

Abstract
Biologically active small molecules have long proven useful in the exploration of cell biology. While many early compounds were byproducts of drug development efforts, recent increased small molecule screening efforts in academia have expanded the repertoire of biological processes investigated to include areas of biology that are not of immediate pharmaceutical interest. Many of these new bioassays score for small molecule-induced phenotypic changes at the cellular or even organismal level and thus have been described as ‘chemical genetic’ screens. However, this analogy with traditional genetic screens is misleading; whereas each gene has roughly an equivalent chance of being mutated in a traditional genetic screen, the amount of ‘proteomic space’ that a chemical genetics approach can reach using current small molecule libraries is considerably smaller. Thus, new chemical biology methodologies are needed to target the remaining ‘undruggable proteome’ with small drug-like molecules.
Magic Bullets
Over the last 40 years, the development of new antiviral, antitumor, antibiotic and Central Nervous System (CNS)-targeted drugs has had an immense impact on life expectancy and quality of life (Munos, 2009). In addition to these direct benefits, drug development over the past four decades has indirectly benefited the basic research community by generating new small molecule probes for basic biological studies. As detailed in the other reviews in this special issue of Chemistry & Biology, these small molecules have had a profound impact on many basic biological investigations and are among the impetuses for the burgeoning field of Chemical Biology. For example, while the natural product phosphoinositide 3-kinase (PI3K) inhibitor wortmannin has played a key role in identifying contributions for PI3K in biological processes as diverse as cell survival, histamine release, glucose uptake, and phagocytosis (Lennartz, 1999; Nakanishi et al., 1995; Ui et al., 1995), this natural product lacks PI3K isoform specificity. Fortunately, recent medicinal chemistry efforts have yielded more isoform-selective inhibitors that are helping to define PI3K function in specific cellular contexts (Siragusa et al., 2010; Soond et al., 2010; Sturgeon et al., 2008; Wang et al., 2008a). Other examples of useful biological probes that resulted from drug development efforts include inhibitors for the serine/threonine kinases MEK, JNK, and GSK-3β (Katsanakis et al., 2002; Saporito et al., 2002; Takahashi-Yanaga and Sasaguri, 2009; Wang et al., 2004).
Despite these many successes, there still are many instances where the right small molecule probe is lacking. As a chemical biologist who approaches the chemistry:biology interface from the biological side, there have been several occasions when I have wished for a ‘magic bullet’ to allow for specific regulation of a biological process of interest. Here I discuss the impact of academic screening efforts on new probe development, the shortcomings of ‘chemical genetic’ approaches and the currently unfulfilled need for novel libraries of small molecules capable of controlling intracellular protein function independent of protein class.
Developing Small Molecule Biological Probes: Design versus Serendipity
The availability and diversity of new bioactive probes for research has exploded in the past two decades, primarily due to the increase in academic small molecule screening facilities (Wu and Schultz, 2009). Previously, biologically active compounds were either direct byproducts of the pharmaceutical industry’s efforts to develop novel drugs or an indirect consequence of these efforts, e.g., natural product screens for drug target inhibitors. Thus, not surprisingly, these early small molecule probes had biological properties (e.g., anti-cancer, anti-inflammatory, anti-angiogenic, etc.) that were of interest to large pharma. Despite their pharmaceutical origins, many of these compounds have proven extremely useful as probes in basic research such as the immunosuppressive natural products FK506 and rapamycin that were instrumental in the exploration of immune cell signaling pathways (Cardenas et al., 1998). In addition, the use of these probes in basic research studies has generated new leads for novel drug targets, thus generating renewed interest in their therapeutic potential. For example, the identification of methionine aminopeptidase-2 (MetAP-2) as the target of the anti-angiogenic microbial metabolite fumagillin has led to the development of novel anti-angiogenic MetAP-2 inhibitors (Kallander et al., 2005; Marino et al., 2007; Sin et al., 1997; Wang et al., 2008b). However useful, these early probes failed to span the breadth of cell biology, thus leaving many areas that lacked small molecule-based research tools.
While small molecule-powered cell biology research proved very successful such as in the use of novel histone deacetylase Inhibitors to explore the role of chromatin structure in gene regulation (Yoshida et al., 2001), most of the early chemical biology probes were limited to those research areas of interest to the pharmaceutical industry. However, once academic labs began to acquire small molecule screening capabilities, new compounds could be identified possessing biological activities unrelated to drug development. This second generation of small molecule probe development relies more on targeted screens looking for compounds with highly specific biological activities as opposed to more general phenotypes such as decreased proliferation (Kawasumi and Nghiem, 2007; Schlueter and Peterson, 2009). Moreover, it has allowed for increased input from the basic biology research community to custom design/select compounds with particular biological characteristics. For example, a high-content cell-based assay for perturbation of mitotic spindle formation yielded monastrol, an inhibitor of the kinesin Eg5 (Mayer et al., 1999). Subsequent studies using monastrol demonstrated the importance of Eg5 in normal spindle body formation (Kapoor et al., 2000; Kapoor and Mitchison, 2001). Blebbistatin is another small molecule probe that resulted from a basic biology-driven screen. Identified as a non-muscle myosin II inhibitor, blebbistatin has been critical in the investigation of cleavage furrow formation during mitosis and cytokinetic contractile ring assembly (Straight et al., 2003). Likewise, new assays have been developed to screen for inhibitors of the Wnt/β-catenin pathway, which is a major developmental biology signaling pathway. In a creative fusion of a small molecule and RNA interference (RNAi) screening, the Moon lab recently identified Bruton’s Tyrosine Kinase (BTK) as an Wnt/β-catenin pathway inhibitor (James et al., 2009). These targeted proactive approaches to small molecule probe identification contrast with the more serendipitous nature of how traditional chemical biology probes were discovered and continues to have a major impact on biology through the identification of useful research reagents.
Chemical Genetics: An Unrealized Dream
This growth in novel bioassays has generated many new research tools as well as excitement about the potential for small molecule-based biological discovery, in general. Indeed, use of a combination of new bioassay development and compound library screening to identify novel bioactive molecules have become commonplace on campuses today (Sachinidis et al., 2008; Soderholm et al., 2006; Specht and Shokat, 2002; Wheeler and Brandli, 2009). Many of these new screens are phenotype-based, i.e., assays screening small molecule-induced changes in cellular context or even in whole organisms such as zebrafish, Drosophila, or nematodes. Since these screens score for a change in phenotype without regard a priori to a given target protein, this approach has been compared to a traditional ‘forward’ genetic screen, leading to the sobriquet ‘chemical genetics’ to describe these small molecule screens. However semantically appealing this analogy with traditional genetic screening may be, it is grossly misleading; whereas each gene has an equivalent chance of being mutated in a traditional genetic screen (ignoring mutagenic hotspots for the sake of argument), the amount of ‘proteomic space’ that a chemical genetics approach can reach using small molecule perturbagens is considerably smaller. Put another way, the oft-stated goal of ‘a small molecule inhibitor for every protein’ has yet to be realized.
Today’s Challenge: Targeting the Undruggable Proteome
Both academic and pharmaceutical screening efforts have been inherently limited in the types of proteins that are targeted using small molecules, i.e., the segment of proteome that is characterized by the presence of well-defined small molecule binding pockets such as ion channels, nuclear receptors, GPCRs or enzymes (Overington et al., 2006). Collectively, these protein families are but a fraction of the entire proteome and, thus, this exclusive focus leaves as ‘undruggable’ many other types of proteins that cannot be controlled using small molecules such as transcription factors, non-enzymatic proteins, regulatory/scaffolding proteins, etc. (Arakaki et al., 2006; Verdine and Walensky, 2007). The challenge, therefore, is how can one develop a methodology that targets this ‘undruggable’ proteome? Is it possible to make every protein equally susceptible to small molecule control? I argue that a true chemical genetic screen will require a small molecule library that targets both ‘traditional’ drug targets as well as the 80% of the proteome lacking a catalytic site or a small molecule binding site that controls protein function when occupied.
Wanted: Controlling Protein Function Irrespective of Protein Class
Given the incomplete coverage of the proteome by current compound libraries, new methodologies are needed to control protein function using small molecules. One possible solution is to employ the cell’s own quality control mechanisms to induce the degradation of targeted proteins and thus modulate intracellular protein concentrations. For example, a recent report described the use of Heat Shock Cognate protein HSC70 peptide binding motifs to recruit proteins to the lysosome for degradation (Figure 1A) (Bauer et al., 2010). By harnessing HSC70, a chaperonin protein responsible for either the refolding or targeted degradation of misfolded proteins, this approach selectively induced the degradation of mutant huntingtin, the protein responsible for Huntington’s disease and, moreover, ameliorated disease in an animal model.
Figure 1. Strategies for employing cellular protein degradation machinery to control intracellular protein levels.
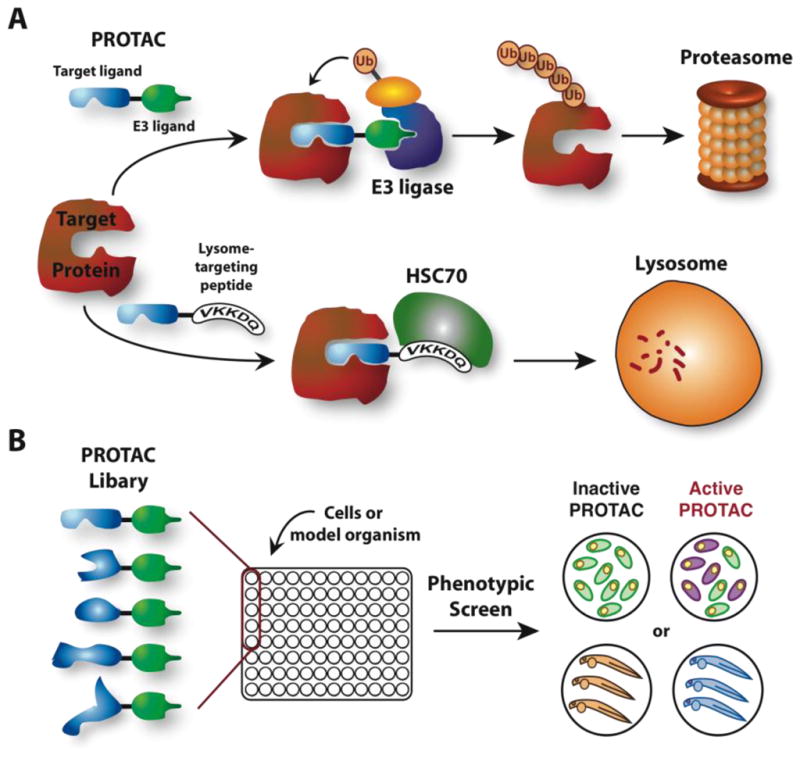
A) Schematic of Inducing Protein Degradation via Recruitment to the Proteasome or Lysosome. B) Design of a PROTAC-based Library Targeting Protein Function Independently of Protein Class
A similar approach to employ cellular protein degradation machinery to control intracellular protein levels was developed in my lab, in collaboration with Ray Deshaies (CalTech). Whereas the approach described above recruits targeted proteins to the lysosome for degradation, Proteolysis Targeting Chimeras (PROTACs) recruit targeted proteins to E3 ubiquitin ligases (Rodriguez-Gonzalez et al., 2008; Schneekloth and Crews, 2005) as a first step in their induced degradation. E3 ubiquitin ligases, together with E2 conjugating enzymes, are responsible for coupling the 76 amino acid tag ubiquitin to lysine ε-amino groups on the surface of proteins, thus targeting them for degradation by the major intracellular proteolytic complex, the 26S proteasome. As heterobifunctional compounds composed of a target protein binding ligand and an E3 ubiquitin ligase ligand (Figure 1), PROTACs induce proteasome-mediated degradation of selected proteins via their recruitment to E3 ubiquitin ligase and subsequent ubiquitination (Bargagna-Mohan et al., 2005). In several proof of concept studies, this methodology has been shown to induce intracellular protein degradation with greater temporal and dosage control than that offered by RNAi-mediated gene knockdown (Puppala et al., 2008; Sakamoto et al., 2003; Schneekloth et al., 2004). In addition, unlike the lysosomal targeting approach using peptidic HSC70 binding motifs, it is possible to target proteins for degradation using a non-peptidic, ‘all small molecule’-based PROTAC (Itoh et al., 2010; Schneekloth et al., 2008).
Affinity-based HTS screens: An Unbiased Approach to Identify a Ligand for Every Protein
Whether targeting proteins for proteasomal- or lysosomal-mediated degradation, these approaches for controlling intracellular protein levels face the same major challenge, namely, the identification of ligands for proteins to be targeted for degradation. Fortunately, new advances in high-throughput affinity-based screening are helping to address this challenge (Zhu and Cuozzo, 2009). Unlike common functional assay based HTS, which identifies compounds based on their ability to elicit a biological consequence upon binding, affinity-based HTS instead focuses only on identifying compounds that bind their protein targets, irrespective of their protein class. While several low and medium throughput affinity strategies are available, (e.g., calorimetry-based, surface plasmon resonance (SPR)-based, NMR/X-ray structure-based, mass spectrometry-based, small molecule microarrays), newer methodologies offer the ability to screen readily hundreds of thousands to millions of compounds (Zhu and Cuozzo, 2009). For example, several related DNA-tagged small molecule libraries (Gartner et al., 2004; Melkko et al., 2007) have been used to identify protein ligands from libraries as large as 108 compounds (Clark et al., 2009). As these technologies continue to mature, low and medium throughput affinity based approaches should become more amenable to HTS. Hence, functionally unbiased affinity-based HTS screens hold the promise of identifying a ligand to each protein in the proteome. Although ligands identified for each protein by these methods may not have an inherent biological activity, when they are coupled to an approach such as PROTACs, they will ultimately allow for the elusive all-encompassing chemical genetic screen.
The Molecules of My Dreams: A Truly Comprehensive Small Molecule Library
In theory, coupling protein ligands identified in affinity-based screens to a HSC70 ligand or an E3 ligase ligand (e.g., the MDM2 binding compound nutlin) would generate compounds capable of targeting any desired protein for intracellular degradation (Schneekloth et al., 2008). While all of the technology is in place to generate a degradation-inducing compound for every protein, I question if this ‘reverse’ genetic approach is the best? Instead of starting with individual proteins and identifying novel targeting ligands to them for incorporation into such molecules, why not take a ‘forward’ genetic approach to score a cellular phenotype based on loss of protein function? A library of PROTACs could be generated in which all compounds possess the same E3 ubiquitin ligase ligand, but each is coupled to a different chemical diversity element (Figure 1B). Such a naïve PROTAC library of sufficient size might be capable of binding to (and inducing the degradation of) every protein within the proteome irrespective of protein function or class. However, there are several limitations to this strategy. First, some proteins are naturally unstable and thus, would be difficult to control via a PROTAC. Likewise, it may be difficult to find a small molecule ligand capable of binding to a target protein with the requisite affinity and specificity for use in a PROTAC. Despite these potential limitations, the generation of a PROTAC-based compound collection would represent the first truly comprehensive small molecule library that could be used to perform proteome-wide chemical genetic screens for induction or modulation of a given cellular phenotype.
The development of new research strategies has permitted small molecule bioassays and screens to evolve significantly in the past two decades. This has enabled researchers to investigate more basic biological phenomena beyond those areas mandated by clinical needs. Nevertheless, much remains to be done before the dream of a small molecule perturbagen for every protein is realized. In my opinion, the largest unmet need in bioprobe development today is the ability to modulate protein function independent of protein class. Although several current technologies offer possible solutions to this challenge, chemical genetics will not truly be on par with traditional genetics until this challenge is overcome.
Acknowledgments
I thank members of my lab for their thoughtful comments on this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arakaki AK, Tian W, Skolnick J. High precision multi-genome scale reannotation of enzyme function by EFICAz. BMC Genomics. 2006;7:315. doi: 10.1186/1471-2164-7-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargagna-Mohan P, Baek SH, Lee H, Kim K, Mohan R. Use of PROTACS as molecular probes of angiogenesis. Bioorg Med Chem Lett. 2005;15:2724–2727. doi: 10.1016/j.bmcl.2005.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer PO, Goswami A, Wong HK, Okuno M, Kurosawa M, Yamada M, Miyazaki H, Matsumoto G, Kino Y, Nagai Y, et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol. 2010;28:256–263. doi: 10.1038/nbt.1608. [DOI] [PubMed] [Google Scholar]
- Cardenas ME, Sanfridson A, Cutler NS, Heitman J. Signal-transduction cascades as targets for therapeutic intervention by natural products. Trends Biotechnol. 1998;16:427–433. doi: 10.1016/s0167-7799(98)01239-6. [DOI] [PubMed] [Google Scholar]
- Clark MA, Acharya RA, Arico-Muendel CC, Belyanskaya SL, Benjamin DR, Carlson NR, Centrella PA, Chiu CH, Creaser SP, Cuozzo JW, et al. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat Chem Biol. 2009;5:647–654. doi: 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR. DNA-templated organic synthesis and selection of a library of macrocycles. Science. 2004;305:1601–1605. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein Knockdown Using Methyl Bestatin-Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J Am Chem Soc. 2010 doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- James RG, Biechele TL, Conrad WH, Camp ND, Fass DM, Major MB, Sommer K, Yi X, Roberts BS, Cleary MA, et al. Bruton’s tyrosine kinase revealed as a negative regulator of Wnt-beta-catenin signaling. Sci Signal. 2009;2:ra25. doi: 10.1126/scisignal.2000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallander LS, Lu Q, Chen W, Tomaszek T, Yang G, Tew D, Meek TD, Hofmann GA, Schulz-Pritchard CK, Smith WW, et al. 4-Aryl-1,2,3-triazole: a novel template for a reversible methionine aminopeptidase 2 inhibitor, optimized to inhibit angiogenesis in vivo. J Med Chem. 2005;48:5644–5647. doi: 10.1021/jm050408c. [DOI] [PubMed] [Google Scholar]
- Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol. 2000;150:975–988. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor TM, Mitchison TJ. Eg5 is static in bipolar spindles relative to tubulin: evidence for a static spindle matrix. J Cell Biol. 2001;154:1125–1133. doi: 10.1083/jcb.200106011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsanakis KD, Owen C, Zoumpourlis V. JNK and ERK signaling pathways in multistage mouse carcinogenesis: studies in the inhibition of signaling cascades as a means to understand their in vivo biological role. Anticancer Res. 2002;22:755–759. [PubMed] [Google Scholar]
- Kawasumi M, Nghiem P. Chemical genetics: elucidating biological systems with small-molecule compounds. J Invest Dermatol. 2007;127:1577–1584. doi: 10.1038/sj.jid.5700853. [DOI] [PubMed] [Google Scholar]
- Lennartz MR. Phospholipases and phagocytosis: the role of phospholipid-derived second messengers in phagocytosis. Int J Biochem Cell Biol. 1999;31:415–430. doi: 10.1016/s1357-2725(98)00108-3. [DOI] [PubMed] [Google Scholar]
- Marino JP, Jr, Fisher PW, Hofmann GA, Kirkpatrick RB, Janson CA, Johnson RK, Ma C, Mattern M, Meek TD, Ryan MD, et al. Highly potent inhibitors of methionine aminopeptidase-2 based on a 1,2,4-triazole pharmacophore. J Med Chem. 2007;50:3777–3785. doi: 10.1021/jm061182w. [DOI] [PubMed] [Google Scholar]
- Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- Melkko S, Dumelin CE, Scheuermann J, Neri D. Lead discovery by DNA-encoded chemical libraries. Drug Discov Today. 2007;12:465–471. doi: 10.1016/j.drudis.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Munos B. Lessons from 60 years of pharmaceutical innovation. Nat Rev Drug Discov. 2009;8:959–968. doi: 10.1038/nrd2961. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Yano H, Matsuda Y. Novel functions of phosphatidylinositol 3-kinase in terminally differentiated cells. Cell Signal. 1995;7:545–557. doi: 10.1016/0898-6568(95)00033-l. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Puppala D, Lee H, Kim KB, Swanson HI. Development of an aryl hydrocarbon receptor antagonist using the proteolysis-targeting chimeric molecules approach: a potential tool for chemoprevention. Mol Pharmacol. 2008;73:1064–1071. doi: 10.1124/mol.107.040840. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, Deshaies RJ, Sakamoto KM. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201–7211. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachinidis A, Sotiriadou I, Seelig B, Berkessel A, Hescheler J. A chemical genetics approach for specific differentiation of stem cells to somatic cells: a new promising therapeutical approach. Comb Chem High Throughput Screen. 2008;11:70–82. doi: 10.2174/138620708783398322. [DOI] [PubMed] [Google Scholar]
- Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, Deshaies RJ. Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation. Mol Cell Proteomics. 2003;2:1350–1358. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- Saporito MS, Hudkins RL, Maroney AC. Discovery of CEP-1347/KT-7515, an inhibitor of the JNK/SAPK pathway for the treatment of neurodegenerative diseases. Prog Med Chem. 2002;40:23–62. doi: 10.1016/s0079-6468(08)70081-x. [DOI] [PubMed] [Google Scholar]
- Schlueter PJ, Peterson RT. Systematizing serendipity for cardiovascular drug discovery. Circulation. 2009;120:255–263. doi: 10.1161/CIRCULATIONAHA.108.824177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett. 2008;18:5904–5908. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth JS, Jr, Crews CM. Chemical approaches to controlling intracellular protein degradation. Chembiochem. 2005;6:40–46. doi: 10.1002/cbic.200400274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth JS, Jr, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. Journal of the American Chemical Society. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc Natl Acad Sci U S A. 1997;94:6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siragusa M, Katare R, Meloni M, Damilano F, Hirsch E, Emanueli C, Madeddu P. Involvement of phosphoinositide 3-kinase gamma in angiogenesis and healing of experimental myocardial infarction in mice. Circ Res. 2010;106:757–768. doi: 10.1161/CIRCRESAHA.109.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderholm J, Uehara-Bingen M, Weis K, Heald R. Challenges facing the biologist doing chemical genetics. Nat Chem Biol. 2006;2:55–58. doi: 10.1038/nchembio0206-55. [DOI] [PubMed] [Google Scholar]
- Soond DR, Bjorgo E, Moltu K, Dale VQ, Patton DT, Torgersen KM, Galleway F, Twomey B, Clark J, Gaston JS, et al. PI3K p110{delta} regulates T-cell cytokine production during primary and secondary immune responses in mice and humans. Blood. 2010;115:2203–2213. doi: 10.1182/blood-2009-07-232330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht KM, Shokat KM. The emerging power of chemical genetics. Curr Opin Cell Biol. 2002;14:155–159. doi: 10.1016/s0955-0674(02)00317-4. [DOI] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Sturgeon SA, Jones C, Angus JA, Wright CE. Advantages of a selective beta-isoform phosphoinositide 3-kinase antagonist, an anti-thrombotic agent devoid of other cardiovascular actions in the rat. Eur J Pharmacol. 2008;587:209–215. doi: 10.1016/j.ejphar.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Takahashi-Yanaga F, Sasaguri T. Drug development targeting the glycogen synthase kinase-3beta (GSK-3beta)-mediated signal transduction pathway: inhibitors of the Wnt/beta-catenin signaling pathway as novel anticancer drugs. J Pharmacol Sci. 2009;109:179–183. doi: 10.1254/jphs.08r28fm. [DOI] [PubMed] [Google Scholar]
- Ui M, Okada T, Hazeki K, Hazeki O. Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem Sci. 1995;20:303–307. doi: 10.1016/s0968-0004(00)89056-8. [DOI] [PubMed] [Google Scholar]
- Verdine GL, Walensky LD. The challenge of drugging undruggable targets in cancer: lessons learned from targeting BCL-2 family members. Clin Cancer Res. 2007;13:7264–7270. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
- Wang J, Knight ZA, Fiedler D, Williams O, Shokat KM, Pearce D. Activity of the p110-alpha subunit of phosphatidylinositol-3-kinase is required for activation of epithelial sodium transport. Am J Physiol Renal Physiol. 2008a;295:F843–850. doi: 10.1152/ajprenal.90348.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tucker LA, Stavropoulos J, Zhang Q, Wang YC, Bukofzer G, Niquette A, Meulbroek JA, Barnes DM, Shen J, et al. Correlation of tumor growth suppression and methionine aminopetidase-2 activity blockade using an orally active inhibitor. Proc Natl Acad Sci U S A. 2008b;105:1838–1843. doi: 10.1073/pnas.0708766105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Ma C, Mao Z, Li M. JNK inhibition as a potential strategy in treating Parkinson’s disease. Drug News Perspect. 2004;17:646–654. doi: 10.1358/dnp.2004.17.10.873916. [DOI] [PubMed] [Google Scholar]
- Wheeler GN, Brandli AW. Simple vertebrate models for chemical genetics and drug discovery screens: lessons from zebrafish and Xenopus. Dev Dyn. 2009;238:1287–1308. doi: 10.1002/dvdy.21967. [DOI] [PubMed] [Google Scholar]
- Wu X, Schultz PG. Synthesis at the interface of chemistry and biology. J Am Chem Soc. 2009;131:12497–12515. doi: 10.1021/ja9026067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Furumai R, Nishiyama M, Komatsu Y, Nishino N, Horinouchi S. Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemother Pharmacol. 2001;48(Suppl 1):S20–26. doi: 10.1007/s002800100300. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Cuozzo J. Review article: high-throughput affinity-based technologies for small-molecule drug discovery. J Biomol Screen. 2009;14:1157–1164. doi: 10.1177/1087057109350114. [DOI] [PubMed] [Google Scholar]