

Abstract
Quercetin (Qu) is currently being investigated as a chemopreventive agent for a number of cancers including non-melanoma skin cancer induced by ultraviolet (UV) light. We previously reported that Qu degradation has important consequences on signaling and cell biology. In the current study, we report that Qu induces c-Fos mRNA and protein expression through activation of p38 and CREB, and Qu potentiates UVB-induced c-Fos expression. Inclusion of ascorbic acid (AA) in cell culture medium stabilizes Qu and completely prevents both Qu- and UVB- induced p38 and CREB activation, leading to a blockade of c-fos gene expression through reduced CREB/CRE binding. AA stabilizes c-Fos mRNA, increasing steady state levels even when c-fos gene expression is suppressed, but this has no effect on c-Fos protein levels in either mock- or UVB-irradiated cells. We report that Qu blocks mTOR signaling and inhibits c-Fos protein expression directly through this mechanism, since co-treatment with Qu and AA resulted in complete suppression of UVB-induced c-Fos protein expression even in the presence of significantly increased mRNA levels. We further confirmed that this was not due to increased protein turnover since inhibition of proteasome activity with MG-132 did not raise c-Fos protein levels in Qu+AA treated cells. Together, these data indicate that while Qu has been reported to have some beneficial properties as a chemopreventive agent, it is also capable of inducing c-fos expression, a cellular event important for the promotion phase of tumor development, if it is not stabilized.
Keywords: quercetin, c-Fos, ultraviolet light, p38, mTOR
Introduction
Currently, there is a major focus on the use of natural products as chemopreventive agents for non-melanoma skin cancer (NMSC) and other types of cancer because of their anti-oxidant and anti-inflammatory properties in addition to their potential to inhibit numerous signaling pathways involved in cell proliferation, transformation, migration, and survival (1–3). Flavonoids represent one of the most actively studied classes of molecules for their potential to prevent cancer. However, the cellular effects of these compounds are often very complicated due to the general lack of specificity for any particular intracellular target, and the end effect on cell fate is often the combination of multiple cellular activities. In the current study, we investigated some of the cellular effects of quercetin (Qu; 3,3’,4’,5,7-pentahydroxyflavone) on ultraviolet B (UVB)-induced signaling in human keratinocytes. Our original interest in Qu was with regard to the compound’s known phosphatidylinositide 3-kinase (PI3-K) inhibitory activity, and we initially reported that a concentration of 50 µM was required for consistent suppression of UVB-induced PI3-K activation in HaCaT human keratinocytes due to the cell-independent rapid degradation of Qu in aqueous cell culture medium (4). At this concentration, we demonstrated that Qu actually displays pro-oxidant activity, an effect that could be inhibited by including 1 mM ascorbic acid (AA) in the growth medium to stabilize Qu. However, this reduced the pro-apoptotic effect of Qu, leading to the interpretation that the generation of reactive species during Qu degradation could be a beneficial action through increased killing of initiated cells. On the contrary, our current findings suggest that there are significant cellular consequences to Qu degradation that could lead to elevated promotion of the initiated cells that survive Qu treatment, which raises questions regarding the chemopreventive efficacy in UV-induced skin cancer.
Chronic exposure to ultraviolet light is the primary cause of NMSC, the most common type of cancer in the U.S. (5). UVB (280–320 nm) comprises between 1% and 10% of the total UV light that reaches the Earth’s surface. UVB is known to act as a complete carcinogen, and our laboratory and others have demonstrated that UVB-induced activation of the transcription factor activator protein-1 (AP-1) plays a functional role in UVB-induced tumor promotion (6). AP-1 stimulates cell proliferation through regulation of cell cycle protein expression [reviewed in (7)] and also controls cellular transformation since inhibition of AP-1 has been shown to block the effect of tumor promoting agents (6, 8–12).
The AP-1 transcription factor complex is comprised of dimers of Jun and Fos protein family members. The exact constituents depend on the stimulus or the physiological condition, and the different dimer pairs can activate different sets of genes [reviewed in (13)]. In HaCaT keratinocytes, the UVB-activated AP-1 complex is composed of c-Fos and JunD (14). While JunD expression is unaffected by UVB irradiation, c-Fos levels have been shown to increase with UVB treatment in a manner that correlates with AP-1 activation, suggesting that alterations in c-Fos protein levels are a major driving factor behind the level of AP-1 activity. In addition, c-Fos expression has been shown to be required for malignant progression of skin tumors (15). Increased cellular oxidative stress can lead to p38 activation and ultimately c-fos gene expression by stimulating cyclic AMP response element binding (CREB) protein binding to the c-fos promoter (16–18). In the current study we worked to determine the effect of Qu treatment on c-Fos protein levels in UVB-irradiated HaCaT keratinocytes. We report that Qu actually increased c-Fos protein levels and potentiated the UVB response. This effect could be prevented completely by stabilizing Qu with AA, since treatment with Qu+AA completely inhibited the UVB-induced increase in c-fos promoter activity and also inhibited protein synthesis by suppressing mTOR signaling. These findings demonstrate an important cellular effect of Qu resulting from its pro-oxidant activity. Although we previously reported that Qu treatment may be beneficial by killing initiated cells (4), failure to stabilize Qu could have detrimental effects that limit any chemopreventive efficacy of the compound. Even stabilization of Qu with AA was shown to be a transient event, which might not prevent the tumor promoting event of elevated c-Fos expression that occurs as Qu degrades. Importantly, these findings highlight some of the complexities of working with natural products and suggest that some of these compounds may require a significant amount of characterization to validate them as chemopreventive agents and avoid deleterious side effects.
Materials and Methods
Materials
Quercetin dihydrate and ascorbic acid were purchased from Sigma-Aldrich (St. Louis, MO.). Phospho-specific antibodies for p38 (T180/Y182)), CREB (S133), Akt (S473), mTOR (S2448), p70S6K (T389), and 4EBP (S65) were from Cell Signaling Technology (Danvers, MA.). c-Fos antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Real-time PCR probes (c-Fos: Hs00170630_m1 and GAPDH: Hs99999905_m1) and TaqMan reagents were purchased from Applied Biosystems (Foster City, CA). Actinomycin D and MG-132 were from EMD Biosciences/Calbiochem (San Diego, CA).
Cells
The human keratinocyte cell line, HaCaT, was established from cells obtained from adult sun damaged skin and have been described previously (19–21). HaCaT cells contain UV-signature mutations and express mutant dysfunctional p53 and a defective NF-κB signaling pathway, which are common findings in UV-initiated keratinocytes in human skin. However, these cells maintain p38, PI3-K and AP-1 signaling pathway functionality compared to normal human keratinocytes and were thus chosen as an appropriate cell line to study UVB-induced c-Fos expression and upstream signaling events. FL-30 cells were developed from HaCaT cells and stably express firefly luciferase driven by a full length c-fos promoter element (14). The cells were cultured in Dulbecco’s modified Eagle’s Medium (DMEM) with 10% fetal bovine serum and 100 units/ml penicillin/streptomycin at 37°C and in 5% CO2. The cells were cultured to 80–90% confluence and maintained in serum-free DMEM (SFM) for 24 h prior to UVB exposure.
UVB irradiation of HaCaT cells
HaCaT cells were pretreated with quercetin and/or ascorbate for 1 hr prior to UVB irradiation. The concentration of 50 µM quercetin was chosen based on our previously published study (4) and the fact that this concentration is feasible for chemoprevention studies using topical Qu. After incubation, HaCaTs were washed once in PBS and irradiated with a dose of 250 J/m2 using a bank of two SF20 UVB lamps (National Biological Corp.) providing a peak emission of 313 nm. Control cells were treated in the same manner and mock irradiated. Following irradiation, HaCaT cells were again washed with PBS and returned to DMEM containing appropriate drug treatments. In experiments using MG-132, there was no initial pretreatment and MG-132 was only used following exposure to UVB.
Western blotting
Cells were lysed in RIPA buffer and protein concentration was determined as previously described (4). A total of 40 µg protein was resolved by SDS-PAGE and transferred to a PVDF membrane. Membranes were blocked in Tris buffered saline containing 0.1% Tween-20 (TBST) and either 5% nonfat dry milk or 5% BSA (for phospho-specific antibodies). After washing in TBST, membranes were incubated with HRP-conjugated secondary antibodies and then washed extensively in TBST. Antigen-antibody complexes were detected using Amersham ECL Detection Reagent (GE Healthcare, Buckinghamshire, UK). Densitometric analysis was performed using NIH ImageJ software and density values were normalized to the untreated control, which was assigned a value of 1.0.
c-fos-Luciferase assay
FL-30 cells were treated and UVB-irradiated in triplicate for each independent experiment. Six hours after exposure to UVB, cells were lysed and a total of 20 µg protein per sample replicate were assayed for luciferase activity according to the manufacturer’s instructions for the Luciferase Assay System (Promega, Madison, WI) using a TD 20/20 luminometer (Turner Designs, Sunnyvale, CA). The experiment triplicates were averaged and the means from each independent experiment were subsequently averaged and analyzed by Student’s t test for statistical significance.
RNA extraction and real-time PCR analysis
For the measurement of steady state RNA levels, cells were treated as indicated above. In RNA stability assays, cells were pretreated for 30 min with 5µg/ml actinomycin D prior to treatment with Qu and AA for the indicated times. Cellular RNA was isolated from treated HaCaT cells by phenol-chloroform extraction using the Ambion ToTally RNA Kit (Applied Biosystems, Foster City, CA) according to manufacturer’s instructions. Extracted RNA was treated with DNase using the Ambion DNA-free Kit and purified using the RNeasy MinElute Cleanup Kit from Qiagen (Valencia, CA). Purified RNA was then reverse transcribed using the High Capacity cDNA Reverse Transcription Kit from Applied Biosystems. Real-time PCR was performed using an ABI PRISM 7700 Sequence Detection System and TaqMan probes specific for human c-fos and GAPDH. Relative expression was calculated to be 2−ΔΔCt, where ΔCt is equal to the difference in cycle number (Ct) between c-Fos and GAPDH for each sample and ΔΔCt is equal to the difference between ΔCt for each treatment and the untreated control. Each individual assay was performed in triplicate for each gene product and a total of 3 independent experiments were performed.
Electromobility shift assay
After treatment and UVB-irradiation, nuclear extracts were isolated from HaCaT cells as described previously (22). The following sequences derived from the human c-fos promoter containing the CRE were used to generate double stranded oligos for EMSA analysis: CRE1: 5’-GAGCCCGTGACGTTTACACT-3’, and CRE2: 5’-TGAGTGTAAACGTCACGG-3’. They were annealed and 5’-overhangs were labeled with [32P]dCTP (Perkin Elmer, Waltham, MA). For gel shift assays, 5 µg of nuclear protein extracts were incubated at room temperature for 20 min with a mixture containing 10 mM Hepes (pH 7.9), 0.1 mM EDTA, 50 mM KCl, 2.5 mM DTT, 10% glycerol, 0.5% Triton X-100, and 0.05 mg/ml polydIdC. Labeled probe was added and the reactions were incubated at room temperature for another 30 min. Reactions were then fractionated on a non-denaturing 6% polyacrylamide gel in 0.25 X Tris borate EDTA. The gels were dried and visualized with an MD Storm phosphorimager (Molecular Dynamics, Sunnyvale, CA).
Results
Quercetin induces p38 and CREB phosphorylation leading to c-fos promoter activity unless stabilized by ascorbic acid
We previously demonstrated that quercetin (Qu) is unstable in DMEM, but can be stabilized for several hours by inclusion of 1 mM ascorbic acid (AA) in the medium (4). Western analysis of protein from HaCaT keratinocytes indicated that 250 J/m2 UVB induced p38 phosphorylation (Figure 1A). Treatment with 50 µM Qu increased p38 phosphorylation in both mock irradiated and UVB-irradiated HaCaT cells compared to the controls. When 1 mM AA was included in the growth medium, Qu treated cells displayed no increase in phospho-p38 levels, and the response to UVB was completely lost. CREB, a substrate downstream of p38 signaling, displayed a nearly identical response to UVB and Qu as p38, where there were increases in CREB phosphorylation with both UVB and Qu alone and in combination and a significant reduction in the UVB response when cells were treated in the presence of AA (Figure 1B). Total p38 and CREB levels were unaffected by Qu, AA or UVB treatment.
Figure 1. Quercetin induces p38 and CREB phosphorylation unless stabilized by ascorbic acid.
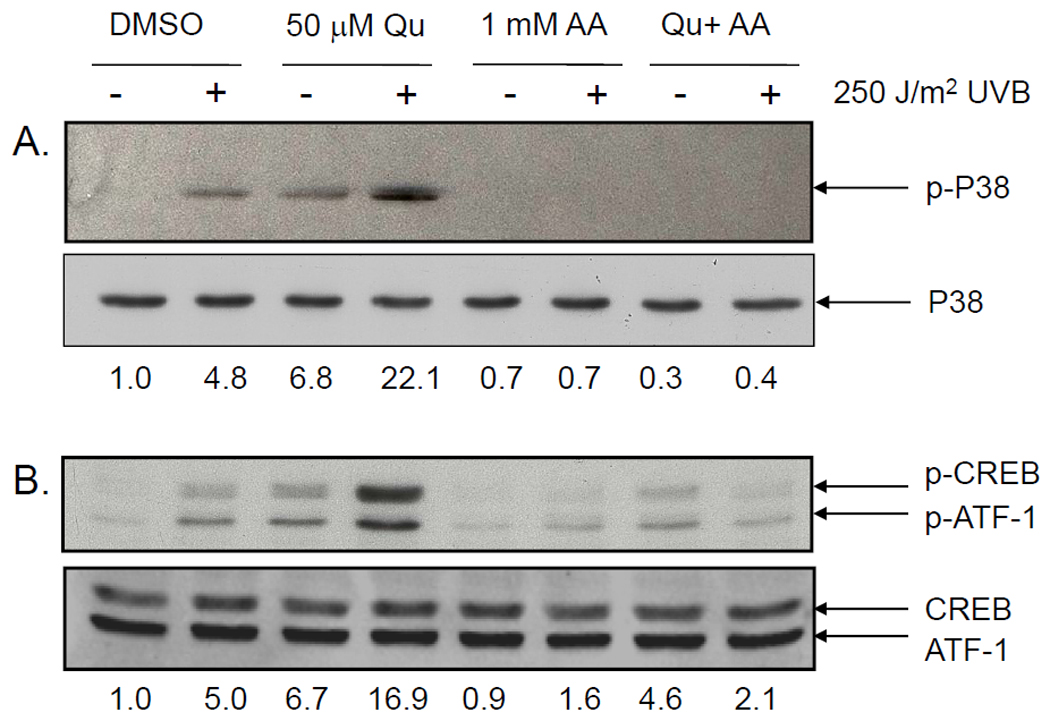
HaCaT cells were treated with 50 µM Qu for 1 hr prior to exposure to 250 J/m2 UVB irradiation and for 2 hr post UVB. Cells were either treated in normal DMEM or in DMEM supplemented with 1 mM ascorbic acid. (A) p38 phosphorylation was induced by UVB and Qu alone, and the response was potentiated in cells treated with both UVB and Qu. Treatment with 1 mM ascorbic acid completely inhibited the UVB- and Qu-induced increase in p38 phosphorylation. (B) CREB phosphorylation in UVB-irradiated and Qu-treated HaCaT cells was similar to that seen with p38 phosphorylation. Both Qu and UVB treatment individually increased CREB phosphorylation and the combined treatment potentiated the response. Again, AA significantly reduced CREB phosphorylation in response to both UVB and Qu and prevented the potentiated response seen in cells treated with both Qu and UVB. The antibodies used for both p-CREB and CREB recognize p-ATF-1 and ATF-1, respectively, as well. Relative band densities are represented numerically immediately below each set of blots.
Transcriptional activation of c-fos is primarily driven by binding of CREB to the cyclic AMP response element (CRE) in the promoter region (17). We examined the effects of Qu on c-fos promoter activity using FL-30 cells, a HaCaT cell line that has been stably transfected with a firefly luciferase reporter construct where expression of luciferase is driven by a full length human c-fos promoter. Irradiation of FL-30 cells with 250 J/m2 UVB resulted in a 5.1-fold increase in luciferase expression over control, while Qu treatment alone induced a 2.7-fold increase in luciferase expression (Figure 2A). Qu treatment of UVB-irradiated HaCaT cells significantly reduced luciferase expression compared to UVB alone, although luciferase levels were still significantly increased over the untreated control (2.1-fold over control). In this assay system, luciferase expression is dependent on both gene induction and protein translation, and the potential for Qu to act on both of these pathways will be addressed below.
Figure 2. Quercetin stimulates c-fos promoter activity and CRE binding.
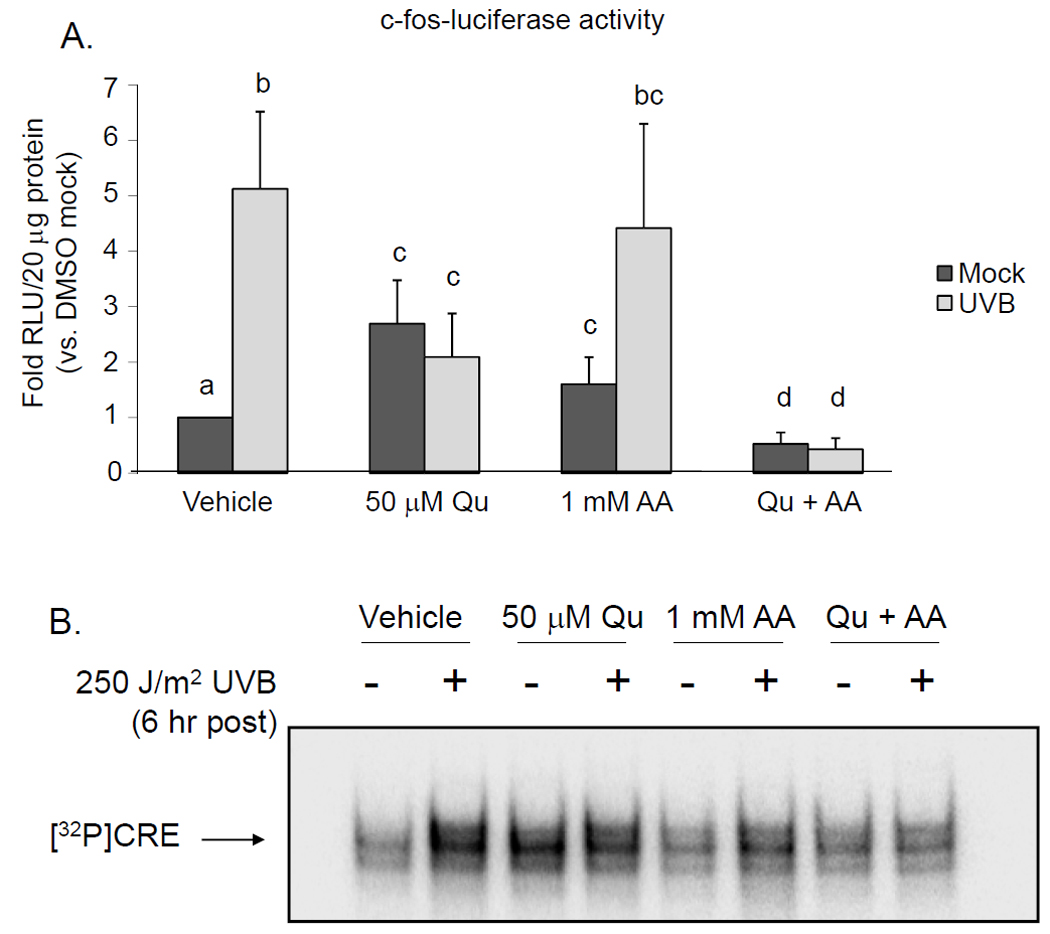
Cells were treated with 50 µM Qu +/− 1 mM AA for 1 hr prior to irradiation with 250 J/m2 UVB and following irradiation for 6 hr. (A) UVB induced a 5.1-fold increase in c-fos-luciferase activity over untreated controls. Treatment with Qu induced a 2.7-fold increase, but also inhibited UVBinduced c-fos-luciferase activity by 74%. AA treatment significantly increased the basal level of c-fos-luciferase activity by 1.6-fold, but did not affect the response to UVB. However, supplementation with AA and treatment with Qu suppressed basal c-fos-luciferase activity and completely inhibited the response to UVB. Data represent the mean from 4 independent experiments and statistically significant differences between groups (p < 0.05) are indicated by labels containing different letters. Any two treatment conditions with a label containing the same letter are not significantly different. (B) CRE binding was assessed using nuclear lysates from Qu-treated HaCaT cells and a double stranded oligo containing the CREB binding sequence. CRE binding was increased in lysates extracted from UVB-irradiated cells and from Qu-treated cells. Qu apparently did not affect CRE binding induced by UVB, as it was not different from Qu alone. AA had little to no effect on CRE binding, but Qu+AA completely suppressed basal and UVB-induced CRE binding. These data are representative of 3 independently performed experiments.
The effects of Qu on the c-fos promoter were further analyzed by performing an EMSA using probe containing the CRE from the c-fos promoter. We determined that UVB irradiation, treatment with Qu alone, and combining Qu treatment with UVB irradiation all increased CRE binding and that CRE binding was highest in nuclear extracts from UVB-irradiated HaCaT cells (Figure 2B). The level of CRE binding that resulted from these treatments followed a similar trend to that seen in the c-fos-luciferase assay in Figure 2A. Similar to p38 and CREB phosphorylation, inclusion of AA in the medium during the time of treatment abrogated the effect of Qu on CRE binding. Taken together, these data indicate that Qu induces c-fos promoter activity to levels greater than that seen in untreated cells, but partially blocks UVB-induction of the c-fos promoter. Furthermore, these results suggest that co-treatment with Qu and AA (Qu+AA) suppresses both basal and UVB-induced promoter activity.
Quercetin increases c-fos mRNA, but stabilization with ascorbic acid prevents UVB-induced c-Fos mRNA expression
Total cellular RNA was isolated from Qu- and UVB-treated HaCaT cells 2 hr after exposure to UVB, and reverse transcribed cDNA was analyzed by real-time PCR with a probe specific for human c-fos. UVB-irradiated cells displayed an 8.4-fold increase in c-fos message compared to mock irradiated controls (Figure 3A). Qu treatment also increased the level of c-fos mRNA by 50-fold over control, a level much greater than that induced by UVB and potentiated the UVB response (126-fold over control). Interestingly, AA supplementation also increased steady state mRNA levels in both mock- and UVB-irradiated HaCaT cells, a finding that differed from the effect of AA on c-fos promoter activity. Although this increase was not statistically significant compared to control levels, treatment with both Qu+AA did in fact result in a moderate increase.
Figure 3. Quercetin treatment increases c-fos mRNA expression and potentiates UV-Binduced c-Fos protein production.
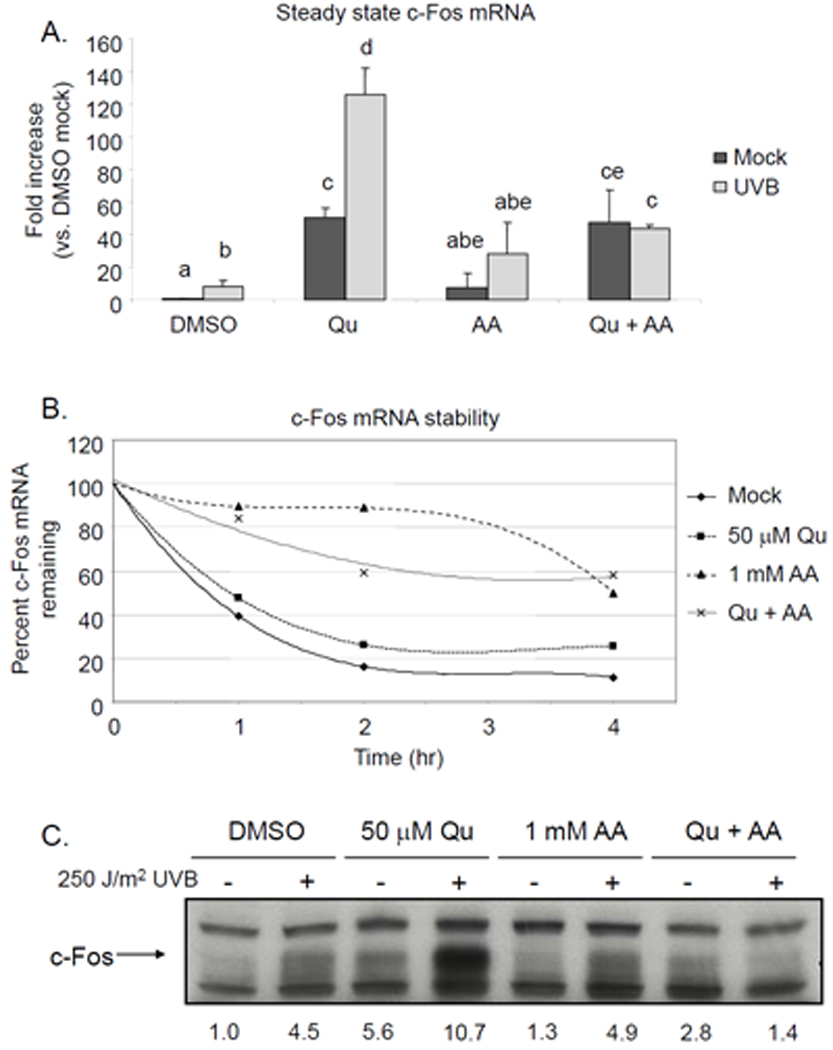
(A) HaCaT cells were pretreated with 50 µM Qu +/− 1 mM AA 1 hr prior to irradiation with 250 J/m2 UVB and for 2 hr post-UVB, at which point RNA was extracted and purified. Real time RT-PCR was performed using probes specific for c-fos and GAPDH. The fold increase of steady state c-fos mRNA was determined as indicated in Materials and Methods. UVB induced an 8.4-fold increase in c-fos mRNA and Qu alone induced a 50-fold increase. Qu treatment in UVB-irradiated cells resulted in a potentiated response, where the induction of c-fos mRNA was equal to 126-fold over control cells. AA treatment alone raised steady state c-fos mRNA levels by 7.8-fold and the UVB-induced increase was 28-fold. AA combined with Qu treatment raised steady state c-fos levels 47-fold over control and the UVB response in these cells was completely inhibited, with c-fos levels remaining at 43-fold over control. These data are means from 3 independently performed experiments and statistically significant differences between groups (p < 0.05) are indicated by labels containing different letters. Any two treatment conditions with a label containing the same letter are not significantly different. (B) HaCaT cells treated with Qu, AA or both were concurrently treated with actinomycin D to prevent the synthesis of new mRNA. At the indicated time points, mRNA was extracted and real time RT-PCR was performed and data were analyzed as indicated above. Data were normalized to 0 hr, set to 100% for each treatment condition. AA stabilized c-fos mRNA, which likely contributed to the increase in steady state c-fos mRNA detected in the AA treated samples in (A). Qu did not stabilize c-fos mRNA and reduced the stabilizing effect of AA. These data are a representative sample from 2 independently performed experiments. (C) HaCaT cells were pretreated with 50 µM Qu +/− 1 mM AA for 1 hr prior to irradiation with 250 J/m2 UVB and following irradiation for 6 hr, at which point protein lysates were extracted and Western analysis was performed using specific antibodies for c-Fos. Treatment with either Qu or UVB irradiation alone induced c-Fos protein expression over control, but cells treated with both Qu and UVB irradiation displayed a potentiated increase in c-Fos protein. Supplementation with AA had no effect on c-Fos protein levels in mock and UVB-irradiated HaCaT cells. However, Qu treatment in AA-supplemented cells completely blocked the UVB-induced increase in c-Fos protein levels. Relative band densities are represented numerically immediately below the blot. Data are representative of 3 independently performed experiments.
Since this increase in steady state c-fos mRNA caused by supplementation of DMEM with AA could not have been the result of increased transcriptional activity, we next investigated the possibility that AA was increasing the stability of c-fos mRNA. HaCaT cells were treated with actinomycin D to inhibit mRNA synthesis and subsequently treated with Qu, AA, or both. Through real-time PCR analysis, we determined that AA supplementation alone resulted in a marked reduction in the rate of c-fos mRNA decay compared to control (Figure 3B). Qu treatment had no significant effect on mRNA stabilization alone, but actually reduced the effect of AA.
Quercetin potentiates UVB-induced c-Fos protein expression in HaCaT keratinocytes, and ascorbate-stabilized quercetin inhibits increases in c-Fos protein levels
Our laboratory has previously reported that UVB irradiation increases the levels of c-Fos protein (14, 17, 18). We analyzed c-Fos protein levels by Western analysis and confirmed that 250 J/m2 UVB did increase expression over the level expressed in mock irradiated HaCaT cells (Figure 3C). Treatment with Qu alone also increased c-Fos protein to a level comparable to that induced by UVB, and treatment of cells with both Qu and UVB resulted in level of c-Fos that was substantially increased over either Qu or UVB treatment alone. These data corroborate our previous finding for the Qu and UVB effects on mRNA expression. Contrary to the effect of AA on steady state mRNA levels, there was no effect of AA treatment alone on c-Fos protein expression in either mock- or UVB-irradiated cells compared to their respective controls. Interestingly, treatment with Qu+AA showed only a slight increase in c-Fos protein expression in mock-irradiated cells, and a complete suppression of c-Fos protein expression even when cells were irradiated with UVB, suggesting that the stabilization of c-fos mRNA did not translate to increased protein.
Quercetin blocks mTOR signaling and inhibits c-Fos protein translation
One explanation for the apparent disconnect between mRNA and protein levels is that Qu independently affects protein synthesis and mRNA expression. To address this possibility, we investigated the effects of Qu on mTOR signaling since this pathway is known to regulate de novo protein synthesis and since we and others have reported that Qu has inhibitory activity against PI3-K, a kinase upstream of Akt and mTOR activation (4, 23, 24). We determined that mTOR phosphorylation at Ser2448, an activating phosphorylation, was slightly increased with UVB (Figure 4A), as was phosphorylation of the downstream mTOR effectors 4EBP (S65; Figure 4B) and p70S6K (T389; Figure 4C). Qu suppressed phosphorylation of each of these proteins in both mock and UVB-irradiated cells, and when the cells were treated with Qu in the presence of AA, the level of suppression was increased, a finding that is consistent with our previous report that AA stabilizes Qu in aqueous medium (4).
Figure 4. Quercetin inhibits mTOR signaling, but only blocks c-Fos protein synthesis when stabilized by AA.
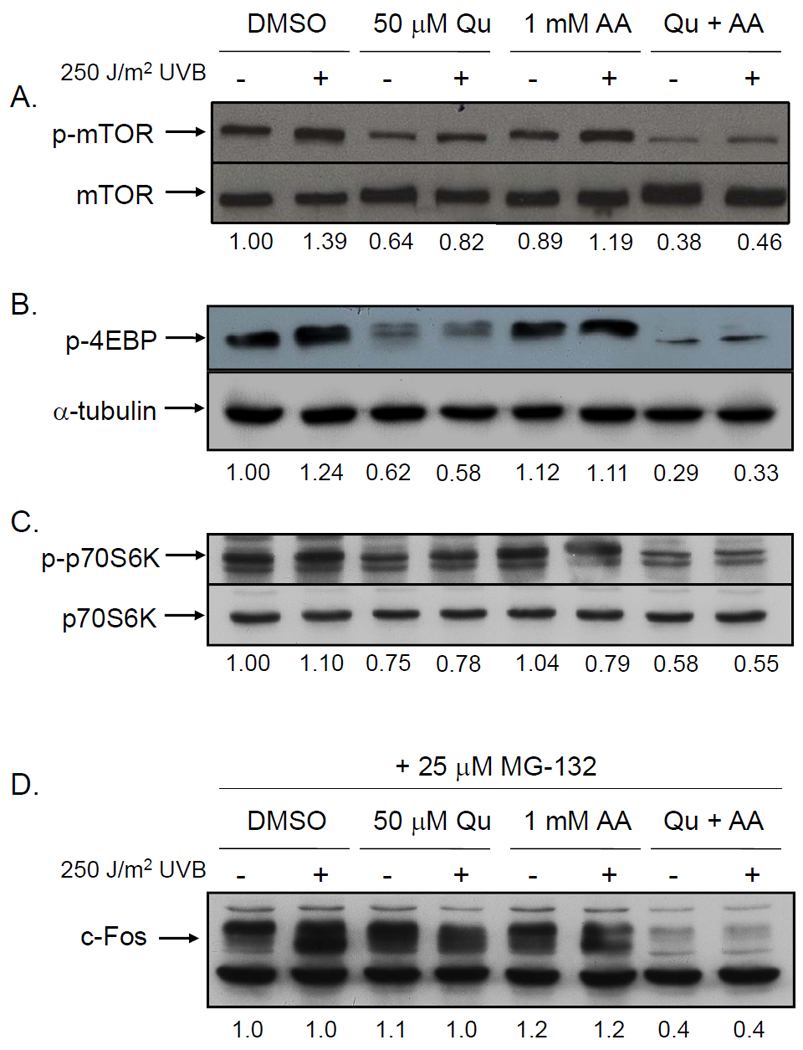
HaCaT cells were pretreated with 50 µM Qu +/− 1 mM AA for 1 hr prior to irradiation with 250 J/m2 UVB and following irradiation for 2 hr. Protein lysates were extracted and Western analysis was performed using specific antibodies for (A) phospho-mTOR, (B) phospho-4EBP, and (C) phospho-p70S6K. UVB irradiation increased phosphorylation of each of these signaling intermediates, whereas treatment with Qu suppressed basal and UVB-induced phosphorylation. AA supplementation of DMEM alone had no effect on either mock or UVB-induced phosphorylation. However, inhibition of phosphorylation by Qu was enhanced in the presence of AA for mTOR, 4EPB, and p70S6K. Total mTOR and α-tubulin levels were unchanged across all treatment conditions. Relative band densities are represented numerically immediately below each set of blots. Data are representative of 3 independently performed experiments. (D) HaCaT cells were pretreated with 50 µM Qu +/− 1 mM AA for 1 hr prior to irradiation with 250 J/m2 UVB and following irradiation for 6 hr. Immediately following exposure to UVB, cells were treated with 25 µM MG-132 to inhibit proteasome activity. Following the treatment period, cell lysates were prepared and Western analysis was performed to determine the effect of treatment on c-Fos protein levels. Treatment with MG-132 increased c- Fos levels in all treatment conditions compared to cells with no MG-132 treatment (Figure 4) except for in cells treated with Qu+AA, in which there was no increase. Relative band densities are represented numerically immediately below the blot. Data are representative of 3 independently performed experiments.
Lastly, we treated HaCaT cells with Qu+AA in the presence of the proteasome inhibitor MG-132 in order to prevent c-Fos turnover and more specifically address the effects of stabilized Qu on c-Fos protein synthesis (Figure 4D). HaCaT cells treated with MG-132 displayed significantly increased c-Fos protein levels due to inhibition of protein degradation, which resulted in a reduction of the effect of Qu on UVB-induced c-Fos protein levels. However, cells treated with Qu+AA still displayed low levels of c-Fos protein in the presence of MG-132 resulting from a sustained reduction of c-Fos translation.
Discussion
Non-melanoma skin cancer (NMSC) is the most commonly diagnosed of all human malignancies and is the cause of 2,000 deaths annually and millions of dollars in health care costs. Clearly, new treatment strategies are needed to prevent the development of precancerous actinic keratoses and squamous cell tumors caused by prolonged exposure to ultraviolet light. Quercetin is currently being evaluated as a potential chemopreventive agent in multiple types of cancer in part because it has established PI3-K and MAPK inhibitory activities [for review, see (25)]. Our initial interest in Qu was specifically due to this activity, since it has been shown that the PI3-K and MAPK signaling pathways are activated in response to UVB irradiation and both PI3-K and p38 are upstream of c-Fos expression and AP-1 activation (16, 26, 27). Natural products and small molecule inhibitors of these pathways would protect against promotion of initiated cells by reducing AP-1 activation and could be potentially useful in cancer prevention.
The findings from the current study are summarized in Figure 5. We determined that Qu potentiated the UVB-induced c-fos gene expression, a direct result of increased p38 and CREB phosphorylation. The inhibitory effect of Qu on PI3-K was not sufficient to prevent the elevation of c-Fos protein levels that directly resulted from increased gene expression. These effects of Qu could be prevented by stabilizing the compound with AA, which resulted in a complete inhibition of UVB-induced c-fos mRNA and protein expression and all upstream signaling. These findings exemplify the difficulties that can arise when working with natural products.
Figure 5. Proposed mechanism of quercetin-mediated induction and inhibition of c-Fos mRNA and protein expression.
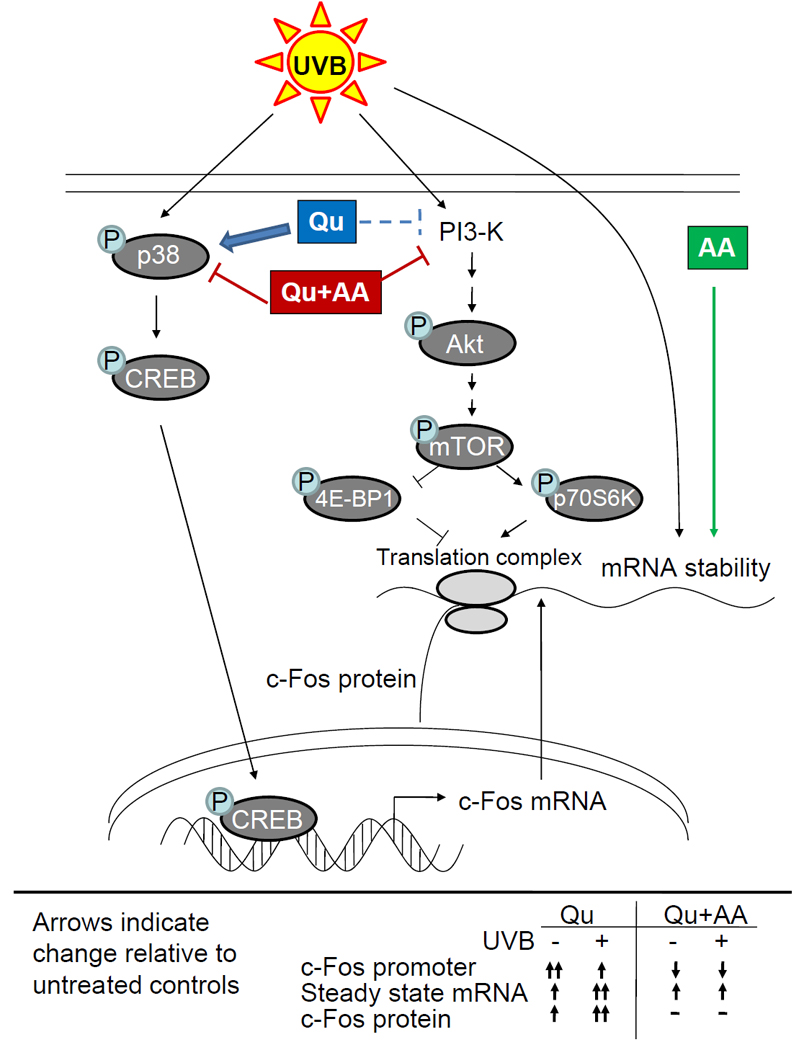
Quercetin induces c-fos gene expression through upstream activation of p38 and CREB and potentiates the response to UVB, leading to an increase in steady state c-fos mRNA levels. Co-treatment with Qu and ascorbic acid blocks p38 and CREB activation and reduces c-fos promoter activity (confirmed by the observation that CREB/CRE binding is reduced). c-fos mRNA levels are actually increased after treatment with Qu+AA as a result of increased c-fos mRNA stability, but the response to UVB is completely inhibited. PI3-K, also activated by UVB and upstream of mTOR, is known to be inhibited by Qu and with greater efficacy by Qu+AA. Simultaneous inhibition of mTOR signaling by Qu+AA prevents c- Fos protein synthesis even in the presence of increased mRNA levels. With Qu alone, inhibition of this pathway is less efficacious and more transient as a result of Qu degradation, leading to increased c-Fos protein driven by significantly increased levels of c-fos mRNA.
We previously reported that Qu could potentially act as a chemopreventive agent due to its ability to kill initiated cells through PI3-K inhibition and pro-oxidant effects (4). However, considering the results of the current study, Qu degradation could potentially have the consequence of increasing AP-1 activity and negating the chemopreventive effect. Dietary Qu has been shown to be effective in preventing cancer in animal models in colon (28–33), breast (34), and lung tissues (35), as well as squamous cell carcinoma (SCC) of the tongue (36). In NMSC models, the effects of Qu are unclear. In one study, dietary Qu had no effect on UV-induced SCC (37), while topically-applied Qu decreased papilloma formation in a chemically-induced skin cancer mouse model (38). We have tested topical Qu in a UVB-induced skin cancer model and found no significant reduction of SCC incidence, tumor multiplicity, or tumor burden compared to vehicle-treated controls (data not shown). There are several possible explanations for the differences in these reported results. First, orally administered Qu may not adequately distribute to the skin to elicit any preventative effect. Second, it is possible that Qu is more effective against chemically-induced skin cancer, although Kato et al. (38) used a topical dose that was 5 times more concentrated than the dose we used (5% vs. 1%, respectively). Interestingly, a recent study evaluating myricetin, a flavonol with great structural similarity to Qu, demonstrated significant efficacy in preventing UVB-induced skin tumor formation when much lower amounts were used (0.001–0.003% myricetin/treatment vs. 1% quercetin/treatment in our study) (39). There have been no reports describing myricetin stability, but we have evidence that myricetin does not induce c-fos expression in cultured HaCaT cells when used at the same concentration as Qu in the current study (data not shown). Lastly, the formulation used to deliver Qu topically may affect the long-term stability of Qu in the skin, and as demonstrated in the current study, Qu degradation can affect cell signaling and the cellular response to UVB. Overall, these studies suggest that the effectiveness of Qu is dependent on the specific type of cancer and the method of administration. Furthermore, our findings suggest that any aqueous-based topical formulation of Qu developed for human use would have to contain a Qu stabilizing agent that significantly slowed Qu degradation in order to prevent the detrimental effects of Qu degradation. If Qu is to be pursued as a topical chemopreventive agent for non-melanoma skin cancer, further studies are needed to determine if there are optimal conditions under which Qu can be effective.
Our findings, combined with the large number of reported cellular effects of various natural products, highlight the complexities and the careful considerations required to properly evaluate these compounds for their chemopreventive potential, particularly with regard to the proper dosing required to achieve desired experimental or clinical outcomes. Some of these studies highlight an important conflict in the reports of in vitro and in vivo effects of Qu. For example, pioneering studies by Nagao et al. (40) identified numerous flavanoids, including quercetin, with mutagenic potential. However, oral Qu has been demonstrated to display no carcinogenic effect in rats (41), despite the known relationship between mutagenicity and carcinogenicity. In addition, other studies using different in vitro models demonstrate conflicting findings to our own. Ying, et al. (42) recently published a study demonstrating that 10 µM Qu blocks ICAM expression in part through inhibition of c-fos induction. As previously stated, we had determined that this dose was ineffective at preventing UVB-induced PI3-K activity, our primary goal, but raising the concentration to an effective level caused increased sensitization to UVB-induced c-fos expression. This property may be detrimental to the chemopreventive effect of Qu, but might be useful for other purposes, like increasing the sensitivity of tumor cells to agents like cisplatin as reported by Sharma, et al. (43). These facts all indicate that Qu is capable of eliciting different effects that need to be well characterized to fully understand how the compound might be useful in preventing or treating cancer. Quercetin is an excellent example of how natural products need to be thoroughly examined before drawing premature conclusions regarding their preventive or therapeutic efficacy.
Acknowledgements
The authors thank Anne Cione for administrative assistance.
This work was supported by: NIH grants: Arizona Cancer Center Support Grant CA23074, Chemoprevention of Skin Cancer Program Project Grant CA27502, R25T Cancer Prevention and Control Postdoctoral Fellowship CA78447, Southwest Environmental Health Sciences Center Support Grant ES06694.
REFERENCES
- 1.Lee KW, Kang NJ, Heo YS, et al. Raf and MEK protein kinases are direct molecular targets for the chemopreventive effect of quercetin, a major flavonol in red wine. Cancer research. 2008;68(3):946–955. doi: 10.1158/0008-5472.CAN-07-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin CW, Hou WC, Shen SC, et al. Quercetin inhibition of tumor invasion via suppressing PKC delta/ERK/AP-1-dependent matrix metalloproteinase-9 activation in breast carcinoma cells. Carcinogenesis. 2008;29(9):1807–1815. doi: 10.1093/carcin/bgn162. [DOI] [PubMed] [Google Scholar]
- 3.Xavier CP, Lima CF, Preto A, Seruca R, Fernandes-Ferreira M, Pereira-Wilson C. Luteolin, quercetin and ursolic acid are potent inhibitors of proliferation and inducers of apoptosis in both KRAS and BRAF mutated human colorectal cancer cells. Cancer letters. 2009;281(2):162–170. doi: 10.1016/j.canlet.2009.02.041. [DOI] [PubMed] [Google Scholar]
- 4.Olson ER, Melton T, Dong Z, Bowden GT. Stabilization of quercetin paradoxically reduces its proapoptotic effect on UVB-irradiated human keratinocytes. Cancer prevention research (Philadelphia, Pa. 2008;1(5):362–368. doi: 10.1158/1940-6207.CAPR-08-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA: a cancer journal for clinicians. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 6.Cooper SJ, MacGowan J, Ranger-Moore J, Young MR, Colburn NH, Bowden GT. Expression of dominant negative c-jun inhibits ultraviolet B-induced squamous cell carcinoma number and size in an SKH-1 hairless mouse model. Mol Cancer Res. 2003;1(11):848–854. [PubMed] [Google Scholar]
- 7.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20(19):2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein LR, Colburn NH. AP1/jun function is differentially induced in promotion-sensitive and resistant JB6 cells. Science (New York, NY. 1989;244(4904):566–569. doi: 10.1126/science.2541502. [DOI] [PubMed] [Google Scholar]
- 9.Dong Z, Huang C, Brown RE, Ma WY. Inhibition of activator protein 1 activity and neoplastic transformation by aspirin. The Journal of biological chemistry. 1997;272(15):9962–9970. doi: 10.1074/jbc.272.15.9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang C, Ma WY, Dawson MI, Rincon M, Flavell RA, Dong Z. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(11):5826–5830. doi: 10.1073/pnas.94.11.5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson EJ, MacGowan J, Young MR, Colburn N, Bowden GT. A dominant negative c-jun specifically blocks okadaic acid-induced skin tumor promotion. Cancer research. 2002;62(11):3044–3047. [PubMed] [Google Scholar]
- 12.Young MR, Li JJ, Rincon M, et al. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(17):9827–9832. doi: 10.1073/pnas.96.17.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaminska B, Pyrzynska B, Ciechomska I, Wisniewska M. Modulation of the composition of AP-1 complex and its impact on transcriptional activity. Acta neurobiologiae experimentalis. 2000;60(3):395–402. doi: 10.55782/ane-2000-1358. [DOI] [PubMed] [Google Scholar]
- 14.Chen W, Borchers AH, Dong Z, Powell MB, Bowden GT. UVB irradiation-induced activator protein-1 activation correlates with increased c-fos gene expression in a human keratinocyte cell line. The Journal of biological chemistry. 1998;273(48):32176–32181. doi: 10.1074/jbc.273.48.32176. [DOI] [PubMed] [Google Scholar]
- 15.Saez E, Rutberg SE, Mueller E, et al. c-fos is required for malignant progression of skin tumors. Cell. 1995;82(5):721–732. doi: 10.1016/0092-8674(95)90469-7. [DOI] [PubMed] [Google Scholar]
- 16.Bachelor MA, Cooper SJ, Sikorski ET, Bowden GT. Inhibition of p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase decreases UVB-induced activator protein-1 and cyclooxygenase-2 in a SKH-1 hairless mouse model. Mol Cancer Res. 2005;3(2):90–99. doi: 10.1158/1541-7786.MCR-04-0065. [DOI] [PubMed] [Google Scholar]
- 17.Gonzales M, Bowden GT. Ultraviolet B (UVB) induction of the c-fos promoter is mediated by phospho-cAMP response element binding protein (CREB) binding to CRE and c-fos activator protein 1 site (FAP1) cis elements. Gene. 2002;293(1–2):169–179. doi: 10.1016/s0378-1119(02)00723-0. [DOI] [PubMed] [Google Scholar]
- 18.Gonzales M, Bowden GT. The role of PI 3-kinase in the UVB-induced expression of c-fos. Oncogene. 2002;21(17):2721–2728. doi: 10.1038/sj.onc.1205366. [DOI] [PubMed] [Google Scholar]
- 19.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. The Journal of cell biology. 1988;106(3):761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehrhart JC, Gosselet FP, Culerrier RM, Sarasin A. UVB-induced mutations in human key gatekeeper genes governing signalling pathways and consequences for skin tumourigenesis. Photochem Photobiol Sci. 2003;2(8):825–834. doi: 10.1039/b302281a. [DOI] [PubMed] [Google Scholar]
- 21.Lehman TA, Modali R, Boukamp P, et al. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis. 1993;14(5):833–839. doi: 10.1093/carcin/14.5.833. [DOI] [PubMed] [Google Scholar]
- 22.Rosenberger SF, Finch JS, Gupta A, Bowden GT. Extracellular signal-regulated kinase 1/2-mediated phosphorylation of JunD and FosB is required for okadaic acid-induced activator protein 1 activation. The Journal of biological chemistry. 1999;274(2):1124–1130. doi: 10.1074/jbc.274.2.1124. [DOI] [PubMed] [Google Scholar]
- 23.Granado-Serrano AB, Martin MA, Bravo L, Goya L, Ramos S. Quercetin induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI-3-kinase/Akt and ERK pathways in a human hepatoma cell line (HepG2) The Journal of nutrition. 2006;136(11):2715–2721. doi: 10.1093/jn/136.11.2715. [DOI] [PubMed] [Google Scholar]
- 24.Gulati N, Laudet B, Zohrabian VM, Murali R, Jhanwar-Uniyal M. The antiproliferative effect of Quercetin in cancer cells is mediated via inhibition of the PI3K-Akt/PKB pathway. Anticancer research. 2006;26(2A):1177–1181. [PubMed] [Google Scholar]
- 25.Murakami A, Ashida H, Terao J. Multitargeted cancer prevention by quercetin. Cancer letters. 2008;269(2):315–325. doi: 10.1016/j.canlet.2008.03.046. [DOI] [PubMed] [Google Scholar]
- 26.Tang Q, Gonzales M, Inoue H, Bowden GT. Roles of Akt and glycogen synthase kinase 3beta in the ultraviolet B induction of cyclooxygenase-2 transcription in human keratinocytes. Cancer research. 2001;61(11):4329–4332. [PubMed] [Google Scholar]
- 27.Tullai JW, Chen J, Schaffer ME, Kamenetsky E, Kasif S, Cooper GM. Glycogen synthase kinase-3 represses cyclic AMP response element-binding protein (CREB)-targeted immediate early genes in quiescent cells. The Journal of biological chemistry. 2007;282(13):9482–9491. doi: 10.1074/jbc.M700067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deschner EE, Ruperto J, Wong G, Newmark HL. Quercetin and rutin as inhibitors of azoxymethanol-induced colonic neoplasia. Carcinogenesis. 1991;12(7):1193–1196. doi: 10.1093/carcin/12.7.1193. [DOI] [PubMed] [Google Scholar]
- 29.Deschner EE, Ruperto JF, Wong GY, Newmark HL. The effect of dietary quercetin and rutin on AOM-induced acute colonic epithelial abnormalities in mice fed a high-fat diet. Nutrition and cancer. 1993;20(3):199–204. doi: 10.1080/01635589309514287. [DOI] [PubMed] [Google Scholar]
- 30.Dihal AA, de Boer VC, van der Woude H, et al. Quercetin, but not its glycosidated conjugate rutin, inhibits azoxymethane-induced colorectal carcinogenesis in F344 rats. The Journal of nutrition. 2006;136(11):2862–2867. doi: 10.1093/jn/136.11.2862. [DOI] [PubMed] [Google Scholar]
- 31.Gee JM, Hara H, Johnson IT. Suppression of intestinal crypt cell proliferation and aberrant crypt foci by dietary quercetin in rats. Nutrition and cancer. 2002;43(2):193–201. doi: 10.1207/S15327914NC432_10. [DOI] [PubMed] [Google Scholar]
- 32.Matsukawa Y, Nishino H, Okuyama Y, et al. Effects of quercetin and/or restraint stress on formation of aberrant crypt foci induced by azoxymethane in rat colons. Oncology. 1997;54(2):118–121. doi: 10.1159/000227674. [DOI] [PubMed] [Google Scholar]
- 33.Volate SR, Davenport DM, Muga SJ, Wargovich MJ. Modulation of aberrant crypt foci and apoptosis by dietary herbal supplements (quercetin, curcumin, silymarin, ginseng and rutin) Carcinogenesis. 2005;26(8):1450–1456. doi: 10.1093/carcin/bgi089. [DOI] [PubMed] [Google Scholar]
- 34.Verma AK, Johnson JA, Gould MN, Tanner MA. Inhibition of 7,12-dimethylbenz(a)anthracene- and N-nitrosomethylurea-induced rat mammary cancer by dietary flavonol quercetin. Cancer research. 1988;48(20):5754–5758. [PubMed] [Google Scholar]
- 35.Khanduja KL, Gandhi RK, Pathania V, Syal N. Prevention of N-nitrosodiethylamine-induced lung tumorigenesis by ellagic acid and quercetin in mice. Food Chem Toxicol. 1999;37(4):313–318. doi: 10.1016/s0278-6915(99)00021-6. [DOI] [PubMed] [Google Scholar]
- 36.Makita H, Tanaka T, Fujitsuka H, et al. Chemoprevention of 4-nitroquinoline 1-oxide-induced rat oral carcinogenesis by the dietary flavonoids chalcone, 2-hydroxychalcone, and quercetin. Cancer research. 1996;56(21):4904–4909. [PubMed] [Google Scholar]
- 37.Steerenberg PA, Garssen J, Dortant P, et al. Quercetin prevents UV-induced local immunosuppression, but does not affect UV-induced tumor growth in SKH-1 hairless mice. Photochemistry and photobiology. 1997;65(4):736–744. doi: 10.1111/j.1751-1097.1997.tb01918.x. [DOI] [PubMed] [Google Scholar]
- 38.Kato R, Nakadate T, Yamamoto S, Sugimura T. Inhibition of 12-O-tetradecanoylphorbol-13-acetate-induced tumor promotion and ornithine decarboxylase activity by quercetin: possible involvement of lipoxygenase inhibition. Carcinogenesis. 1983;4(10):1301–1305. doi: 10.1093/carcin/4.10.1301. [DOI] [PubMed] [Google Scholar]
- 39.Jung SK, Lee KW, Byun S, et al. Myricetin suppresses UVB-induced skin cancer by targeting Fyn. Cancer research. 2008;68(14):6021–6029. doi: 10.1158/0008-5472.CAN-08-0899. [DOI] [PubMed] [Google Scholar]
- 40.Nagao M, Morita N, Yahagi T, et al. Mutagenicities of 61 flavonoids and 11 related compounds. Environmental mutagenesis. 1981;3(4):401–419. doi: 10.1002/em.2860030402. [DOI] [PubMed] [Google Scholar]
- 41.Utesch D, Feige K, Dasenbrock J, et al. Evaluation of the potential in vivo genotoxicity of quercetin. Mutation research. 2008;654(1):38–44. doi: 10.1016/j.mrgentox.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Ying B, Yang T, Song X, et al. Quercetin inhibits IL-1 beta-induced ICAM-1 expression in pulmonary epithelial cell line A549 through the MAPK pathways. Mol Biol Rep. 2009;36(7):1825–1832. doi: 10.1007/s11033-008-9386-1. [DOI] [PubMed] [Google Scholar]
- 43.Sharma H, Sen S, Singh N. Molecular pathways in the chemosensitization of cisplatin by quercetin in human head and neck cancer. Cancer Biol Ther. 2005;4(9):949–955. doi: 10.4161/cbt.4.9.1908. [DOI] [PubMed] [Google Scholar]