

Abstract
The protein transduction domain of HIV-1 TAT, TAT(48-60), is an efficient cell-penetrating peptide (CPP) that diffuses across the lipid membranes of cells despite eight cationic Arg and Lys residues. To understand its mechanism of membrane translocation against the free energy barrier, we have conducted solid-state NMR experiments to determine the site-specific conformation, dynamics, and lipid interaction of the TAT peptide in anionic lipid bilayers. We found that TAT(48-60) is a highly dynamic and nearly random-coil peptide in the lipid bilayer, and inserts into the membrane-water interface near the glycerol backbone region. Arg-phosphate salt bridge interaction was revealed by short guanidinium-phosphate distances and restricted dynamics of the guanidinium. Together with the observation of strong peptide-water cross peaks in 1H spin diffusion spectra, these results indicate that TAT binding to the membrane-water interface is stabilized not only by electrostatic attraction to the anionic lipids, but also by intermolecular hydrogen bonding with the lipid phosphates and water, which may take the role of intramolecular hydrogen bonds in canonical secondary structures. The random-coil structure of TAT and another CPP, penetratin, suggests that the lack of amphipathic structure is essential for rapid translocation of these Arg-rich CPPs across the lipid membrane without causing permanent damages to the membrane integrity.
Keywords: Cell-penetrating peptide, solid-state NMR, guanidinium-phosphate interaction, conformational dynamics, random coil
Cell-penetrating peptides (CPPs) are highly cationic and Arg-rich peptides that are able to cross lipid membranes into cells both alone and in conjugation with large macromolecular cargos (1, 2). Therefore, CPPs are potentially important molecules for drug delivery and for studying macromolecular structures inside living cells (3). CPPs have been discovered from diverse origins such as the TAT protein of HIV-1 (4), penetratin from the Drosophilia Antennapedia homeodomain (5), and synthetic polyarginine peptides (6, 7). In contrast to antimicrobial peptides (AMPs), which are similarly Arg-rich sequences but which kill bacterial cells by disrupting their lipid membranes, CPPs appear to enter eukaryotic cells without causing long-lasting damage to the integrity of the cell membrane.
The physical basis for the membrane translocation of CPPs has been much debated and still not well understood (8). It is well known that the low-dielectric interior of cell membranes presents a high-energy barrier to the direct unassisted diffusion of charged ions and molecules. The free energies of transfer of amino acid residues from the aqueous to a nonpolar solvent have been measured experimentally and are highly positive for Arg and Lys (9). It is thus puzzling how short peptides containing a high density of Arg residues are able to cross the membrane. Two models, an inverse micelle model (10) and an electroporation model (11), had been proposed to account for membrane translocation of CPPs, but neither was supported by recent solid-state NMR studies of penetratin. Specifically, 31P NMR lineshapes indicated the absence of isotropic entities in the membrane, thus ruling out the inverse micelle model (12). Paramagnetic relaxation enhancement experiments using Mn2+ ions bound to the outer surface of lipid bilayers versus both surfaces indicated that penetratin was distributed equally in the two leaflets of the membrane even at low peptide concentrations (12). This finding contradicted the electroporation model, which posits that asymmetric association of CPPs with the outer leaflet of the membrane causes an electric field that alters the lateral and curvature stress of the membrane, eventually causing electroporation-like perforation of the membrane (11). Instead, site-specific distances between penetratin and the lipid headgroups indicated tight guanidinium-phosphate associations (13), suggesting that ion pair interaction between the Arg residues and the anionic lipid headgroups may play a significant role in the membrane translocation of CPPs.
Despite many biophysical studies of CPPs in membrane-mimetic environments, little site-specific high-resolution structural information of membrane-bound CPPs is yet available. Our recent studies of membrane-bound penetratin through solid-state NMR 13C chemical shifts suggested an unusual turn-rich conformation at physiological temperature (14), whose functional role is still unclear. Moreover, the penetratin conformation was found to depend on the temperature: in the gel phase of the membrane, penetratin adopts a β-strand conformation, while in the liquid-crystalline phase random-coil chemical shifts were observed for most labeled sites except for an Arg (14). To further elucidate how conformation underlies the mechanism of CPPs, and to examine the diversity of the structure-function relation for this class of peptides, we have now investigated the structure and dynamics of the hexa-Arg CPP domain of the HIV TAT protein (residues 48-60), whose potent CPP activities have been extensively characterized (15-17). Using solid-state NMR, we have determined the conformation, dynamics, depth of insertion, and lipid interaction of TAT(48-60) in anionic lipid membranes, which led us to propose a structural basis for the translocation mechanism of this peptide.
Materials and Methods
Membrane sample preparation
TAT(48-60) (GRKKR RQRRR PPQ-CONH2) was synthesized using standard solid phase Fmoc chemistry and purified by HPLC to >95% purity. Uniformly 13C, 15N-labeled Lys, Gln, Arg, and Pro were incorporated at residues 4, 7, 8, and 11 of the peptide, respectively, in singly labeled samples. A 13C, 15N-Ile3 labeled penetratin sample (RQIKI WFQNR RMKW KK-CONH2) bound to DMPC/DMPG bilayers (13) was also used for the 1H T1ρ experiment to compare with the TAT peptide. All lipids, including DMPC, DMPG, POPC, POPE and POPG, were purchased from Avanti Polar Lipids (Alabaster, AL). Most experiments were conducted on hydrated DMPC/DMPG (8:7 mole ratio) membranes. The high-melting DMPC/DMPG mixture allows the peptide to be immobilized at moderate low temperatures to facilitate distance and chemical shift measurements. The low-melting unsaturated lipids (POPC, POPE and POPG) were used in aligned membrane samples since they are more easily hydrated and aligned at ambient temperature. We chose phosphatidylglycerol (PG) instead of phosphatidylserine (PS), which is the anionic lipid of eukaryotic cell membranes, for our samples in order to facilitate comparison with the large literature of CPP studies in model membranes. Eukaryotic cell membranes contain ~10% anionic lipids (18), which is much less than the ~50% charge density used in this work. However, the cationic TAT is most likely clustered in the anionic-lipid-rich regions of the biological membrane during translocation, so the current model membrane is still relevant for understanding the translocation mechanism.
Hydrated membrane samples were prepared by aqueous phase mixing. DMPC and DMPG (8 : 7 mole ratio) lipids were mixed in chloroform and dried under a stream of N2 gas. The lipid mixture was further lyophilized overnight to obtain a homogeneous dry powder, which was then suspended in 2 mL phosphate buffer (5.8 mM NaH2PO4 and 4.2 mM Na2HPO4, pH = 7.0) and freeze-thawed 8 times. The resulting lipid vesicle solution was added to the peptide solution to obtain a peptide : lipid molar ratio of 1 : 15. After incubation overnight, the solution was centrifuged at 55,000 rpm for 4 hrs at 4-6 °C to obtain a wet pellet. The pellet was slowly dried to a hydration level of ~40 wt%, then packed into a 4 mm MAS rotor for NMR experiments. Over 95% of the peptide was bound to the lipids as checked by UV-VIS spectrometry. The POPE/POPG bound TAT sample was prepared similarly.
Oriented membranes were prepared on thin glass plates using an organic-solvent protocol described before (19). The dry lipid/peptide film (~1 mg) on each glass plate was first hydrated by directly dropping 1 μl water on each plate, giving a hydration level of ~50%. The hydrated glass plates were then kept in a 97% humidity chamber containing saturated K2SO4 solution at room temperature for 4-5 days before NMR measurements. Samples with TAT concentrations at 1%, 2%, 4% and 8% were prepared to determine the degree of membrane disorder induced by the peptide. Three series of lipid membranes, neutral POPC, anionic POPC/POPG (8:7), and anionic POPE/POPG (8:7), were prepared.
Solid-state NMR experiments
All solid-state NMR experiments were carried out on a Bruker DSX-400 (9.4 T) spectrometer (Karlsruhe, Germany) operating at 400.5 MHz for 1H, 100.7 MHz for 13C and 162.1 MHz for 31P. A triple-resonance 1H/13C/31P 4 mm magic-angle spinning (MAS) probe was used for 13C-31P REDOR experiments, while a double-resonance 1H/X MAS probe was used for other MAS experiments. Low temperature was achieved using a Kinetics Thermal System XR air-jet sample cooler (Stone Ridge, NY). All experimental temperatures refer to the sample temperature indicated by the probe thermocouple, and are estimated to be within 1° of the actual sample temperature since no fast spinning nor high-salt samples were used in this work. Typical 90° pulse lengths of 13C, 15N and 31P were 5 μs and typical 1H decoupling field strengths were 75 kHz at low temperature and 50 kHz at ambient temperature. 13C and 15N chemical shifts were calibrated, respectively, to the α-Gly 13C’ resonance at 176.49 ppm on the TMS scale and 15N-acetyl-valine (NAV) at 122 ppm on the ammonium scale. 31P chemical shifts were referenced to the hydroxyapatite signal at 2.73 ppm for unoriented samples and to the signal of 98% phosphoric acid at 0 ppm for oriented samples.
13C and 15N cross-polarization (CP) MAS experiments were conducted using CP contact times of 0.5 – 1.5 ms. One-dimensional double-quantum (DQ) filtered 13C MAS spectra were measured at 233 K using SPC-5 for 13C-13C dipolar recoupling (20). When extracting full widths at half maximum, we took care to use appropriate line broadening that is less than the intrinsic linewidths of the signals at the specific temperatures.
Two 2D 13C-13C correlation experiments, dipolar-assisted rotational resonance (DARR) (21) and the DQ dipolar INADEQUATE experiment (22), were used to assign the TAT 13C chemical shifts at low temperature. The INADEQUATE experiment suppresses the natural abundance lipid 13C signals and gives relatively well-resolved peptide 13C resonances. The SPC-5 sequence was used for DQ excitation and reconversion. The INADEQUATE spectra were measured at 233 K under 5333 Hz MAS.
2D 13C-1H heteronuclear correlation (HETCOR) experiments without 1H spin diffusion were used to assign 1H and 13C chemical shifts of TAT at ambient temperatures. No 1H decoupling was applied during the evolution period, thus only signals of highly dynamic species can survive in the spectra. The experiments were conducted either with or without 1JCH decoupling for the t1 period, by the presence or absence of a 13C 180° pulse in the middle of the t1 period. The pulse sequence of 1JCH-decoupled 2D HETCOR experiment was shown in Fig. 2a. The non-1JCH decoupled HECTOR was conducted at 5 kHz MAS while the 1JCH decoupled HETCOR experiments were carried out under 7-8 kHz MAS at 303 K, in the liquid-crystalline phase of the DMPC/DMPG membrane. For 1H spin diffusion experiments probing the interaction of TAT with lipids and water, we used a similar HETCOR sequence but with the addition of a 1H spin diffusion mixing period after t1 (23). These experiments were carried out under 5 kHz MAS at 303 K.
Fig. 2.
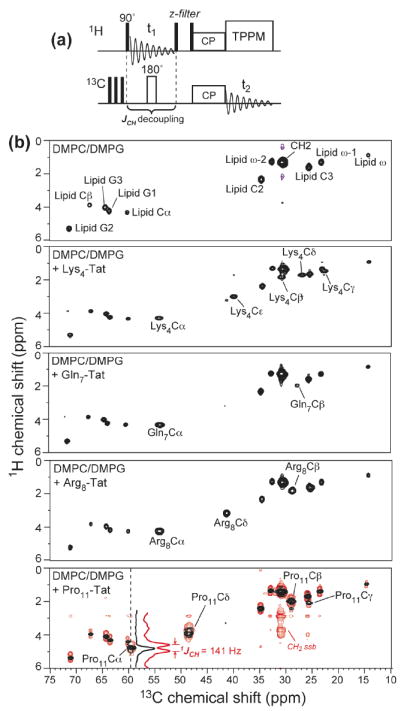
2D 1H-13C HETCOR spectra of TAT in DMPC/DMPG bilayers at 303 K. (a) Pulse sequence of 1JCH-decoupled 2D HETCOR experiment. No 1H homonuclear decoupling was applied during t1, thus only highly dynamic molecules can exhibit 1H-13C cross peaks. (b) 1JCH-decoupled HETCOR spectra (black) of the DMPC/DMPG membrane without and with various site-specifically labeled TAT. The spectrum in red in the bottom panel is the HETCOR spectrum of Pro11-TAT without 1JCH decoupling. A 141 Hz 1JCH splitting of Pro11Cα peak was observed. The Cα 1H cross section was compared between the 1JCH-decoupled and undecoupled spectra.
13C-1H and 15N-1H order parameters were measured using the 2D dipolar-chemical-shift correlation (DIPSHIFT) experiment with dipolar doubling (24, 25). The experiments were conducted at 303 K under slow spinning rates of 2800 Hz and 3401 Hz. 1H homonuclear decoupling was achieved using semi-windowless MREV-8 with a 1H 105° pulse length of 4.0 μs (26). Small asymmetry in the t1 curves was fit using an apparent T2 relaxation factor. The best-fit couplings were divided by the theoretical scaling factor, 0.47, of semi-windowless MREV-8 and a factor of 2 for doubling to give the true X-H dipolar couplings. The order parameters, SXH, were calculated as the ratio of the true coupling with the rigid-limit one-bond X-H dipolar coupling. The rigid-limit value was taken as 22.7 kHz for C-H and 10.6 kHz for N-H dipolar coupling, respectively.
1H rotating-frame spin-lattice relaxation times (T1ρ ), which reflect microsecond-timescale motions that are important for biological membranes, were measured using a 13C-detected 1H Lee-Goldburg spin-lock sequence with an effective 1H field strength of 61.2 kHz (27). The experiments were conducted at 303 K under 7 kHz MAS.
13C-31P rotational-echo double-resonance (REDOR) experiments were carried out under 4 kHz MAS at 230 K, where both the lipids and the peptide were immobilized. A 31P 180° pulse length of 9 μs was used to achieve complete inversion of the broad 31P spectral width. A frequency-selective version of REDOR (28) was used to measure the distance of Cα to 31P. A 1 ms 13C Gaussian 180° pulse centered at the 13C resonance of interest was used to remove the J-coupling between the on-resonance Cα and its directly bonded 13C spins. Two-spin simulations were used to fit the 13C-31P REDOR data. Although multiple 31P spins are present on the membrane surface, the average 31P density on the membrane surface (10 Å spacing between 31P atoms) is sufficiently low that when the REDOR dephasing is fast, corresponding to two-spin 13C-31P distances of ~5 Å or less, two-spin simulations report the correct distance of the 13C to a single 31P (29). When the dephasing is slow, comparison of multi-spin and two-spin simulations indicated that the two-spin distance is effectively the vertical distance between the 13C and the 31P plane, thus reporting the depth of the 13C spin (29).
Due to overlap with the lipid 13CO signals, the 13CO-31P REDOR data were corrected for the natural abundance lipid background (13) using the equation (S/S0)observed = 0.79(S/S0)peptide + 0.21(S/S0)lipid, where the weight fractions were calculated based on the peptide/lipid molar ratio of 1 : 15. The lipid 13CO-31P REDOR dephasing was previously measured using POPC lipids (29). The Pro11 Cα REDOR was similarly corrected for the small percentage of overlapping natural abundance lipid Cα signal, whose distance to 31P is fixed at 2.9 Å. To fit the Arg Cζ REDOR data, which shows heterogeneous couplings, we used two distances in the simulation, where the long and short distances were each incremented at 0.1 Å steps within physically allowed ranges (13). The best fit was obtained as the lowest RMSD between the data and simulations. Table S1 summarizes the main conditions of all the experiments carried out in this work.
Results
Random coil conformation and high mobility of TAT in lipid bilayers
We first examined the conformation of TAT in DMPC/DMPG membranes using 13C and 15N chemical shifts. Four uniformly 13C, 15N-labeled residues, Lys4, Gln7, Arg8 and Pro11, were singly incorporated into the peptide. Fig. 1 shows representative 2D 13C-13C correlation spectra of Lys4- and Arg8-labeled TAT in the gel phase of the membrane at 233 K. Broad Cα and Cβ peaks with linewidths of 4.0 – 5.1 ppm were observed, indicating significant conformational heterogeneity. When the lipid 13C signals were removed by a double-quantum (DQ) filter, more than one set of 13C chemical shifts was found for some of the 13C sites in Lys4, Gln7, and Arg8 (Fig. 1b), suggesting that the conformational distribution may be more complex than a single Gaussian. Chemical shift multiplicity for the same domain in the intact TAT protein was also observed in solution, and was postulated to result from transient folding events of the protein (30). When the chemical shifts of the dominant peaks of membrane-bound TAT were compared with random coil values from protein databases (31), we found most residues to exist in a random-coil state (Fig. 1c). The only exception is Pro11, which is known to be predisposed to β-strand conformation by the imine sidechain.
Fig. 1.
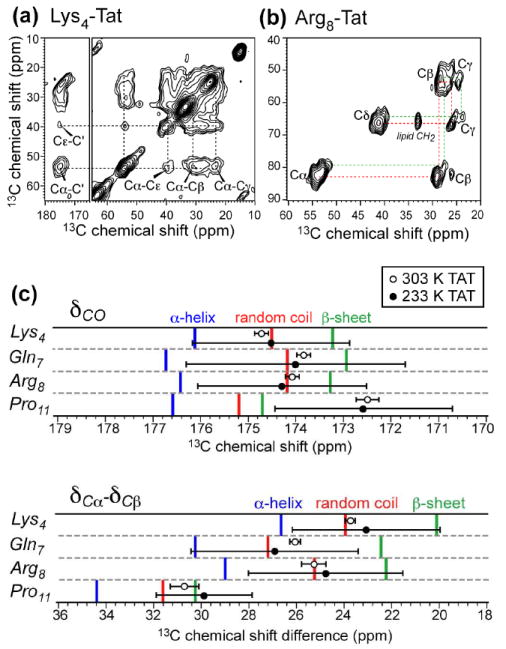
13C chemical shifts of HIV TAT(48-60) in DMPC/DMPG (8:7) bilayers. (a) 2D 13C-13C DARR spectrum of Lys4-TAT at 233 K, measured with a mixing time of 30 ms. (b) 2D INADEQUATE spectrum of Arg8-TAT at 233 K. (c) TAT 13C chemical shifts at 303 K (open circles) and 233 K (closed circles) compared to database values for various secondary structures (31). The Cα-Cβ chemical shift difference was used as a marker of secondary structure (58).
To examine if the random coil chemical shifts of TAT at low temperature were caused by the gel-phase disorder of the membrane, we measured the TAT 13C chemical shifts in the liquid-crystalline phase of the membrane at 303 K using 2D 1H-13C correlation experiments. Even in the absence of 1H-1H homonuclear decoupling, we observed extremely narrow 1H and 13C linewidths and resolved 1JCH splittings in the 1H dimension (Fig. 2b): the 13C linewidths were 0.3-0.4 ppm while the undecoupled 1H linewidths were 0.15-0.30 ppm. Thus, fast isotropic motion largely averaged the 1H-1H dipolar couplings, giving well-resolved 1H peaks even without homonuclear decoupling. As a comparison, in the HETCOR spectrum (data not shown) of Ile3-labeled penetratin with 1JCH-decoupling, no peptide 1H peaks survived in the spectra, indicating penetratin has slower motion than TAT. Most importantly, the TAT 13C chemical shifts remained at the random coil positions, close to the center of the broad peaks at low temperature (Fig. 1c). Thus, TAT undergoes near-isotropic motion at high temperature, with an average conformation approaching the random coil. This result is consistent with circular dichroism data of TAT in POPC/POPG vesicles, which also suggested a random-coil structure similar to that in buffer (32).
To observe how the narrow linewidths of TAT at high temperature transition to broad linewidths at low temperature, we measured the 1D 13C and 15N spectra of TAT as a function of temperature. Fig. 3a, b shows 13C and 15N spectra of Arg8-labeled TAT bound to DMPC/DMPG bilayers from 303 K to 233 K. The Arg8 signals were sharp at 303 K: the limiting 13C linewidths were sufficiently small that 13C-13C scalar-splittings of ~40 Hz were observed for many sites such as Cα. The peptide signals became undetectable around 283 K, and became broad and strong at 233 K, with 3.5 – 4.8 ppm linewidths for 13C and a 12 ppm linewidth for Nα. Similar trends were observed for the other labeled residues. For the backbone Cα, the low-temperature linewidth of Arg8 covers the entire range of helix to sheet conformations (Fig. 3c). We further compared the TAT linewidths to several other membrane peptides, including penetratin, the β-sheet antimicrobial peptide PG-1 (29), and the α-helical influenza M2 transmembrane peptide (33). Among these peptides, TAT exhibits the narrowest linewidths at high temperature and the largest linewidths at low temperature (Fig. 3d), indicating that TAT undergoes faster exchange among a wider range of conformations at high temperature, thus giving rise to sharp signals averaged at the random coil chemical shifts. Lowering the temperature freezes the motion and captures all conformation-dependent chemical shifts. Thus, the broad low-temperature linewidths reflect the large conformational space sampled by TAT. While it is difficult to quantify the exact conformational distribution, compared to the structurally defined β-sheet PG-1 and α-helical M2, the TAT structure is almost completely random. Comparison with PG-1 and M2 also indicates that the contribution of gel-phase membrane disorder is minor compared the intrinsic disorder of the peptide. Furthermore, the possibility that freezing changed the TAT conformation can be ruled out, since the average chemical shift frequencies were unchanged between high and low temperatures (with the slight exception of Pro11 Cα).
Fig. 3.
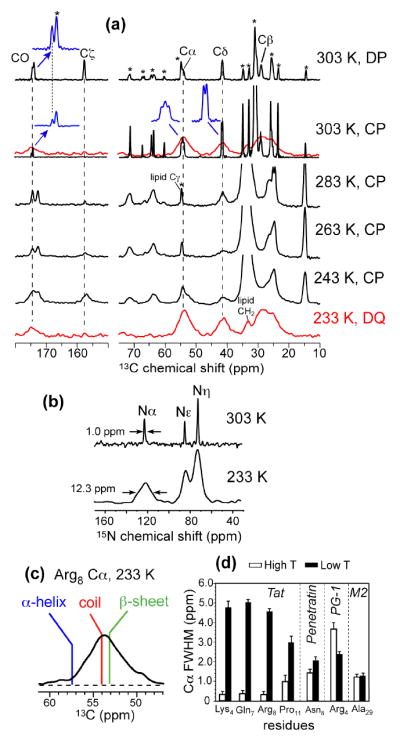
Temperature-dependent TAT linewidths in DMPC/DMPG membranes. (a) 13C MAS spectra from 303 K to 233 K for Arg8-TAT. Stars denote lipid peaks. Most spectra were measured by cross polarization (CP). A 303 K spectrum was measured by 13C direct polarization (DP). A 233 K 13C DQ-filtered spectrum gives pure peptide peaks and linewidths. 13C-13C J splittings were resolved for some peaks at 303 K, as shown in expanded regions in blue. (b) 15N spectra of Arg8-TAT at 303 K (by DP) and 233 K (by CP). (c) The Cα peak of Arg8 at 233 K from the DQ spectrum spans the chemical shift range for canonical secondary structures. (d) Comparison of Cα linewidths of TAT, penetratin, PG-1, and the influenza M2 peptide in lipid bilayers at high and low temperatures.
To quantify the dynamics of TAT in the lipid membrane, we measured 13C-1H and 15N-1H dipolar couplings and the corresponding order parameters at 303 K using 2D DIPSHIFT experiments. Order parameters indicate the amplitude of motion. A maximum order parameter of 1 corresponds to the rigid limit while a minimum order parameter of 0 indicates large-amplitude, isotropic motion. Fig. 4 shows representative dipolar coupling data of the membrane-bound TAT. Low order parameters of 0.14-0.20 were found for the peptide backbone (Table 2), comparable to the order parameters of the middle portion of the lipid molecules (34), indicating that the peptide undergoes large-amplitude motion. Interestingly, the Arg8 order parameters decrease from Nα (SNH = 0.20) to Cδ (SCH = 0.08) but then reverse the trend and increase to Nη (SNH = 0.30), indicating that two relatively rigid ends of the residue flank a more mobile aliphatic middle (Fig. 4c). The larger order parameters of the guanidinium moiety immediately suggest stabilizing interactions with the lipid headgroups. TAT also exhibits long 1H T1ρ relaxation times of 10 – 30 ms (Table 3), indicating that the large-amplitude motions occur at rates far exceeding 106 s−1.
Fig. 4.
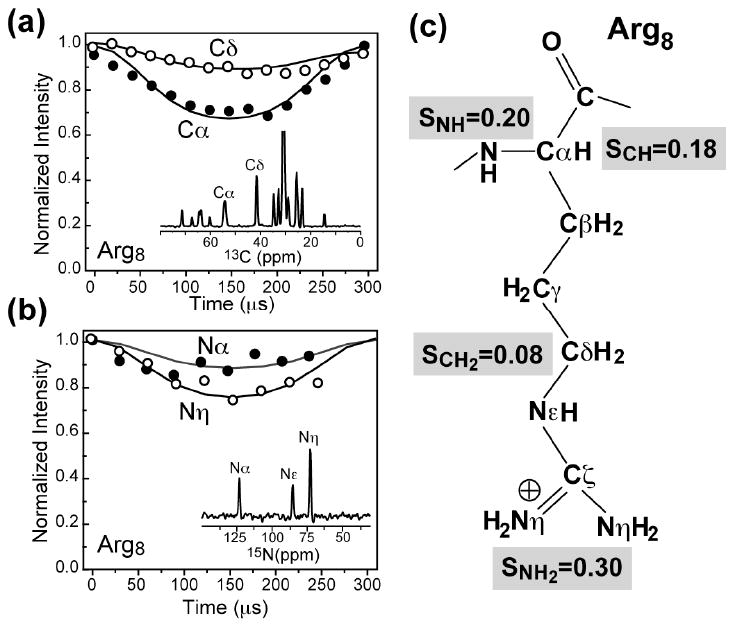
C-H and N-H dipolar couplings of Arg8-TAT in DMPC/DMPG membranes at 303 K. (a) Cα and Cδ C-H DIPSHIFT curves. (b) Nα and Nη N-H DIPSHIFT curves. (c) Arg8 order parameters from the measured dipolar couplings. The lowest order parameters were observed for the middle of the sidechain, while the guanidinium moiety and the backbone exhibit higher order parameters.
Table 2.
13C-1H and 15N-1H dipolar couplings (kHz) and order parameters of TAT in DMPC/DMPG membranes at 303 K.
| Residue | Site | δ (ppm) | XHn | ωXHa | SXH |
|---|---|---|---|---|---|
| Arg8 | Nα | 123.1 | NH | 2.13 | 0.20 |
| Nη | 73.0 | NH2 | 3.19 | 0.30 | |
| Cα | 54.2 | CH | 4.09 | 0.18 | |
| Cβ | 28.9 | CH2 | 3.41 | 0.15 | |
| Cδ | 41.4 | CH2 | 1.82 | 0.08 | |
| Pro11 | Cα | 59.7 | CH | 3.18 | 0.14 |
| Cβ | 28.9 | CH2 | 3.86 | 0.17 | |
| Cδ | 48.7 | CH2 | 4.54 | 0.20 | |
| Lipids | Cα | 60.3 | CH2 | 2.72 | 0.12 |
| G2 | 71.4 | CH | 4.31 | 0.19 | |
| C2 | 34.7 | CH2 | 4.77 | 0.21 | |
| (CH2)n | 31.0 | CH2 | 5.68 | 0.25 |
ωXH is the true dipolar couplings after taking into account various scaling factors in the pulse sequence. The rigid-limit couplings used for calculating the order parameters are 10.6 kHz for N-H and 22.7 kHz for C-H dipolar couplings.
Table 3.
1H T1ρ relaxation times (ms) of TAT and penetratin in DMPC/DMPG membranes at 303 K, measured with a 1H effective spin-lock field of 61.2 kHz.
| Peptide | Residue | Site | T1ρ (ms) |
|---|---|---|---|
| TAT (48-60) | Arg8 | Cα | 11.6 |
| Cβ | 11.3 | ||
| Cδ | 29.3 | ||
| Pro11 | Cα | 14.9 | |
| Cβ | 8.8 | ||
| Cδ | 7.0 | ||
| Penetratin | Ile3 | Cα | 3.0 |
| Cδ | 4.9 | ||
| Cε | 6.2 |
Interaction of TAT with lipid membranes
To probe whether TAT binding causes membrane disorder, we measured 31P spectra of oriented lipid bilayers of several compositions in the presence of increasing amounts of the peptide. Fig. 5 shows the effects of TAT on both the neutral zwitterionic POPC membrane and two anionic membranes, POPC/POPG (8:7) and POPE/POPG (8:7). Up to 8 mol% peptide, the POPC and POPC/POPG membranes remain largely well ordered, as shown by the high 0° peak at ~30 ppm and the low 90° peak of the powder pattern at -15 ppm, which is indicative of misalignment of the membrane. The lack of an isotropic peak at 0 ppm indicates that the membrane remains lamellar and intact. For the POPE/POPG membrane, higher powder intensities were observed compared to the other two membranes, but the isotropic peak is still absent. The lack of membrane disruption was further confirmed by static 31P spectra of unoriented DMPC/DMPG and POPE/POPG membranes containing 6% TAT, where regular uniaxial powder lineshapes were observed (Fig. 5b). The minor isotropic peaks were attributed to the phosphate buffer. The lack of isotropic disorder is consistent with previously reported 31P NMR spectra of unoriented POPC/POPG (3:1) membranes (32) containing TAT, and is also consistent with the ability of this cell-penetrating peptide to cross the membrane without damaging its integrity.
Fig. 5.
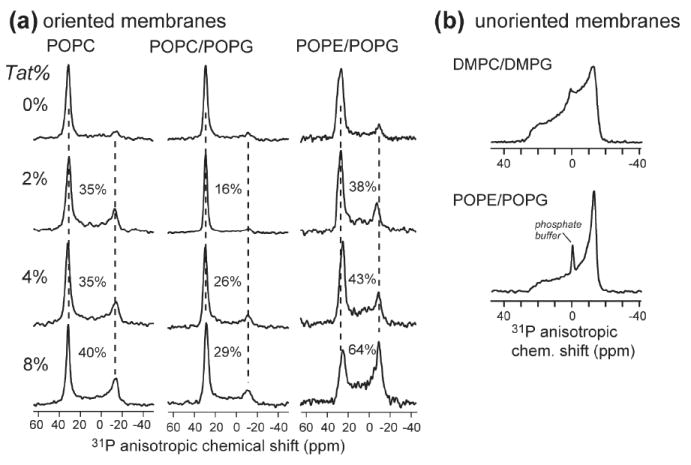
Effects of TAT on the membrane structure. (a) Static 31P spectra of glass-plate oriented lipid membranes with varying mole concentrations of TAT. Three membrane compositions, POPC, POPC/POPG (8:7) and POPE/POPG (8:7), were examined. The percentage of intensity in the 12 to -27 ppm range is indicated for each spectrum to denote the amount of membrane disorder. No isotropic signal near 0 ppm was observed in any spectra. (b) Static 31P spectra of unoriented DMPC/DMPG (8:7) membrane and POPE/POPG (8:7) membranes containing 6 mol% TAT. The isotropic peak at 1.6 ppm is the phosphate buffer peak. All 31P spectra were measured at 296 K.
Depth of insertion of TAT in lipid bilayers from 1H spin diffusion
To determine how deeply TAT is inserted into the lipid bilayer we carried out 13C-detected 1H spin diffusion experiments from water and lipids to the peptide (23). The distance between the source protons and protein protons determines the intensities of the intermolecular cross peaks. Since TAT is similarly mobile as lipids at ambient temperature, the dipolar-mediated 1H spin diffusion process is similarly inefficient in the peptide as in the soft lipid matrix. Therefore, spin diffusion cross peak intensities depend on the distances of individual residues from the source protons. The site-specific nature of the depth information for the mobile TAT differs from the case of large immobile membrane proteins, where rapid spin diffusion within the protein largely removes the site-resolution of the depth, and only the shortest distance between the protein and the source protons can be obtained (23).
Fig. 6 shows representative 13C-detected 1H spin diffusion spectra of Arg8- and Pro11-TAT, measured with a 1H spin diffusion mixing time of 225 ms and 144 ms, respectively. Weak cross peaks were observed between the lipid chain CH2 protons at 1.3 ppm and the peptide 13C signals. The mixing time of 100-200 ms is modest for the highly dynamic TAT with low spin diffusion coefficients. For comparison, immobilized DNA bound to the surface of cationic lipid membranes showed no cross peaks with the lipid chain protons until after ~400 ms, even though the spin diffusion coefficient of the rigid DNA rods is much larger than TAT (23). Thus, if TAT were similarly surface-bound as DNA, ~20 Å from the center of the membrane, then it will not exhibit any cross peaks with the lipid chain protons in the short mixing times used here. Therefore, the presence of the lipid-TAT cross peaks within the modest mixing time is strong qualitative evidence that TAT binds inside the lipid bilayer rather than lying on the membrane surface.
Fig. 6.
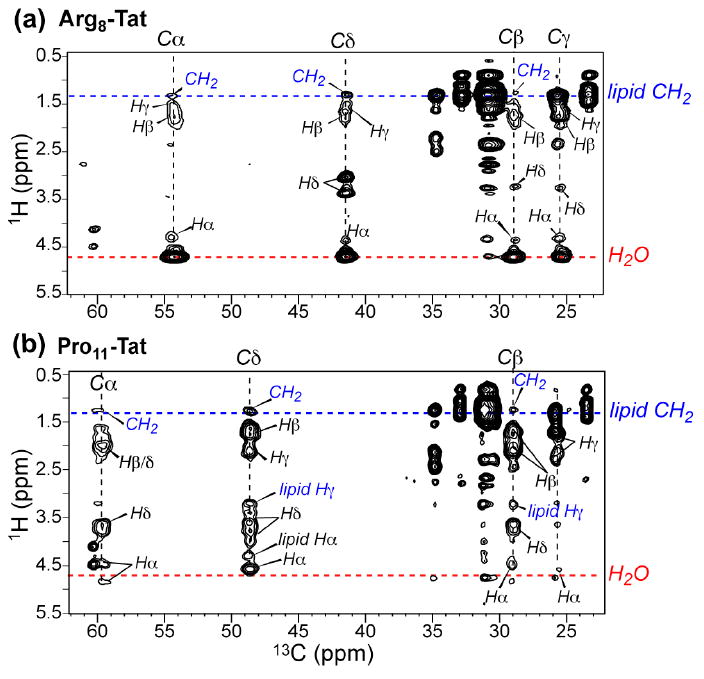
(a) 2D 13C-detected 1H spin diffusion spectra of DMPC/DMPG-bound TAT at 303 K. (a) Arg8-TAT spectrum measured with 225 ms spin diffusion. Water and lipid CH2 cross peaks are colored. (b) Pro11-TAT spectrum with a mixing time of 144 ms. No water cross peaks (37) but clear lipid CH2 cross peaks were detected.
Fig. 6 shows strong water cross peaks at 4.7 ppm for Arg8 but not Pro11. Fig. 7 compares the water region of the 13C-1H 2D spectra for all sites studied. Interestingly, all polar residues containing labile protons – Lys4, Gln7 and Arg8 - exhibited water cross peaks, while the hydrophobic Pro11 without any exchangeable protons did not. This difference indicates that hydrogen exchange is necessary for water-protein spin diffusion cross peaks to occur, consistent with many recent studies of the mechanism of water-protein magnetization transfer (35-37). Thus, Pro11 is not necessarily further away from water than the other residues. Overall, the strong water cross peaks with the peptide indicate that TAT is not deeply inserted into the bilayer.
Fig. 7.

2D 13C-detected 1H spin diffusion spectra of DMPC/DMPG (8:7) membranes with and without TAT. (a) Without TAT. (b) With Lys4-labeled TAT. (c) With Gln7-labeled TAT. (d) With Arg8-labeled TAT. Water-peptide cross peaks were detected in (b-d). (e) With Pro11-labeled TAT with a mixing time of 225 ms. No Pro11-water cross peaks were observed.
The coexistence of water and lipid cross peaks with TAT is most consistent with a location of the peptide in the glycerol backbone and headgroup region of the membrane. This interfacial location inside the membrane is further supported by the fact that TAT enhanced the water-to-lipid spin diffusion of the POPE/POPG membrane. Fig. 8 shows that the lipid headgroup Cα and acyl chain C2 have stronger cross peaks with water in the presence of TAT than in its absence, which is not possible if TAT were on the surface of the membrane.
Fig. 8.
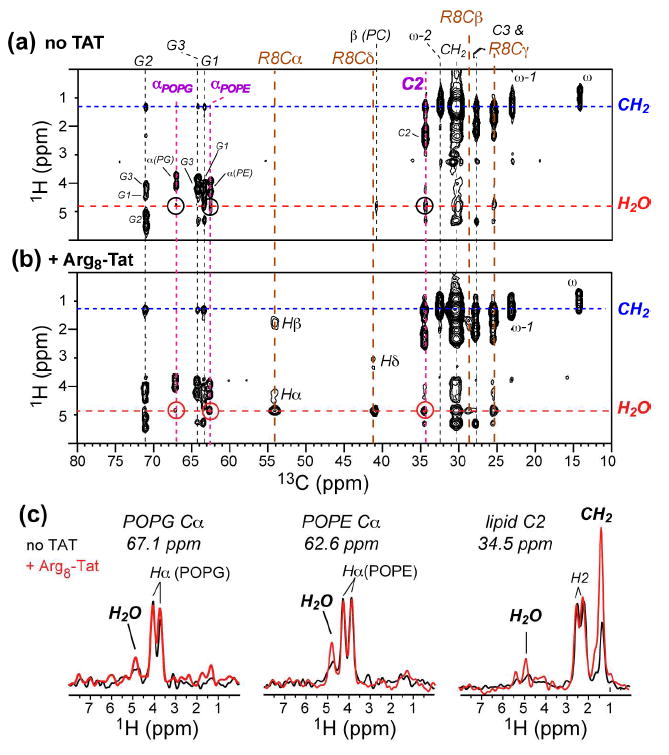
2D 13C-detected 1H spin diffusion spectra of POPE/POPG (8:7) membranes with a mixing time of 144 ms. (a) Without TAT. Only very weak water-lipid cross peaks were observed. (b) With Arg8-labeled TAT (P/L = 1:15), showing stronger water-lipid cross peaks. (c) 1H cross sections at several lipid 13C frequencies. The TAT-free spectrum is in black and the TAT-containing spectrum is in red. TAT enhances the water-lipid cross peaks, indicating that the peptide is inserted into the membrane. All spectra were measured at 303 K under 5 kHz MAS.
The exact distance of TAT to the center of the membrane cannot be quantified by observing cross peak buildup intensities as a function of mixing time due to the lack of well-calibrated spin diffusion coefficients for dynamic molecules. Nevertheless, one can obtain some insights based on the relative intensities of the water-peptide and lipid-peptide cross peaks. In ~40% hydrated membrane samples, the number of water protons is about twice the number of lipid CH2 protons. Previous calibrations of water and lipid-chain spin diffusion coefficients indicated that the water spin diffusion coefficient is about 10-fold larger than the lipid chain diffusion coefficient because of the faster translational motion of water (23, 38). Thus, the water-peptide cross peak should be higher than the lipid-peptide cross peak even if a residue is at comparable distances to the membrane surface water and to the hydrophobic chains. Thus, the presence of a lipid-TAT cross peak at all is a strong indication of the membrane-immersed nature of TAT.
Site-specific distances to the membrane surface from 13C-31P REDOR
To confirm the interfacial location of TAT, we carried out 13C-31P distance measurements at low temperature using REDOR. If TAT were inserted into the hydrophobic core of the bilayer, long 13C-31P distances or slow dipolar dephasing would be expected, as is the case for the lipid chain 13C signal. The REDOR experiments were conducted at ~230 K to completely freeze the dynamics of both the DMPC/DMPG membrane and the peptide. The 31P spectra at this temperature give a span of 197 ppm (Fig. S1), which is the rigid limit for the 31P chemical shift tensor (29). Fig. 9 shows the 13C-31P REDOR data of the backbone and sidechain sites of Arg8 and Pro11. Relatively short backbone 13C-31P distances of 5.7 – 6.6 Å were observed, consistent with the 1H spin diffusion result that the peptide backbone lies in the glycerol backbone region of the membrane-water interface. Natural abundance lipid signals were corrected for both the Arg8 CO peak and the Pro11 Cα peak. For the guanidinium Cζ of Arg8, a 1 : 1 combination of 4.1 Å and 5.5 Å distances fit the data well (Fig. 9b). The former is at the lower limit of the possible distance between the guanidinium and the lipid phosphate, and places the two groups within hydrogen-bonding contact of each other (29, 39). Thus, the Arg residues in TAT interact with the negatively charged lipid headgroups in a similar fashion to Arg-rich antimicrobial peptides (29, 40). Since the superposition of two distances fits the Cζ data well, there is no sign of a broad distance distribution, suggesting that guanidinium-phosphate ion pair interaction, rather than the peptide backbone conformation, is the determining factor for the Arg sidechain conformation.
Fig. 9.
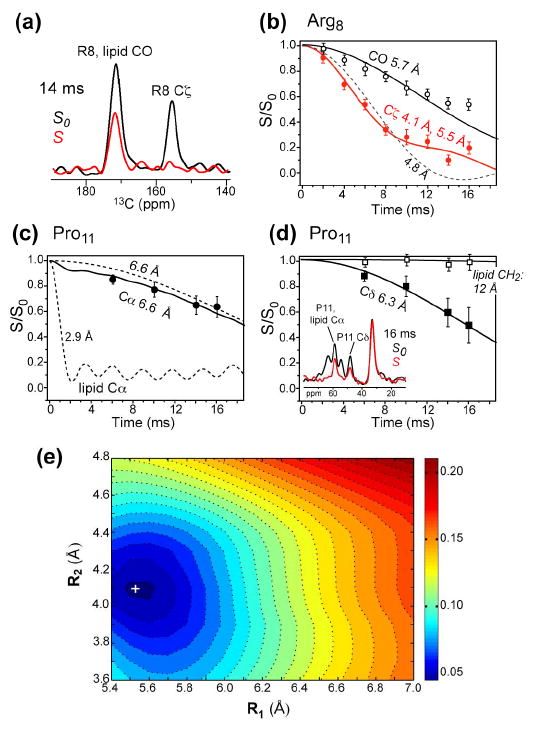
13C-31P REDOR data of DMPC/DMPG-bound TAT at 230 K. (a) Representative REDOR control (S0) and dephased (S) spectra. (b) REDOR data and simulation for Arg8 CO and Cζ. The Cζ data is best-fit fit by a 1:1 ratio of 4.1 Å and 5.5 Å. (c) REDOR data and best fit for Pro11 Cα. The lipid Cα contribution (7% of full intensity) was taken into account by a fixed distance of 2.9 Å. (d) REDOR data and best fit for Pro11 Cδ. (e) RMSD between the simulated and experimental REDOR curves of Arg8 Cζ to obtain the best-fit distances for Cζ in (b).
Discussion
Depth and thermodynamics of insertion of TAT into lipid membranes
The 1H spin diffusion and 13C-31P distance data indicate that TAT binds inside the DMPC/DMPG (8:7) bilayer in the glycerol backbone and headgroup region of the membrane-water interface (Fig. 10). This location is consistent with the temperature dependence of the 13C and 15N spectra (Fig. 3), since exchange broadening of TAT signals coincided with the membrane phase transition. This depth is also consistent with molecular dynamics simulations of TAT and the related CPP penetratin (41, 42).
Fig. 10.
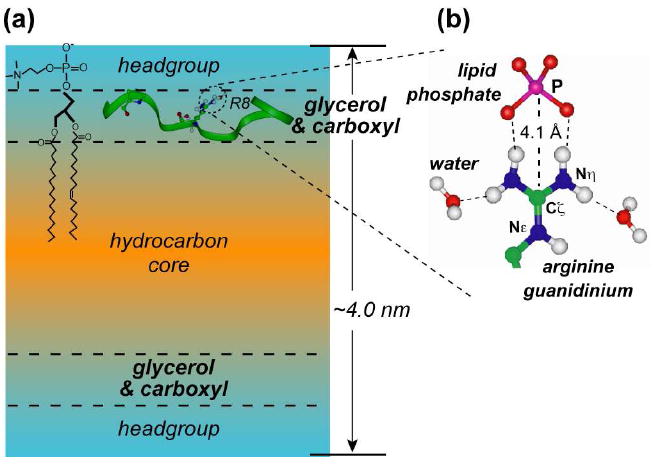
Model of TAT structure and dynamics in DMPC/DMPG bilayers. The random coil peptide is inserted into the bilayer at the glycerol backbone region, outside the hydrophobic core, and is stabilized by intermolecular hydrogen bonds with lipid phosphates and water. The glycerol region has intermediate dielectric constant and accommodates TAT through the polar phosphate and carboxyl groups, which can act as hydrogen bond acceptors for the guanidinium.
It is interesting to consider the free energies of TAT insertion into the membrane-water interface. Using the Wimley-White interfacial scale developed for zwitterionic POPC bilayers (43), an energy barrier of about 9 kcal/mol against insertion would be predicted. However, the interfacial scale derived from neutral POPC bilayers does not account for strong electrostatic attraction between cationic peptides and highly anionic membranes as used in this study. Using isothermal titration calorimetry experiments, Seelig and coworkers characterized the thermodynamics of TAT binding to anionic membranes in detail, and found that the free energy of binding of TAT to 25% anionic membranes in 100 mM salt solution was about 80% electrostatic and 20% hydrophobic, with a total Gibbs free energy of -5.2 kcal/mol (32). The NMR samples used here contain less salt (10 mM) and more (~50%) anionic lipids, thus the binding energy should be even more negative or favorable. Similar to TAT, oligoarginine peptides also showed a significant electrostatic component in their binding energies (44).
In addition to the electrostatic attraction, 13C-31P REDOR experiments found tight association of the Arg8 guanidinium group with the lipid phosphates, similar to a number of other Arg-rich CPPs and antimicrobial peptides (12, 29, 40). A Cζ-P distance of 4-5 Å indicates hydrogen bonding between the guanidinium N-H and the lipid P-O groups. The importance of this hydrogen bonding was demonstrated for the antimicrobial peptide PG-1, where mutation of the guanidinium to dimethylated guanidinium weakened the antimicrobial activity by 3-fold and changed the oligomeric structure and orientation of the peptide in the lipid bilayer (40). Each guanidinium ion can form up to five hydrogen bonds with lipid phosphates or water. Thus, the hexa-Arg TAT can form as many as thirty hydrogen bonds from the Arg sidechains. If each hydrogen bond formation gives a favorable free energy change of -0.5 kcal/mol (45), then the total free energy change can be as much as -15 kcal/mol in favor of binding, which would greatly stabilize TAT at the membrane-water interface. Therefore, the combined electrostatic attraction and hydrogen bonding effects should be more than sufficient to overcome Born repulsion and facilitate TAT insertion into the glycerol backbone region of the membrane-water interface. An increasing number of MD simulations also pointed out the importance of guanidinium-phosphate and guanidinium-water interactions for reducing the free energy cost of inserting Arg residues into the lipid membrane (42, 46-48).
The location of TAT is deeper than polylysine peptides, which was found by monolayer pressure and area measurements to lie on the surface of anionic lipid monolayers (49). Differences in the experimental conditions as well as the intrinsic membrane affinities of the two peptides most likely account for the observed depth difference. The polylysine measurements were carried out in 33% negatively charged membranes in 100 mM salt (49), while the bilayer samples in the present NMR study contained ~50% anionic lipids and only 10 mM salt. A lower fraction of acidic lipids reduces the electrostatic attraction while a higher salt concentration shields the electrostatic attraction between the peptide and the membrane. Thus the polylysine peptides are expected to have weaker membrane binding than the TAT peptide under the conditions of the two studies. The sensitivity of membrane binding to salt concentration is well known, and is exemplified by the fact that the free energies of polylysine binding decreased from -1.5 kcal/mol to -7 kcal/mol from 500 mM to 50 mM salt (49). The polylysine binding study also used lipid monolayers, where the lack of a distal membrane surface, with its concomitant anionic lipid headgroups, may weaken the propensity of the polylysines to insert into the membrane. In addition to these experimental differences, there is also a real difference in the affinity of Arg-rich and Lys-rich cationic peptides for the membrane interior. It is well known that Lys substitution for Arg in cell-penetrating peptides abolishes the membrane translocation ability (50, 51). An increasing number of recent studies of CPPs and antimicrobial peptides suggested that the structural basis for this difference may lie in the ability of guanidinium to form hydrogenbond-stabilized bidentate complexes with lipid phosphates (12, 29, 40, 52), which cannot be achieved by the mobile amine of the Lys sidechains.
Taken together, the interfacial location of the Arg-rich TAT in the lipid bilayer correlates well with the membrane translocation ability of the peptide, and can be attributed to the strong electrostatic attraction between TAT and the anionic lipid membranes as well as hydrogen-bond stabilization of the Arg residues by the lipid phosphates and water.
TAT perturbation of lipid membranes
31P NMR spectra of both neutral and anionic lipid membranes in the presence of TAT showed little isotropic disorder. The observation is reproduced in both aligned membranes and unoriented vesicle samples (Fig. 5). This finding suggests that TAT binding does not significantly perturb the lamellar structure of the lipid bilayer. Interestingly, a detailed 31P NMR characterization of TAT binding as a function of membrane composition showed that the isotropic disorder is a sensitive function of the acyl chain saturation and temperature (53). Low-melting lipids such as DLPC and POPC did not exhibit membrane disruption by TAT, while high-melting DMPC and DPPC bilayers were. Moreover, the non-lamellar disorder was only present in zwitterionic bilayers and not in anionic bilayers. The present use of unsaturated lipids in all three membrane series for the oriented 31P NMR experiments thus precluded the observation of any potential isotropic features. Our depth and conformational experiments were conducted in negatively charged lipid membranes, thus similarly precluding the observation of membrane disorder. When we applied two cycles of heating and cooling to the TAT-containing DMPC/DMPG (8:7) membrane sample, we did not observe an isotropic peak (Fig. S2), which confirmed the lack of significant membrane disorder induced by thermal history.
Interestingly, an MD simulation of HIV TAT in a neutral DOPC bilayer as a function of peptide/lipid molar ratios found transient pores at high peptide concentrations (42). Based on these simulations, Herce and Garcia proposed that the mechanism of translocation involves membrane thinning, reorientation of the lipids near the peptide, and attraction of TAT to lipid headgroups on the distal (far) side of the membrane. The 13C-31P distances measured here support the latter two proposals. However, the lack of isotropic 31P signals indicates the absence of long-lasting pores, although our data, which describes the equilibrium state of the peptide, does not rule out the existence of transient pores during translocation. One possibility is that the Arg-phosphate salt bridge interaction may slow down the fast lipid lateral diffusion necessary for observing an isotropic signal, so that membrane defects may be present but undetectable in the 31P NMR spectra. However, we do not think this scenario is likely, because the salt bridge interaction is too weak (with a guanidinium Nη order parameter of 0.30) to retard the lipid diffusion. Moreover, similar Arg-phosphate salt bridge interactions were observed in antimicrobial peptides such as PG-1 (29), which caused pronounced 31P isotropic features (39, 54).
Random coil conformation of TAT in lipid bilayers
The temperature-independent random coil chemical shifts of TAT and the temperature-dependent linewidth changes provide a rare example of a truly dynamic random coil peptide in lipid membranes. The 2D INADEQUATE spectra at low temperatures (Fig. 1) exhibited fine structures for some sidechain peaks. However, the chemical shift differences between the multiple peaks of each site are small compared to the linewidths, thus they do not change the qualitative conclusion that the TAT adopts a random coil conformation in the lipid membrane.
While TAT is known to be unstructured in solution (30), the finding that it remains a random coil in the lipid bilayer is still surprising, since it is generally believed that unstructured peptides in solution acquire hydrogen-bonded canonical conformations upon membrane binding to reduce the number of polar N-H and C=O groups exposed to the membrane (45). In light of the lipid-peptide interaction data here, we propose that TAT remains random coil by substituting intramolecular hydrogen bonds by intermolecular ones with the lipid phosphates and water, and by residing in the membrane-water interface, which has a higher dielectric constant than the hydrocarbon region of the bilayer. The intermolecular Arg-phosphate interaction is not only revealed by the short guanidinium-phosphate distance but also by the fact that the guanidinium exhibits higher order parameters than the rest of the Arg sidechain. This lipid anchoring effect dovetails a recent solution NMR and MD simulation study of Arg sidechains in ribonuclease H, where the dynamics of the guanidinium Nε was found to be primarily governed by salt bridges and was decoupled from the dynamics of the rest of the aliphatic sidechain (55).
The random coil backbone should facilitate TAT interaction with multiple lipid headgroups, whose positions in the fluid bilayer are highly disordered. The completeness of the Arg8 Cζ-31P REDOR dephasing (Fig. 9) confirms that all peptide molecules, instead of just a fraction, experience guanidinium-phosphate interactions. Finally, a random coil backbone reduces the hydrophobic surface of TAT and prevents any defined polar/apolar separation. In this way, the peptide may rapidly cross the bilayer instead of being retained permanently in the membrane, as is the case for amphipathic antimicrobial peptides. Since the intermolecular lipid-TAT and water-TAT hydrogen bonds can be formed for any phospholipid membranes, TAT is expected to adopt similar random coil structures in any anionic membranes. This prediction is supported by the fact that the same random coil chemical shifts were found in both DMPC/DMPG membranes and POPE/POPG membranes, even though PE membranes appear to permit more efficient translocation of TAT than PC membranes (52).
Conclusion
In summary, using solid-state NMR we found that HIV TAT(48-60) adopts a random-coil conformation and inserts into the glycerol backbone region of the membrane-water interface in anionic lipid bilayers. This structure is thermodynamically stabilized by electrostatic attraction with the anionic lipids, guanidinium-phosphate salt bridges and guanidinium-water hydrogen bonding. The highly dynamic TAT does not cause permanent damages to the membrane integrity, but may cause transient defects, as suggested by short guanidinium-phosphate distances. These results suggest that TAT translocation is an extremely rapid phenomenon that relies on transient interactions of the Arg sidechains with lipid phosphates of the distal leaflet. The equilibrium structure observed by solid-state NMR, showing the peptide lying at the membrane-water interface of both leaflets, is the structure after translocation. Catching the peptide “in action”, during translocation, may require independent experiments of the kind that has been used to probe transient macromolecular interactions and structural changes in solution (56, 57). We postulate that the role of the random coil conformation is to prevent the peptide from permanently residing in the hydrocarbon core of the lipid bilayer through hydrophobic interactions. The dynamic nature of TAT, together with the previously reported dynamic turn-rich conformation of penetratin, suggests that the Arg-rich CPPs differ from the Arg-rich amphipathic AMPs mainly by the presence or absence of intramolecular hydrogen-bonded conformations (14).
Supplementary Material
Table 1.
1H, 13C and 15N chemical shifts (ppm) of labeled residues of TAT in DMPC/DMPG bilayers at 303 K and 233 K.
| Residue | Site | 303 K | 233 K | Random coil values (31) |
|---|---|---|---|---|
| Lys4 | CO | 174.7 | 174.5 | 174.5 |
| Cα | 54.4 | 54.3 | 54.7 | |
| Cβ | 30.9 | 31.3 | 30.8 | |
| Cγ | 22.8 | 22.9 | - | |
| Cδ | 27.2 | 26.3 | - | |
| Cε | 40.2 | 39.7 | - | |
| Hα | 4.29 | - | 4.28 | |
| Hβ | 1.82 | - | - | |
| Hγ | 1.45 | - | - | |
| Hδ | 1.69 | - | - | |
| Gln7 | CO | 173.8 | 174.0 | 174.2 |
| Cα | 53.8 | 53.8 | 54.2 | |
| Cβ | 27.8 | 26.7 | 27.0 | |
| Cγ | 32.0 | 33.3 | - | |
| Cδ | 178.4 | 177.9 | - | |
| Hα | 4.37 | - | 4.26 | |
| Hβ | 2.01 | - | - | |
| Arg8 | Nα | 123.1 | 122.2 | 118.9 |
| Nε | 85.4 | 83.2 | - | |
| Nη | 73.0 | 71.3 | - | |
| CO | 174.1 | 174.3 | 174.2 | |
| Cα | 54.2 | 53.4 | 54.3 | |
| Cβ | 28.9 | 28.7 | 28.8 | |
| Cγ | 25.2 | 25.4 | - | |
| Cδ | 41.5 | 41.0 | - | |
| Cζ | 157.7 | 156.9 | - | |
| HN | 8.40 | - | 8.17 | |
| Hα | 4.31 | - | 4.33 | |
| Hβ | 1.87 | - | - | |
| Hγ | 1.64 | - | - | |
| Hδ | 3.22 | - | - | |
| Pro11 | Nα | 138.3 | 136.9 | - |
| CO | 172.5 | 172.6 | 175.2 | |
| Cα | 59.7 | 58.6 | 61.8 | |
| Cβ | 29.0 | 28.7 | 30.2 | |
| Cγ | 25.4 | 25.5 | - | |
| Cδ | 48.6 | 48.3 | - | |
| Hα | 4.68 | - | 4.41 | |
| Hβ | 1.92 | - | - | |
| Hγ | 2.00 | - | - | |
| Hδ | 3.82 | - | - |
Acknowledgments
Funding information: This work was supported by grant GM066976 from the National Institutes of Health (to M.H.).
Abbreviations
- CPP
cell-penetrating peptide
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine
- DMPG
1,2-dimyristoyl-sn-glycero-3-phosphatidylglycerol
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine
- POPE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol
- MAS
magic-angle spinning
- CP
cross-polarization
- DARR
dipolar-assisted rotational resonance
- INADEQUATE
incredible natural abundance double quantum transfer experiment
- HETCOR
heteronuclear correlation
- DIPSHIFT
dipolar-chemical-shift correlation
- REDOR
rotational-echo double-resonance
- FWHM
full width at half maximum
Footnotes
Supporting Information Static 31P spectra of TAT-containing unoriented DMPC/DMPG membranes under two heating and cooling cycles and a summary table of the solid-state NMR experiments are included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Delivery Rev. 2008;60:548. doi: 10.1016/j.addr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 2.Brooks H, Lebleu B, Vives E. Tat peptide-mediated cellular delivery: back to basics. Adv Drug Delivery Rev. 2005;57:559. doi: 10.1016/j.addr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Inomata K, Ohno A, Tochio H, Isogai S, Tenno T, Nakase I, Takeuchi T, Futaki S, Ito Y, Hiroaki H, Shirakawa M. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature. 2009;485:106–110. doi: 10.1038/nature07839. [DOI] [PubMed] [Google Scholar]
- 4.Vives E, Brodin P, Lebleu B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 5.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 6.Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc. 2004;126:9506–9507. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 7.Rothbard JB, Jessop TC, Wender PA. Adaptive translocation: the role of hydrogen bonding and membrane potential in the uptake of guanidinium-rich transporters into cells. Adv Drug Delivery Rev. 2005;57:495. doi: 10.1016/j.addr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Fischer R, Fotin-Mleczek M, Hufnagel H, Brock R. Break on through to the other side-biophysics and cell biology shed light on cell-penetrating peptides. ChemBioChem. 2005;6:2126–2142. doi: 10.1002/cbic.200500044. [DOI] [PubMed] [Google Scholar]
- 9.Hessa T, Kim H, Bihlmaier K, Lundin C, Boekel J, Andersson H, Nilsson I, White SH, von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 10.Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, Prochiantz A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J Biol Chem. 1996;271:18188–18193. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- 11.Binder H, Lindblom G. Charge-dependent translocation of the Trojan peptide penetratin across lipid membranes. Biophys J. 2003;85:982–995. doi: 10.1016/S0006-3495(03)74537-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su Y, Mani R, Hong M. Asymmetric insertion of membrane proteins in lipid bilayers by solid-state NMR paramagnetic relaxation enhancement: a cellpenetrating Peptide example. J Am Chem Soc. 2008;130:8856–8864. doi: 10.1021/ja802383t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su Y, Doherty T, Waring AJ, Ruchala P, Hong M. Roles of Arginine and Lysine Residues in the Translocation of a Cell-Penetrating Peptide from 13C, 31P, and 19F Solid-State NMR. Biochemistry. 2009;48:4587–4595. doi: 10.1021/bi900080d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su Y, Mani R, Doherty T, Waring AJ, Hong M. Reversible sheet-turn conformational change of a cell-penetrating peptide in lipid bilayers studied by solid-state NMR. J Mol Biol. 2008;381:1133–1144. doi: 10.1016/j.jmb.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frankel AD, Pabo CO. Cellular Uptake of the Tat Protein from Human lmmunodeficiency Virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 16.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci U S A. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eguchi A, Akuta T, Okuyama H, Senda T, Yokoi H, Inokuchi H, Fujita S, Hayakawa T, Takeda K, Hasegawa M, Nakanishi M. Protein transduction domain of HIV-1 Tat protein promotes efficient delivery of DNA into mammalian cells. J Biol Chem. 2001;276:26204–26210. doi: 10.1074/jbc.M010625200. [DOI] [PubMed] [Google Scholar]
- 18.Gennis RB. Biomembranes: Molecular Structure and Function. Springer; New York: 1989. [Google Scholar]
- 19.Hallock KJ, Henzler Wildman K, Lee DK, Ramamoorthy A. An innovative procedure using a sublimable solid to align lipid bilayers for solid-state NMR studies. Biophys J. 2002;82:2499–2503. doi: 10.1016/S0006-3495(02)75592-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hohwy M, Rienstra CM, Jaroniec CP, Griffin RG. Fivefold symmetric homonuclear dipolar recoupling in rotating solids: application to double-quantum spectroscopy. J Chem Phys. 1999;110:7983–7992. [Google Scholar]
- 21.Takegoshi K, Nakamura S, Terao T. C-13-H-1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–637. doi: 10.1063/1.2364503. [DOI] [PubMed] [Google Scholar]
- 22.Hong M. Solid-State Dipolar INADEQUATE NMR Spectroscopy with a Large Double-Quantum Spectral Width. J Magn Reson. 1999;136:86–91. doi: 10.1006/jmre.1998.1631. [DOI] [PubMed] [Google Scholar]
- 23.Huster D, Yao XL, Hong M. Membrane Protein Topology Probed by 1H Spin Diffusion from Lipids Using Solid-State NMR Spectroscopy. J Am Chem Soc. 2002;124:874–883. doi: 10.1021/ja017001r. [DOI] [PubMed] [Google Scholar]
- 24.Hong M, Gross JD, Rienstra CM, Griffin RG, Kumashiro KK, Schmidt-Rohr K. Coupling Amplification in 2D MAS NMR and Its Application to Torsion Angle Determination in Peptides. J Magn Reson. 1997;129:85–92. doi: 10.1006/jmre.1997.1242. [DOI] [PubMed] [Google Scholar]
- 25.Munowitz MG, Griffin RG, Bodenhausen G, Huang TH. Two-dimensional rotational spin-echo nuclear magnetic resonance in solids: correlation of chemical shift and dipolar interactions. J Am Chem Soc. 1981;103:2529–2533. [Google Scholar]
- 26.Rhim WK, Elleman DD, Vaughan RW. Analysis of multiple pulse NMR in solids. J Chem Phys. 1973;59:3740–3749. [Google Scholar]
- 27.Huster D, Xiao LS, Hong M. Solid-State NMR Investigation of the dynamics of colicin Ia channel-forming domain. Biochemistry. 2001;40:7662–7674. doi: 10.1021/bi0027231. [DOI] [PubMed] [Google Scholar]
- 28.Jaroniec CP, Tounge BA, Herzfeld J, Griffin RG. Frequency selective heteronuclear dipolar recoupling in rotating solids: accurate (13)C-(15)N distance measurements in uniformly (13)C,(15)N-labeled peptides. J Am Chem Soc. 2001;123:3507–3519. doi: 10.1021/ja003266e. [DOI] [PubMed] [Google Scholar]
- 29.Tang M, Waring AJ, Hong M. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptide from solid-state NMR. J Am Chem Soc. 2007;129:11438–11446. doi: 10.1021/ja072511s. [DOI] [PubMed] [Google Scholar]
- 30.Shojania S, O’Neil JD. HIV-1 Tat is a natively unfolded protein: the solution conformation and dynamics of reduced HIV-1 Tat-(1-72) by NMR spectroscopy. J Biol Chem. 2006;281:8347–8356. doi: 10.1074/jbc.M510748200. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 2002;11:852–861. doi: 10.1110/ps.3180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ziegler A, LiBlatter X, Seelig A, Seelig J. Protein Transduction Domains of HIV-1 and SIV TAT Interact with Charged Lipid Vesicles. Binding Mechanism and Thermodynamic Analysis. Biochemistry. 2003;42:9185–9194. doi: 10.1021/bi0346805. [DOI] [PubMed] [Google Scholar]
- 33.Cady SD, Hong M. Effects of Amantadine Binding on the Dynamics of Bilayer-Bound Influenza A M2 Transmembrane Peptide Studied by NMR Relaxation. J Biomol NMR. 2009;45:185–196. doi: 10.1007/s10858-009-9352-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong M, Schmidt-Rohr K, Pines A. Measurement of Signs and Magnitudes of C-H Dipolar Couplings in Lecithin. J Am Chem Soc. 1995;117:3310–3311. [Google Scholar]
- 35.Lesage A, Emsley L, Penin F, Böckmann A. Investigation of dipolarmediated water-protein interactions in microcrystalline Crh by solid-state NMR spectroscopy. J Am Chem Soc. 2006;128:8246–8255. doi: 10.1021/ja060866q. [DOI] [PubMed] [Google Scholar]
- 36.Lesage A, Gardiennet C, Loquet A, Verel R, Pintacuda G, Emsley L, Meier BH, Bockmann A. Polarization transfer over the water-protein interface in solids. Angew Chem Int Ed. 2008;47:5851–5854. doi: 10.1002/anie.200801110. [DOI] [PubMed] [Google Scholar]
- 37.Doherty T, Hong M. 2D 1H-31P solid-state NMR studies of the dependence of inter-bilayer water dynamics on lipid headgroup structure and membrane peptides. J Magn Reson. 2009;196:39–47. doi: 10.1016/j.jmr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mani R, Cady SD, Tang M, Waring AJ, Lehrer RI, Hong M. Membrane-dependent oligomeric structure and pore formation of a beta-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc Natl Acad Sci USA. 2006;103:16242–16247. doi: 10.1073/pnas.0605079103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang M, Hong M. Structure and Mechanism of Beta-Hairpin Antimicrobial Peptides in Lipid Bilayers from Solid-State NMR Spectroscopy. Mol Biosys. 2009;5:317–322. doi: 10.1039/b820398a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang M, Waring AJ, Lehrer R, Hong M. Effects of Guanidinium-Phosphate Hydrogen Bonding on the Membrane-Bound Structure and Activity of an Arginine-Rich Membrane Peptide from Solid-State NMR Spectroscopy. Angew Chem Int Ed. 2008;47:3202–3205. doi: 10.1002/anie.200705993. [DOI] [PubMed] [Google Scholar]
- 41.Lensink MF, Christiaens B, Vandekerckhove J, Prochiantz A, Rosseneu M. Penetratin-membrane association: W48/R52/W56 shield the peptide from the aqueous phase. Biophys J. 2005;88:939–952. doi: 10.1529/biophysj.104.052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herce HD, Garcia AE. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc Natl Acad Sci U S A. 2007;104:20805–20810. doi: 10.1073/pnas.0706574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 44.Goncalves E, Kitas E, Seelig J. Binding of Oligoarginine to Membrane Lipids and Heparan Sulfate: Structural and Thermodynamic Characterization of a Cell-Penetrating Peptide. Biochemistry. 2005;44:2692–2702. doi: 10.1021/bi048046i. [DOI] [PubMed] [Google Scholar]
- 45.White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 46.Freites JA, Tobias DJ, von Heijne G, White SH. Interface connections of a transmembrane voltage sensor. Proc Natl Acad Sci USA. 2005;102:15059–15064. doi: 10.1073/pnas.0507618102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li L, Vorobyov I, Allen TW. Potential of mean force and pKa profile calculation for a lipid membrane-exposed arginine side chain. J Phys Chem B. 2008;112:9574–9587. doi: 10.1021/jp7114912. [DOI] [PubMed] [Google Scholar]
- 48.MacCallum JL, Bennett WF, Tieleman DP. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys J. 2008;94:3393–3404. doi: 10.1529/biophysj.107.112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ben-Tal N, Honig B, Peitzsch RM, Denisov G, McLaughlin S. Binding of small basic peptides to membranes containing acidic lipids: theoretical models and experimental results. Biophys J. 1996;71:561–575. doi: 10.1016/S0006-3495(96)79280-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 52.Mishra A, Gordon VD, Yang L, Coridan R, Wong GCL. HIV TAT Forms Pores in Membranes by Inducing Saddle-Splay Curvature: Potential Role of Bidentate Hydrogen Bonding. Angew Chem Int Ed Engl. 2008;47:2986–2989. doi: 10.1002/anie.200704444. [DOI] [PubMed] [Google Scholar]
- 53.Afonin S, Frey A, Bayerl S, Fischer D, Wadhwani P, Weinkauf S, Ulrich AS. The cell-penetrating peptide TAT(48-60) induces a non-lamellar phase in DMPC membranes. ChemPhysChem. 2006;7:2134–2142. doi: 10.1002/cphc.200600306. [DOI] [PubMed] [Google Scholar]
- 54.Mani R, Buffy JJ, Waring AJ, Lehrer RI, Hong M. Solid-State NMR Investigation of the Selective Disruption of Lipid Membranes by Protegrin-1. Biochemistry. 2004;43:13839–13848. doi: 10.1021/bi048650t. [DOI] [PubMed] [Google Scholar]
- 55.Trbovic N, Cho JH, Abel R, Friesner RA, Rance M, Palmer AGr. Protein side-chain dynamics and residual conformational entropy. J Am Chem Soc. 2009;131:615–622. doi: 10.1021/ja806475k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwahara J, Clore GM. Detecting transient intermediates in macromolecular binding by paramagnetic NMR. Nature. 2006;440:1227–1230. doi: 10.1038/nature04673. [DOI] [PubMed] [Google Scholar]
- 57.Lee MK, Gal M, Frydman L, Varani G. Real-time multidimensional NMR follows RNA folding with second resolution. Proc Natl Acad Sci U S A. 2010;107:9192–9197. doi: 10.1073/pnas.1001195107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luca S, Filippov DV, van Boom JH, Oschkinat H, de Groot HJ, Baldus M. Secondary chemical shifts in immobilized peptides and proteins: a qualitative basis for structure refinement under magic angle spinning. J Biomol NMR. 2001;20:325–331. doi: 10.1023/a:1011278317489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.