeed unique and not simply present at a low concentration elsewhere. If some haplotypes are found only in one location while others are spread around the world, then it would suggest that two different mechanisms might be responsible for the dispersion of P. syringae. Both could be linked with the water cycle. But one would be responsible for dispersion over short distances (explaining the high percentage of haplotypes unique to a continent or a sampling site) and one would be responsible for dispersion over large distances (explaining why the same haplotype can be found on different continents). Nevertheless, these unique haplotypes reveal a role for endemicity of water-borne populations of P. syringae, thereby supporting the notion that freshwater habitats are not simply passive reservoirs of P. syringae but play a role in the evolution of this bacterium. Water chemistry had an impact on genetic population structure only when there were marked shifts in chemistry, such as in the early spring in the French Alps. Water chemistry can vary, depending on the residence time of the water emerging from the water table into mountain streams and the geologic context (21). Hence, the specific biogeography portrayed in this study could fluctuate across seasons in some geographic contexts.
Third, there is a link between the phylogeny and pathogenicity of P. syringae. This was revealed by the significant effect of the cts haplotype cluster on aggressiveness on cantaloupe. The strains in cluster 3, in particular, were the most aggressive on cantaloupe. Previously, we demonstrated that aggressiveness on cantaloupe was positively correlated with the number of plant species in P. syringae’s host range (22). The strains most aggressive on cantaloupe were virulent on up to 10 other plant species. The correlation we observed was highly significant (for Pearson’s product-moment correlation, P < 0.0001; not reported in the original publication). We have confirmed this trend with an independent test of the host range of 31 strains of P. syringae on 15 plant species (data not shown; for Pearson’s product-moment correlation, P = 9.1 × 10−7). Hence, the intensity of symptoms induced on cantaloupe (i.e., aggressiveness) is an indication of the relative extent of the host range of strains. The more aggressive strains tend to be more polyvalent in their host range than the less aggressive strains. This leads to the surprising observation that strains in group 2 (which corresponds to cluster 3) are found in a wide diversity of agricultural (crops, irrigation water) and nonagricultural (snow, rain, lakes and rivers, epilithic biofilms, wild plants) substrates and are also effective pathogens with broad host ranges. Among the strains in this group for which we have host range data, CC0094, CFBP1906, and CC0125 were reported to be virulent on 7, 9, and 10 plant species, respectively (22). This suggests that strains of P. syringae in group 2, in particular, have accumulated factors favoring both extended host range and saprophytic ubiquity. It is likely that these factors would be dual-use pathogenicity factors, such as broad-spectrum toxins (7). Based on observations from other pathosystems, we might have expected to observe a fitness cost of aggressiveness and broad host range. For instance, a Turnip mosaic virus isolate avirulent on the turnip line containing the resistance gene TuRB01 outcompeted virulent isolates in coinoculation experiments in a susceptible host background (23). A similar situation was recently well illustrated in the interaction between Potato virus Y and the pepper resistance gene Pvr4 (24). Fitness costs have been reported for fungal plant pathogens such as Leptosphaeria maculans (25) and Melampsora lini (26). For the bacterium Xanthomonas oryzae pv. oryzae, virulent strains obtained through mutagenesis caused shorter lesions than the wild-type strain on a rice line containing no resistance genes (27). However, other authors have reviewed numerous examples where a pathogen’s multiplication is not negatively correlated to its aggressiveness (28). For P. syringae, we observed that a sensitivity to water conductivity above 500 µS might be associated with a fitness cost for high levels of aggressiveness. This might imply that the factors involved in broad host range and general saprophytic fitness in group 2 involve regulation of membrane and cell wall permeability to ions and that this regulation is more constrained in clades with limited host range and environmental distribution. This fitness cost might also be markedly different from the cost of host-specific virulence. In other clades of P. syringae, strains might be aggressive on a very limited number of plant species (such as in group 3) and the underlying mechanisms of virulence could severely preclude their fitness as saprophytes, thereby contributing to their limited spread among habitats. In this light, it is interesting to note the recent emergence of strains of P. syringae in group 3 that are highly aggressive on horse chestnut (29). These strains have a limited host range and fall into a clade that seems to be particularly adapted to woody species.
Fourth, ice nucleation activity and pathogenicity are also linked; viz., there is a very low probability for strains to be aggressive on cantaloupe, and hence to have a broad host range in light of the arguments above, if they are not ice nucleation active. This link is likely the result of dissemination being facilitated by ice nucleation activity. We have previously presented evidence that ice nucleation activity is a sort of active dissemination mechanism ensuring that bacteria, which are otherwise aerodynamically ultralight and tend to remain suspended in the air, are deposited with falling precipitation formed by freezing of atmospheric water drops (13). Airborne dissemination and subsequent fallout would favor encounters with a wide variety of habitats where virulence factors could be acquired or where lines with phenotypes permitting a large host range are positively selected. Initial contact with plant cellular contents might have been facilitated by the induction of frost damage to plants, which would have positively selected for ice nucleation in those strains able to grow on the released nutrients (30). It is interesting that the host range of P. syringae is also positively correlated with the range of carbon sources strains can use for growth (31). This scenario would suppose that the ancestors of modern P. syringae were effectively ice nucleation active but not plant pathogenic. The sequenced alleles of the ice nucleation gene of bacteria are highly conserved and found only in the gammaproteobacteria (32). The known ice nucleation species containing this gene are on disparate branches of the phylogenetic tree of the pseudomonads (33), and there is evidence that the gene derives from a common ancestor of these species (32). The type III secretion system essential for pathogenicity in P. syringae and in many other Gram-negative bacteria is also widely prevalent across divergent lineages of the pseudomonads, as well as outside this group. However, the gene cluster for the type III secretion system is on a pathogenicity island flanked by remnants of mobile genetic elements indicative of acquisition by horizontal gene transfer (14). The sequence diversity between alleles of the genes in this pathogenicity cluster suggests that the clusters evolved independently after acquisition (14). If ice nucleation activity is a driver of diversification of plant pathogenicity in P. syringae, has it played this role in other plant-pathogenic species? Only a very few pathovars in the quintessential plant-associated genus Xanthomonas, for example, are ice nucleation active, and this genus is composed mostly of host-specific strains with virtually no reports of its presence in other substrates. P. fluorescens and Pantoea agglomerans are widespread and frequently associated with plants, yet only a few strains of the former have been reported as ice nucleation active, in contrast to the latter. It is difficult to speculate on the originality of this evolutionary strategy without fully comparative observations of other bacteria. Furthermore, it could also be possible that there are metabolic or regulatory links between ice nucleation activity and pathogenicity that lead to compatibility or incompatibility of these traits in the same individual. However, this question has not been explored.
Taken as a whole, our results suggest that the freshwater habitat has a considerable impact on the biology and population structure of P. syringae. To assess the importance of freshwater habitats in the life history of this bacterium, a comparison of the sizes of the total metapopulation of P. syringae in water relative to that in association with plants would be useful. These population sizes would be indicators of comparative fitness in these two environments. We can only make rough estimates based on existing data on abundance on plant surfaces and in water, neither of which accounts fully for spatial heterogeneity. To make this calculation, we can exploit measures of the total leaf surface on Earth (109 km2) (34), the percentage of this leaf surface in biomes compatible with P. syringae ecology (ca. 10% in cropland plus 30% elsewhere) (35), and the total freshwater reserves in lakes and rivers (175,000 km3) (36). If we use mean values for P. syringae density on nondiseased plants and in water of 100 to 104 cells/cm2 (13) and 1,000 cells/liter (reported here), respectively, then the total planetary population of P. syringae associated with plants would be roughly 1020 to 1022 cells, whereas that in freshwater would be a total of 1020 cells. Disease on plants might have only a small impact on the total plant-associated population if we consider that a hectare of diseased leaves with 108 cells/cm2 of leaf would represent a total of only 1016 P. syringae cells. On the other hand, the size of the population in freshwater might be even greater if we consider that groundwater represents 100 times more volume than surface waters (lakes and rivers) (36, 37), and in many regions groundwater contributes significantly to stream flow and lake volume (38). Glaciers also hold a volume of freshwater 100 times that of surface waters, but it is unknown if they harbor P. syringae populations that fall with snow. These rough calculations suggest that the total planetary population of P. syringae in water is of a size comparable to that on crops.
The considerable effort to sequence the full genomes of numerous strains of P. syringae is motivated by the need to understand the molecular basis of the adaptation of this bacterium to the habitat of plants and its capacity to cause disease (39–42). Intense bioinformatic investigation has been invested in identifying effectors implicated in pathogenesis (43, 44). The fundamental objective of these investigations is to understand the evolutionary history of pathogenicity in P. syringae. The practical objective is to gain insight into targets for methods to control disease. However, as mentioned in the introduction, the epidemiological success of pathogens with significant environmental reservoirs is linked to their capacity to persist in the environment. Furthermore, virulence factors in many human pathogens can have dual uses in both environmental and parasitic fitness, and there are also numerous examples of such dual-use factors in plant pathogens (7). Given the overall abundance and genetic diversity of P. syringae outside its association with diseased plants, it could be argued that pathogenicity per se has a relatively minor role in the fitness of this species and hence in its overall life history. If this is the case, it does not take away from the economic importance of this bacterium as a plant pathogen and the need to control disease. However, this point of view could reorient genomic analyses to be more globally encompassing of the ensemble of mechanisms involved in the epidemiological success of P. syringae.
MATERIALS AND METHODS
Bacterial strains.
P. syringae was isolated from the water samples described in Table 1. All sites except site 22 are at source waters of rivers at altitudes above agriculture or in conservation zones where land was not cultivated. Cultivated fields (pasture) bordered site 22. Up to 1,500 ml of water was collected at each site in sterile plastic bottles that were rinsed once with the site water before the definitive sample was collected. The conductivity of the water at French and U.S. sites was measured on-site with a Consort C561 portable electrochemical analyzer (Consort nv, Turnhout, Belgium), and that of water from New Zealand sites was measured with a Bio-Rad model EG-1 Econo Gradient Monitor (Hercules, CA) in the laboratory. Water was kept in a cooler and transported to the laboratory within 12 h after sampling. To isolate P. syringae, water samples were filtered across nitrocellulose filters (0.22-µm pore diameter), the material collected on the filters was resuspended in filtrate to concentrate the captured bacteria by a factor of 100, and this concentrated suspension was dilution plated (10 replicates per dilution) on KBC medium (45) as described previously (13). One replicate per dilution of the original water sample was also plated on 10% strength tryptic soy agar to estimate the size of the total mesophilic bacterial population as described previously (13). After 4 to 5 days of incubation at 22 to 25°C, fluorescent colonies on KBC medium were randomly collected from a single dilution, purified, and tested for traits compatible with the P. syringae species (absence of cytochrome c oxidase and of the enzyme arginine dihydrolase) (13). According to our previous experience, at least 98% of the strains characterized with these initial tests prove to be P. syringae when subjected to molecular and other biochemical characterizations (13, 15). Strains were stored in nutrient broth containing 40% glycerol at −80°C.
Strain CC0094 of P. syringae from cantaloupe (22) was used as a reference strain for phenotypic tests. For genotypic comparisons, sequences of housekeeping genes (described below) of a wide range of P. syringae strains were collected and are described in the sections concerning phylogenetics and population genetics.
Phenotypic characterization of strains.
Strains were characterized for their pathogenicity and ice nucleation activity. The capacity to induce an HR in tobacco was determined by infiltrating fully developed leaves of plants of Nicotiana tabacum L. cv. Samsun at the 10-leaf stage (bacterial suspensions of 48-h cultures at ca. 1 × 108 CFU/ml). Aggressiveness on cantaloupe plants (Cucumis melo var. cantalupensis Naud. cv. Védrantais) was quantified by injecting 50-µl volumes of bacterial suspensions (prepared in the same way as for hypersensitivity tests) into the junction of the cotyledons of 12 plants at the cotyledon stage (ca. 10 days after sowing). Strains were tested in blocks of up to 30 chosen in random order to distribute the effect of the inoculation date across the different origins of the strains. Strain CC0094 was used as the positive control for all blocks. Plants were incubated for 7 days in a growth chamber at 25°C during the daylight period (16 h) and 18°C at night (8 h). Disease was noted throughout incubation, and disease scores at 7 days were used for analysis. The notation scores were 0 (no symptoms), 1 (one cotyledon wilted or discolored), 2 (both cotyledons wilted or discolored), 3 (both cotyledons completely wilted and upper stem of plant collapsed), and 4 (plant collapsed at base and entire plant fallen on soil surface). Mean absolute aggressiveness and mean aggressiveness relative to that of reference strain CC0094 were calculated for each strain. Ice nucleation activity at −2°C to −8°C was determined for three 30-µl drops of aqueous bacterial suspensions per strain containing a total of 104 and 106 cells per drop as described previously (15). Bacterial suspensions were prepared in the same way as for inoculation of plants and were then kept at 4°C for 1 h before testing. The ice nucleation activity at each concentration and at each temperature was considered positive if at least two of the three drops froze.
Genotyping of strains.
Genotyping was based on sequences of regions of four genes of the core genome, rpoD, gyrB, cts (glt), and gapA (gap1). PCR amplifications were performed as previously described (13), using primers described by Yamamoto et al. (46) for rpoD and gyrB and primers described by Sarkar and Guttman (47) for cts and gapA. For numerous strains, these primers for gapA were not appropriate. Hence, for some strains, the gapA sequence was amplified with primers developed for us by Boris Vinatzer, Virginia Tech, Blacksburg (personal communication). These were gapA230F (5′-GTC AGY GCC ATY CGC AAY C-3′) and gapA942R (5′-AAA ACS CCC AYT CGT TGT C-3′). Amplified products were electrophoresed and purified as described previously (13). Forward and reverse sequencing was conducted by Eurofins MWG Operon (Ebersberg, Germany) and by the Waikato DNA Sequencing Facility (Department of Biological Sciences at the University of Waikato, Hamilton, New Zealand) with the sequencing primers described previously (46, 47).
Phenotypic analyses.
The objective of the phylogenetic analysis was to determine the phylogenetic context of water strains relative to the genetic diversity of P. syringae for the ensemble of habitats from which it has been isolated. We reconstructed phylogenetic relationships for 121 strains of P. syringae, 3 strains of P. fluorescens, and 1 strain of P. viridiflava, including strains used in a previous phylogenetic analysis (13). The present analysis included 44 water strains from this study, as well as strains from crops and all other substrates from which we have isolated this bacterium (see Table S1 in the supplemental material). DNA sequences obtained from the PCR products corresponding to the rpoD, gyrB, cts, and gapA gene fragments were aligned with the ClustalW routine and cut to equal lengths with DAMBE, version 5.1.1 (48). Two approaches were used for the phylogenetic analyses. For the first approach, phylogenetic trees were constructed from the concatenated sequences according to the neighbor-joining method in MEGA, version 3.1 (49), based on the Tamura-Nei model with gamma correction and 1,000 bootstrap replicates as employed by Sarkar and Guttman (47). To choose from among the hundreds of water strains, preliminary trees were constructed based only on cts sequences. We attempted to (i) avoid including multiple strains that were likely to be within the previously described genomic groups, in particular, groups 1 to 5 (47, 50), where we have already demonstrated the presence of strains from water and other substrates (13) and (ii) include strains in new phylogenetic groups. Criteria for choosing strains were based on a preliminary analysis of the variability of each gene sequence within genetic groups 1 to 5. Hence, only a few representatives of water strains that were less than 1.8% divergent in their cts sequence from strains in groups 1 to 5 (and therefore likely to fall into one of these known groups) were included in the phylogenetic tree. For those that were more than 1.8% divergent, the other three housekeeping genes were sequenced and incorporated into the tree. For the second approach, the alignment was analyzed using the DataMonkey Facility, a web server of the HyPhy package (51), to select an appropriate substitution model. Phylogenetic relationships among alleles were then reconstructed using maximum-likelihood methods as implemented in PHYML 3.0 (52), available online (http://www.atgc-montpellier.fr/phyml), using HKY85 as the most appropriate substitution model. All other settings were set to default.
Clustering analyses.
Strains of P. syringae from water and crops were clustered on the basis of their genetic relatedness, in terms of cts sequences, using two different individual-based clustering methods: a Bayesian algorithm and a multivariate analysis. Sequences of 87 strains from crops and 236 strains from water were aligned and cut to a length of 411 bases. Strains from crops were those indicated in Table S1 in the supplemental material under the headings “strains used for phylogenetic analyses” (of crop and agricultural origin) and “strains used for population structure analyses.” Redundant sequences were eliminated, thereby leading to the identification of 88 haplotypes among the total of 323 sequences. The sequences of these haplotypes were converted into the Structure format using xmfa2struct, which was developed by Didelot and Falush and is available at the ClonalFrame website (http://www2.warwick.ac.uk/fac/sci/statistics/staff/research/didelot/clonalframe/).
The Bayesian clustering approach was performed using the software STRUCTURE 2.3 (53, 54). The method assumes that each locus is at Hardy-Weinberg equilibrium and independent of the others, but this Bayesian algorithm has proved to be robust in response to deviations from these assumptions and has been successfully used in partially asexual organisms such as bacteria (55), aphids (56), and fungal and oomycete plant pathogens (57–59). Using the admixture model, we estimated the number of genetic clusters (K) to which the isolates should be assigned to be between k = 1 and k = 10 with 10 repeats. For all simulations, we did not force the model with predefined allele frequencies for source clusters. Runs were based on 200,000 iterations after a burn-in period of 100,000 iterations. We followed the method developed by Evanno et al. (16), which uses the distribution of maximum likelihoods to identify the number of genetically homogeneous clusters (K).
The optimal number of clusters was further evaluated by a PCA using the procedure available in the package adegenet (60) for the statistical freeware R version 2.7.2 (R Foundation for Statistical Computing, 2008). PCA has the advantage over the Bayesian clustering algorithm implemented in STRUCTURE because it does not require assumptions such as the Hardy-Weinberg equilibrium or the absence of linkage disequilibrium between loci (60). PCA was followed by a clustering analysis using the classical Ward method available in R, which is a hierarchical method designed to optimize minimum variance within clusters.
Analysis of population structure.
The genetic structures of the P. syringae populations in water at the different sampling sites were compared based on an FST test (61) using Arlequin software version 3.11 developed by the Population and Molecular Genetics Laboratory at the University of Bern, Bern, Switzerland (http://cmpg.unibe.ch/software/arlequin3/) (62). Calculations of FST were based on the frequency of each of the 88 different 411-base-long haplotypes of the partial cts sequences observed for each of the 13 different dates and sites of sampling (Table 1). AMOVA analyses were conducted to determine the effect of general geographic location (United States, France, New Zealand), of specific location, and of water conductivity (low conductivity, less than 200 µS; medium conductivity, 200 to 500 µS; high conductivity, >500 µS) by assigning each population to the appropriate group. Pairwise comparisons were based on 100 permutations, 100,000 steps in the Markov chain, and 10,000 dememorization steps. The phenotypic structures of P. syringae populations from water were compared via parametric (ANOVA) and nonparametric (χ2) tests using the Statistica software package (StatSoft, Maison-Alfort, France) to determine the effects of geographic location, water chemistry (as indicated above), and genetic cluster.
SUPPLEMENTAL MATERIAL
ACKNOWLEDGMENTS
We thank the American Philosophical Society for a grant to C.E.M. for field work in the United States.
We thank the colleagues who helped with field sampling, including A. Pilgeram and M. Kirkpatrick of Montana State University; C. Eme, Y. Travi, and C. Emblanch of the University of Avignon; A. Buffière at INRA in Avignon; and J. Yu and D. A. Cornish of the New Zealand Institute for Plant and Food Research Limited, Hamilton, New Zealand. We thank B. Moury of INRA for useful scientific discussions that improved this work.
Samples were collected in Grand Teton National Park in accordance with permit GRTE-2007-SCI-0023.
Footnotes
Citation Morris, C. E., D. C. Sands, J. L. Vanneste, J. Montarry, B. Oakley, et al. 2010. Inferring the evolutionary history of the plant pathogen Pseudomonas syringae from its biogeography in headwaters of rivers in North America, Europe, and New Zealand. mBio 1(3):e00107-10. doi:10.1128/mBio.00107-10.
REFERENCES
- 1. Pruzzo C., Vezzulli L., Colwell R. R. 2008. Global impact of Vibrio cholerae interactions with chitin. Environ. Microbiol. 10:1400–1410 [DOI] [PubMed] [Google Scholar]
- 2. Rahman M. H., Biswas K., Hossain M. A., Sack R. B., Mekalanos J. J., Faruque S. M. 2008. Distribution of genes for virulence and ecological fitness among diverse Vibrio cholerae population in a cholera endemic area: tracking the evolution of pathogenic strains. DNA Cell Biol. 27:347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Albert-Weissenberger C., Cazalet C., Buchrieser C. 2007. Legionella pneumophila—a human pathogen that co-evolved with fresh water protozoa. Cell. Mol. Life Sci. 64:432–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oberhardt M. A., Puchałka J., Fryer K. E., Martins dos Santos V. A. P., Papin J. A. 2008. Genome-scale metabolic network analysis of the opportunistic pathogen Pseudomonas aeruginosa PAO1. J. Bacteriol. 190:2790–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Menard A., de los Santos P. E., Graindorge A., Cournoyer B. 2007. Architecture of Burkholderia cepacia complex σ70 gene family: evidence of alternative primary and clade-specific factors, and genomic instability. BMC Genomics 8:308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Casadevall A., Steenbergen J. N., Nosanchuky J. D. 2003. “Ready-made” virulence and “dual use” virulence factors in pathogenic environmental fungi—the Cryptococcus neoformans paradigm. Curr. Opin. Microbiol. 6:332–337 [DOI] [PubMed] [Google Scholar]
- 7. Morris C. E., Bardin M., Kinkel L. L., Moury B., Nicot P. C., Sands D. C. 2009. Expanding the paradigms of plant pathogen life history and evolution of parasitic fitness beyond agricultural boundaries. PLoS Pathog. 5(12):e1000693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Keymer D. P., Miller M. C., Schoolnik G. K., Boehm A. B. 2007. Genomic and phenotypic diversity of coastal Vibrio cholerae strains is linked to environmental factors. Appl. Environ. Microbiol. 73:3705–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khan N. H., Ishii Y., Kimata-Kino N., Esaki H., Nishino T., Nishimura M., Kogure K. 2007. Isolation of Pseudomonas aeruginosa from open ocean and comparison with freshwater, clinical, and animal isolates. Microb. Ecol. 53:173–186 [DOI] [PubMed] [Google Scholar]
- 10. Keymer D. P., Lam L. H., Boehm A. B. 2009. Biogeographic patterns in genomic diversity among a large collection of Vibrio cholerae isolates. Appl. Environ. Microbiol. 75:1658–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khan N. H., Ahsan M., Yoshizawa S., Hosoya S., Yokota A., Kogure K. 2008. Multilocus sequence typing and phylogenetic analyses of Pseudomonas aeruginosa isolates from the ocean. Appl. Environ. Microbiol. 74:6194–6205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cazalet C., Jarraud S., Ghavi-Helm Y., Kunst F., Glaser P., Etienne J., Buchrieser C. 2008. Multigenome analysis identifies a worldwide distributed epidemic Legionella pneumophila clone that emerged within a highly diverse species. Genome Res. 18:431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morris C. E., Sands D. C., Vinatzer B. A., Glaux C., Guilbaud C., Buffière A., Yan S., Dominguez H., Thompson B. M. 2008. The life history of the plant pathogen Pseudomonas syringae is linked to the water cycle. ISME J. 2:321–334 [DOI] [PubMed] [Google Scholar]
- 14. He S. Y., Nomura K., Whittam T. S. 2004. Type III protein secretion mechanism in mammalian and plant pathogens. Biochim. Biophys. Acta 1694:181–206 [DOI] [PubMed] [Google Scholar]
- 15. Morris C. E., Kinkel L. L., Kun X., Prior P., Sands D. C. 2007. A surprising niche for the plant pathogen Pseudomonas syringae. Infect. Genet. Evol. 7:84–92 [DOI] [PubMed] [Google Scholar]
- 16. Evanno G., Regnaut S., Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14:2611–2620 [DOI] [PubMed] [Google Scholar]
- 17. Gross D. C., Cody Y. S., Proebsting E. L., Jr., Radamaker G. K., Spotts R. A. 1984. Ecotypes and pathogenicity of ice-nucleation-active Pseudomonas syringae isolated from deciduous fruit tree orchards. Phytopathology 74:241–248 [Google Scholar]
- 18. Lindemann J., Arny D. C., Upper C. D. 1984. Epiphytic populations of Pseudomonas syringae pv. syringae on snap bean and nonhost plants and the incidence of bacterial brown spot disease in relation to cropping patterns. Phytopathology 74:1329–1333 [Google Scholar]
- 19. Little E. L., Bostock R. M., Kirkpatrick B. C. 1998. Genetic characterization of Pseudomonas syringae pv. syringae strains from stone fruits in California. Appl. Environ. Microbiol. 64:3818–3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saad S. M., Hagedorn D. J. 1972. Relationship of isolate source to virulence of Pseudomonas syringae on Phaseolus vulgaris. Phytopathology 62:678–680 [Google Scholar]
- 21. Jones J. A., Mulholland P. 2000. Streams and ground waters. Academic Press, Orlando, FL [Google Scholar]
- 22. Morris C. E., Glaux C., Latour X., Gardan L., Samson R., Pitrat M. 2000. The relationship of host range, physiology, and genotype to virulence on cantaloupe in Pseudomonas syringae from cantaloupe blight epidemics in France. Phytopathology 90:636–646 [DOI] [PubMed] [Google Scholar]
- 23. Jenner C. E., Wang X., Ponz F., Walsh J. A. 2002. A fitness cost for Turnip mosaic virus to overcome host resistance. Virus Res. 86:1–6 [DOI] [PubMed] [Google Scholar]
- 24. Janzac B., Montarry J., Palloix A., Navaud O., Moury B. 2010. A point mutation in the polymerase of Potato virus Y confers virulence towards the Pvr4 resistance of pepper and a high competitiveness cost in susceptible cultivar. Mol. Plant Microbe Interact. 23:823–830 [DOI] [PubMed] [Google Scholar]
- 25. Huang Y. J., Li Z. Q., Evans N., Rouxel T., Fitt B. D. L., Balesdent M. H. 2006. Fitness cost associated with loss of the AvrLm4 avirulence function in Leptosphaeria maculans (phoma stem canker of oilseed rape). Eur. J. Plant Pathol. 114:77–89 [Google Scholar]
- 26. Thrall P. H., Burdon J. J. 2003. Evolution of virulence in a plant host-pathogen metapopulation. Science 299:1735–1737 [DOI] [PubMed] [Google Scholar]
- 27. Bai J., Choi S. H., Ponciano G., Leung H., Leach J. E. 2000. Xanthomonas oryzae pv. oryzae avirulence genes contribute differently and specifically to pathogen aggressiveness. Mol. Plant Microbe Interact. 13:1322–1329 [DOI] [PubMed] [Google Scholar]
- 28. Sacristán S., García-Arenal F. 2008. The evolution of virulence and pathogenicity in plant pathogen populations. Mol. Plant Pathol. 9:369–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Green S., Studholme D. J., Laue B. E., Dorati F., Lovell H., Arnold D., Cottrel J. E., Bridgett S., Blaxter M., Hultema E., Thwaites R., Sharp P. M., Jackson R. W., Kamoun S. 2010. Comparative genome analysis provides insights into the evolution and adaptation of Pseudomonas syringae pv. aesculi on Aesculus hippocastanum. PLoS One 4:e10224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buttner M. P., Amy P. S. 1989. Survival of ice nucleation-active and genetically engineered non-ice-nucleating Pseudomonas syringae strains after freezing. Appl. Environ. Microbiol. 55:1690–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sands D. C., Schroth M. N., Hildebrand D. C. 1970. Taxonomy of phytopathogenic pseudomonads. J. Bacteriol. 101:9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wolber P. K. 1993. Bacterial ice nucleation. Adv. Microb. Physiol. 34:203–237 [DOI] [PubMed] [Google Scholar]
- 33. Anzai Y., Kim H., Park J. Y., Wakabayashi H., Oyaizu H. 2000. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int. J. Syst. Evol. Microbiol. 50:1563–1589 [DOI] [PubMed] [Google Scholar]
- 34. Morris C. E., Kinkel L. L. 2002. Fifty years of phyllosphere microbiology: significant contributions to research in related fields, p. 353–363. In Lindow S. E., Hecht-Poinar E. I., Elliot V. (eds.), Phyllosphere microbiology. APS Press, Minneapolis, MN. [Google Scholar]
- 35. Whittaker R. H., Likens C. E. 1975. The biosphere and man, p. 305–328. In Lieth H., Whittaker R. H. (eds.), Primary productivity of the biosphere, ecological studies, vol. 14 Springer, Berlin, Germany [Google Scholar]
- 36. Oki T., Kanae S. 2006. Global hydrological cycles and the world water resources. Science 313:1068–1072 [DOI] [PubMed] [Google Scholar]
- 37. Gleik P. H. 2000. The world’s water 2000-2001: the biennial report on freshwater resources. Island Press, Washington, DC [Google Scholar]
- 38. Bailly-Comte V., Jourde H., Pistre S. 2009. Conceptualization and classification of groundwater–surface water hydrodynamic interactions in karst watersheds: case of the karst watershed of the Coulazou River (southern France). J. Hydrol. 376:456–462 [Google Scholar]
- 39. Almeida N. F., Yan S., Lindeberg M., Studholme D. J., Schneider D. J., Condon B., Liu H., Viana C. J., Warren A., Evans C., Kemen E., Maclean D., Angot A., Martin G. B., Jones J. D., Collmer A., Setubal J. C., Vinatzer B. A. 2009. A draft genome sequence of Pseudomonas syringae pv. tomato T1 reveals a type III effector repertoire significantly divergent from that of Pseudomonas syringae pv. tomato DC3000. Mol. Plant Microbe Interact. 22:52–62 [DOI] [PubMed] [Google Scholar]
- 40. Buell C. R., JoarBuell C. R., Joardar V., Lindeberg M., Selengut J., Paulsen I. T., Gwinn M. L., Dodson R. J., Deboy R. T., Durkin A. S., Kolonay J. F., Madupu R., Daugherty S., Brinkac L., Beanan M. J., Haft D. H., Nelson W. C., Davidsen T., Zafar N., Zhou L., Liu J., Yuan Q., Khouri H., Fedorova N., Tran B., Russell D., Berry K., Utterback T., Van Aken S. E., Feldblyum T. V., D’Ascenzo M., Deng W. L., Ramos A. R., Alfano J. R., Cartinhour S., Chatterjee A. K., Delaney T. P., Lazarowitz S. G., Martin G. B., Schneider D. J., Tang X., Bender C. L., White O., Fraser C. M., Collmer A. 2003. The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc. Natl. Acad. Sci. U. S. A. 100:10181–10186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feil H., Feil W., Chain P., Marimer F., DiBartolo G., Copeland A., Lykidis A., Trong S., Nolan M., Goltsman E., Thiel J., Malfatti S., Loper J. E., Lapidus A., Detter J. C., Land M., Richardson P. M., Kyrpides N. C., Ivanova N., Lindow S. E. 2005. Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000. Proc. Natl. Acad. Sci. U. S. A. 102:11064–11069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Joardar V., Lindeberg M., Jackson R. W., Selengut J., Dodson R., Brinkac L. M., Daugherty S. C., DeBoy R., Durkin S., Giglio M. G., Madupu R., Nelson W. C., Rosovitz M. J., Sullivan S., Crabtree J., Creasy T., Davidsen T., Haft D. H., Zafar N., Zhou L. W., Halpin R., Holley T., Khouri H., Feldblyum T., White O., Fraser C. M., Chatterjee A. K., Cartinjour S., Schneider D. J., Mansfield J., Collmer A., Buell C. R. 2005. Whole-genome sequence analysis of Pseudomonas syringae pv. phaseolicola 1448A reveals divergence among pathovars in genes involved in virulence and transposition. J. Bacteriol. 187:6488–6498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lindeberg M., Cartinhour S., Myers C. R., Schechter L. M., Schneider D. J., Collmer A. 2006. Closing the circle on the discovery of genes encoding Hrp regulon members and type III secretion system effectors in the genomes of three model Pseudomonas syringae strains. Mol. Plant Microbe Interact. 19:1151–1158 [DOI] [PubMed] [Google Scholar]
- 44. Lindeberg M., Myers C. R., Collmer A., Schneider D. J. 2008. Roadmap to new virulence determinants in Pseudomonas syringae: insights from comparative genomics and genome organization. Mol. Plant Microbe Interact. 21:685–700 [DOI] [PubMed] [Google Scholar]
- 45. Mohan S. K., Schaad N. W. 1987. An improved agar plating assay for detecting Pseudomonas syringae pv. syringae and P. s. pv. phaseolicola in contaminated bean seed. Phytopathology 77:1390–1395 [Google Scholar]
- 46. Yamamoto S., Kaasai H., Arnold D. L., Jackson R. W., Vivian A., Harayama S. 2000. Phylogeny of the genus Pseudomonas: intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology 146:2385–2394 [DOI] [PubMed] [Google Scholar]
- 47. Sarkar S. F., Guttman D. S. 2004. Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen. Appl. Environ. Microbiol. 70:1999–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xia X., Xie Z. 2001. DAMBE: data analysis in molecular biology and evolution. J. Hered. 92:371–373 [DOI] [PubMed] [Google Scholar]
- 49. Kumar S., Tamura K., Nei M. 1994. MEGA—molecular evolutionary genetics analysis software for microcomputers. Comput. Appl. Biosci. 10:189–191 [DOI] [PubMed] [Google Scholar]
- 50. Hwang M. S. H., Morgan R. L., Sarkar S. F., Wang P. W., Guttman D. S. 2005. Phylogenetic characterization of virulence and resistance phenotypes of Pseudomonas syringae. Appl. Environ. Microbiol. 71:5182–5191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pond K., Frost S. D. W. 2005. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533 [DOI] [PubMed] [Google Scholar]
- 52. Guindon S., Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696–704 [DOI] [PubMed] [Google Scholar]
- 53. Falush D., Stephens M., Pritchard J. K. 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pritchard J. K., Stephens M., Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155:945–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Falush D., Wirth T., Linz B., Pritchard J. K., Stephens M., Kidd M., Blaser M. J., Graham D. Y., Vacher S., Perez-Perez G. I., Yamaoka Y., Mégraud F., Otto K., Reichard U., Katzowitsch E., Wang X., Achtman M., Suerbaum S. 2003. Traces of human migrations in Helicobacter pylori populations. Science 299:1582–1585 [DOI] [PubMed] [Google Scholar]
- 56. Halkett F., Plantegenest M., Prunier-Leterme N., Mieuzet L., Delmotte F., Simon J. C. 2005. Admixed sexual and facultatively asexual aphid lineages at mating sites. Mol. Ecol. 14:325–336 [DOI] [PubMed] [Google Scholar]
- 57. Delmotte F., Giresse X., Richard-Cervera S., M’Bayaa J., Vearb F., Tourvieilleb J., Walserb P., Tourvieille de Labrouheb D. 2008. Single nucleotide polymorphisms reveal multiple introductions into France of Plasmopara halstedii, the plant pathogen causing sunflower downy mildew. Infect. Genet. Evol. 8:534–540 [DOI] [PubMed] [Google Scholar]
- 58. Fournier E., Giraud T. 2008. Sympatric genetic differentiation of populations of a generalist pathogenic fungus, Botrytis cinerea, on two different host plants, grapevine and bramble. J. Evol. Biol. 21:122–132 [DOI] [PubMed] [Google Scholar]
- 59. Gladieux P., Zhang X. G., Afoufa-Bastien D., Valdebenito Sanhueza R. M., Sbaghi M., Le Cam B. 2008. On the origin and spread of the scab disease of apple: out of central Asia. PLoS One 1:e1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jombart T. 2008. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405 [DOI] [PubMed] [Google Scholar]
- 61. Hartl D. L., Clark A. G. 2007. Principles of population genetics, 4th ed Sinauer Associates, Inc., Sunderland, MA [Google Scholar]
- 62. Excoffier L., Laval G., Schneider S. 2005. Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol. Bioinform. Online 1:47–50 [PMC free article] [PubMed] [Google Scholar]
- 63. Sawada H., Suzuki F., Matsuda I., Saitou N. 1999. Phylogenetic analysis of Pseudomonas syringae pathovars suggests the horizontal gene transfer of argK and the evolutionary stability of hrp gene cluster. J. Mol. Evol. 49:627–644 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.