

Abstract
Increasing interest in rodent models for movement disorders has led to an increasing need for more accurate and precise methods for both delineating the nature of abnormal movements and measuring their severity. These studies describe application of simultaneous high-speed video kinematics with multi-channel EMG to characterize the movement disorder exhibited by tottering mutant mice. These mice provide a uniquely valuable model because they exhibit paroxysmal dystonia superimposed on mild baseline ataxia, permitting the examination of these two different problems within the same animals. At baseline with mild ataxia, the mutants exhibited poorly coordinated movements with increased variation of stance and swing times, and slower spontaneous walking velocities. The corresponding EMG showed reduced mean amplitudes of biceps femoris (BF) and vastus lateralis (VL), and poorly modulated EMG activities during the step cycle. Attacks of paroxysmal dystonia were preceded by trains of EMG bursts with doublets and triplets simultaneously in the BF and VL followed by more sustained co-activation. These EMG characteristics are consistent with the clinical phenomenology of the motor phenotype of tottering mice as a baseline of mild ataxia with intermittent attacks of paroxysmal dystonia. The EMG characteristics of ataxia and dystonia in the tottering mice also are consistent with EMG studies of other ataxic or dystonic animals and humans. These studies provide insights into how these methods can be used for delineating movement disorders in mice, and for how they may be compared with similar disorders of humans.
Keywords: Ataxia, Dystonia, Animal Models, Electromyography, Video kinematics
Introduction
Modern advances in the neurosciences have driven increasing interest in rodent models of human neurological diseases. This interest has been accompanied by a growing list of tools for assessing abnormal motor behavior in rodents. Some of the most popular methods include automated activity chambers, the rotarod, and other tests for coordination. These tools provide good measures of functional performance, but they are of limited value in delineating the type of motor disorder in relation to specific human motor syndromes.1
These limitations have led to two different strategies. One involves applying a wide battery of different tests to characterize patterns of performance deficits. This strategy provides a wealth of information on functional impairments, but has little applicability for relating the impairments to diagnostic categories in humans. The other strategy resembles that used in clinical practice, relying first on expert recognition of the motor syndrome, with subsequent application of specific tests to confirm or refute clinical impressions. This approach is straightforward when the motor disorder bears an obvious resemblance to its human counterpart, but diagnostic difficulties arise when the motor syndrome is subtle, or when it does not clearly match that of humans. In these situations, additional tests for discriminating different disorders would be welcome.
Electromyography (EMG) has been valuable for characterizing motor syndromes resulting from dysfunction of peripheral nerves or muscles in rodents. It has been used to identify the electrophysiological signatures associated with neuropathy, myopathy, and myotonia.2–6 In these cases, EMG can be done with single-channel recordings of anesthetized or restrained animals, since defects intrinsic to nerve or muscle can be detected readily. EMG also has the potential to aid in the characterization of more complex motor disorders originating in the central nervous system. For these disorders, animals must be tested awake, because abnormalities often abate during sleep. The animals also must be free to move, so that spontaneously generated movements can be recorded free of artifacts associated with struggling in restraints.7–13 Multi-channel EMG recordings can be especially helpful for identifying abnormal patterns of activity characteristic of central disorders, such abnormal relationships between antagonistic muscles in ataxia or dystonia.2, 14–17
Another method helpful for characterizing central motor syndromes involves video kinematics, which involves recording video and precisely mapping limb and joint positions in time and space. This method permits precise delineation of complex movements by breaking them down into their component parts.15, 18–20 The combination of video kinematics with multi-channel EMG to match muscle activity to specific movements further aids the interpretation of the physiology of movements, by permitting correlation of muscle activity with normal and abnormal movements.11–13
In previous studies we synchronized video kinematic recordings with multi-channel EMG during treadmill locomotion to characterize muscle activity during the natural step cycle in rats and mice.7, 10–12 In the current studies, these methods were used to characterize the motor phenotype of tottering mutant mice. These mice carry a calcium channel mutation resulting in a characteristic syndrome that has been evaluated extensively, including a video demonstration.21–26 At baseline, tottering mutants have a slightly unsteady gait indicative of mild ataxia. Superimposed are transient discrete attacks of severe twisting movements and unnatural postures that last 30–60 minutes, characteristic of paroxysmal dystonia.22 These mice provide a uniquely valuable tool for comparing the video kinematic and EMG characteristics of two very distinct motor syndromes, since both disorders can be studied within a single animal. Our primary goal was to delineate the qualitative features associated with ataxia at baseline and attacks of paroxysmal dystonia in tottering mutant mice.
Materials and Methods
Six female tottering mutant mice (B6.D2-Cacna1atg/J) were examined between 3–6 months of age. In some cases, video kinematic results were compared against a database of information regarding the step cycle of normal mice collected previously under the same conditions.9 All experiments were done in accordance with the animal welfare regulations of Germany, the European Communities Council Directive (86/609/EEC), and the National Institutes of Health Guide for Care and Use of Laboratory Animals.
Synchronized recordings of multi-channel EMG with video kinematics were made 2–14 days following surgical implantation of EMG electrode arrays, as mice walked on a motorized treadmill, as previously described.9, 10 Briefly, mice had 20 trials per day, and each trial lasted 7s. EMG was performed with an implanted array electrode consisting of a silicone pad with 4 electrodes separated by 3 mm, and data were collected with a frequency range of 10–700 Hz, and sampling rate of 4000 Hz. The arrays simultaneously recorded two antagonistic muscles of the rear limb, the vastus lateralis (VL) and the biceps femoris (BF). The EMG amplitude profile (root mean square, RMS) was calculated using a 10 msec moving window. In parallel with EMG, videotapes were collected at a frame rate of 400 Hz to characterize the step phases in relation to the EMG signal. The step patterns were grouped into stance and swing phases, and time-normalized to 100 points with 50 points each in stance and swing phases.9, 10
Statistical comparisons
The stance and swing times of all animals were intra-individually averaged and then summed for the 6 tottering mutants and 11 controls. The RMS amplitude profiles of each step were averaged for each EMG channel and pooled for VL or BF. The RMS was summed over the entire step cycle for mutants versus controls. Mutants and controls were compared using 2-tailed t-tests with p<0.05 as criterion for statistical significance, following confirmation of the normal distribution of data with the Kolmogorov-Smirnov procedure.
Results
Baseline ataxia
The abnormal baseline gait of tottering mutants was visible during treadmill locomotion (Figure 1). They exhibited occasionally irregular or excessive leg motions leading to vertical hip displacement and slowed walking velocities. There also were periods of nearly normal locomotion. Quantitative comparisons of their step characteristics with a database of previously collected data from normal animals revealed significantly longer stance and swing times with increased variability (Table 1). Stance times for mutants averaged more than 3.6 times longer than controls, while swing times averaged 2.8 times longer than controls (Table 1).
Figure 1.

Step cycle in a mutant tottering mouse during treadmill locomotion. The sequence for one step cycle (a–g) begins with foot-down (f-d1, then stance (st1), and foot-up (f-u). The subsequent swing phase (sw) is divided into 4 phases (sw1-1 to sw 1–4). The next step cycle begins again with foot-down (f-d2) and stance (st2). Vertical hip displacements (v.h.d.) can also be seen.
Table 1.
Baseline step characteristics for normal and tottering mice
| Step characteristic | Controls | Tottering | t-test |
|---|---|---|---|
| Step cycle times (msec) | |||
| stance phase | 120±33 | 434±128 | p<0.05 |
| swing phase | 72±14 | 205±76 | p<0.05 |
| Maximum EMG amplitudes (µV) | |||
| biceps femoris | 67±33 | 45±26 | NS |
| vastus lateralis | 88±26 | 35±14 | p<0.05 |
Results show average values for 6 tottering mutants (total 611 steps) and 11 controls (total 648 steps) ± standard deviations. Maximum EMG amplitudes are root mean square (RMS) of bipolar recordings. Comparisons were made via 2-tailed t-test. NS=not significant.
The EMG activity of BF and VL during stance and swing phases also revealed several abnormalities. Unlike normal mice,9 VL EMG amplitudes of mutants varied markedly from stance phase to stance phase (Figure 2). Furthermore, the increase in VL activity began later in the stance phase of the mutants. In comparison to normal controls, the mean EMG amplitudes of the mutants also were lower, but only the amplitudes for VL were significantly different (Table 1). Finally, abnormal BF activity in late swing phase of the mutants could be correlated with abnormal limb flexion on video recordings (Figures 1–2).
Figure 2.
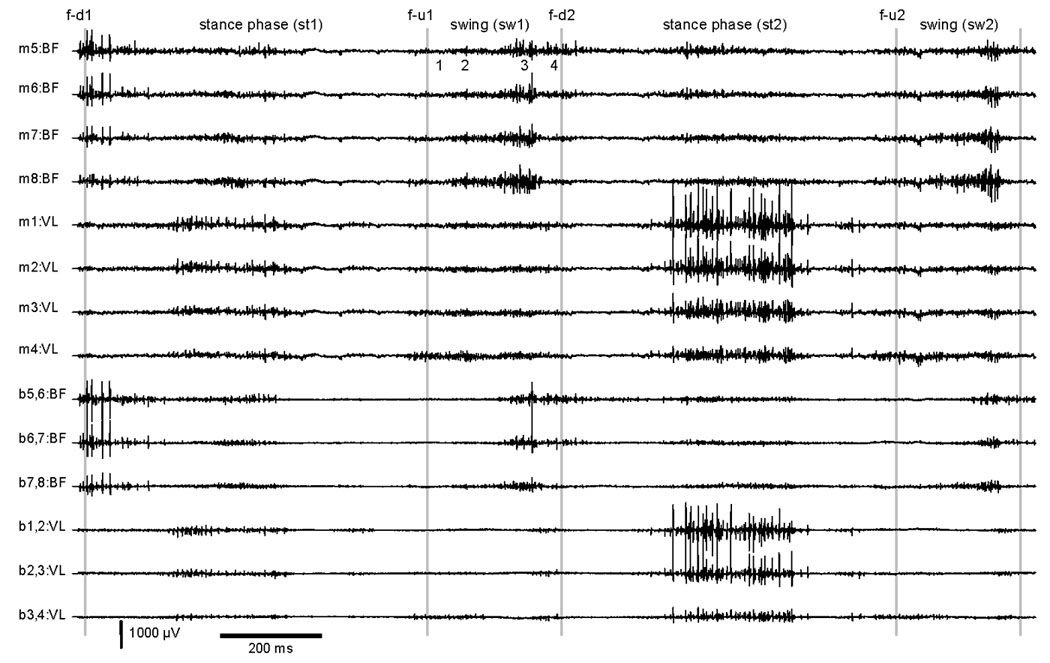
EMG for two step cycles of a tottering mouse during treadmill locomotion. Abbreviations for step cycle follow those used in Figure 1. Four monopolar (m) recordings are shown each for biceps femoris (BF) and vastus lateralus (VL) muscle. Bipolar (b) recordings reflect the difference between the monopolar leads indicated. For example the signal difference between monopolar leads 1 and 2 is designated b1,2. Comparisons of the first stance phase (st1) to the second stance phase (st2) shows abnormal variation of EMG amplitudes for a similar movement.
The abnormal time course of EMG activity during locomotion in tottering mice also was evident in the mean EMG amplitude curves attained by time normalized averaging of bipolar BF and VL activities. During locomotion of mutants, the intra-individual relations between VL and BF EMG activities were different from controls, with occasional abnormal co-activation of these normally antagonistic muscles (Figure 3b–e). As previously described,9 the mean VL and BF EMG activity of controls is modulated during the step cycle, with significant differences between these muscles during the second and last third of stance phase. In mutants this difference between VL and BF was absent, with no significant difference for intra-individual EMG amplitudes.
Figure 3.
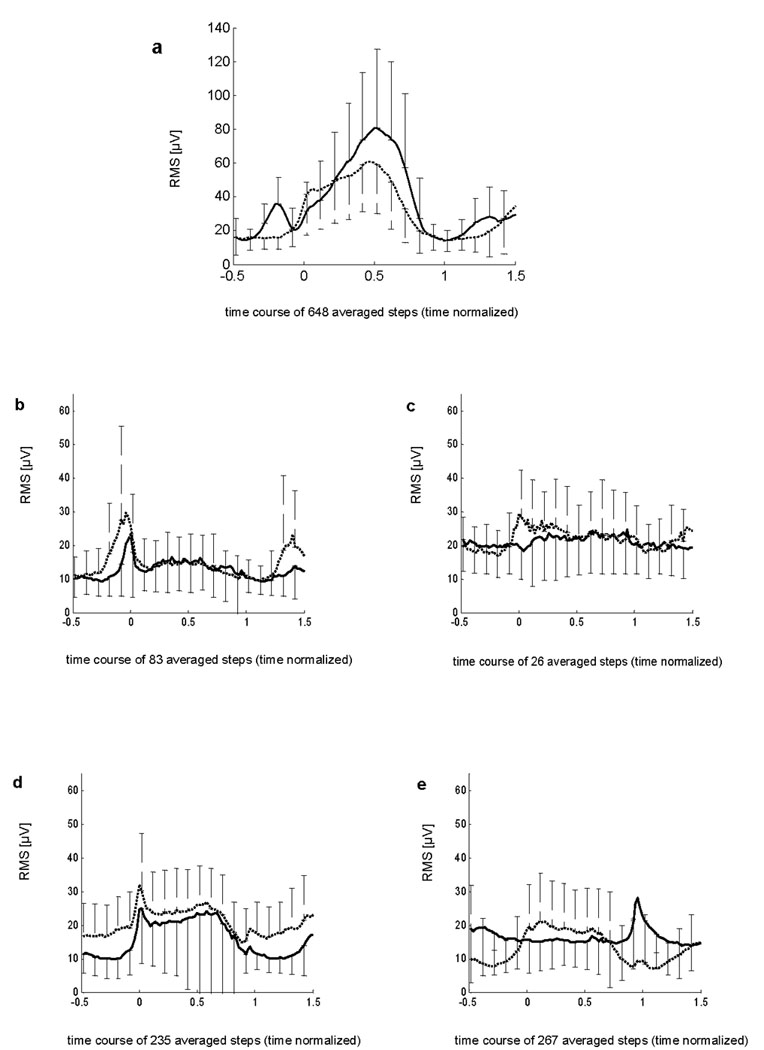
Time course of EMG amplitudes in 11 controls (a) and 4 individual tottering mutants (b–e). Results show bipolar mean amplitudes (RMS) ±SD. The biceps femoris (BF) is shown as a dotted line, while the vastus lateralis (VL) is shown as a solid line. Steps were time-normalized according to prior studies, with foot-down at abscissa (0) and foot-up at 1.
VL activity differed significantly between mutants and controls during all stages of stance phase, while BF activity differed significantly only during the first and second third of stance phase (Table 2). During swing, there were no significant differences at any time. In 4 mutants the BF EMG dominated at the end of the swing phase, a finding not seen in controls where VL extensor activity is needed instead for anti-gravity support (Figure 3).
Table 2.
EMG profiles for normal vs tottering mice
| Step characteristic | t-test |
|---|---|
| EMG amplitude profiles (stance) | |
| biceps femoris | |
| 1st third (0–0.33) | p<0.05 |
| 2nd third (0.34–0.66) | p<0.05 |
| last third (0.67–1.0) | NS |
| vastus lateralis | |
| 1st third (0–0.33) | p<0.05 |
| 2nd third (0.34–0.66) | p<0.05 |
| last third (0.67–1.0) | p<0.05 |
| EMG amplitude profiles (swing) | |
| biceps femoris | NS |
| vastus lateralis | NS |
Results show p values for t-tests comparing EMG amplitude profiles of 6 tottering mutants (total 611 steps) and 11 controls (total 648 steps) during different parts of stance and swing phases. EMG amplitudes are root mean square (RMS) of bipolar recordings. Comparisons were made via 2-tailed t-test. NS=not significant.
Intermittent attacks of dystonia
All 6 tottering mice had intermittent attacks of abnormal muscle contractions with twisting and repetitive movements and abnormal postures lasting 30–60 minutes as previously described.21–26 These attacks were grossly different from baseline motor behavior. The attacks started with jerky limb movements. Later the trunk assumed a hyper-extended posture with intermittent writhing movements. The proximal limbs were flexed abnormally close to the body and the distal limbs were abnormally extended, sometimes with repetitive asynchronous purposeless movements. Symptom resolution was accompanied by recovery of the hind limbs followed by recovery of the fore limbs, followed by return to baseline motor behavior with no-postictal lethargy.
During the attacks, the tottering mutants were no longer capable of walking on the treadmill. As a result, quantitative kinematic recordings of the step cycle were impossible. However, EMG recordings revealed several obvious abnormalities (Figures 4–5). First, during the initial phase of an attack, abnormal activity could be seen with unusually high motor unit discharges up to a frequency of 200 Hz and amplitudes up to 3500 µV (Figure 4a). Second were interposed doublets and triplets (Figure 4a). Third were simultaneous and spontaneous antagonistic discharges in VL and BF, with partially interposed sequences of alternating short bursts (Figure 4b). Finally, there were prolonged semi-rhythmic EMG discharges of both muscles (Figure 5). Spectral power analysis was performed via fast Fourier transformation [after high-pass filtering (20–1000Hz), rectification of EMG signals, then low pass filtering (100 Hz)]. The resulting periodograms were averaged, and a distinct peak with a mean of 4.5 Hz was evident for both BF and VL. This peak was absent during baseline locomotion in tottering mice, and it was never seen in non-mutant animals.
Figure 4.
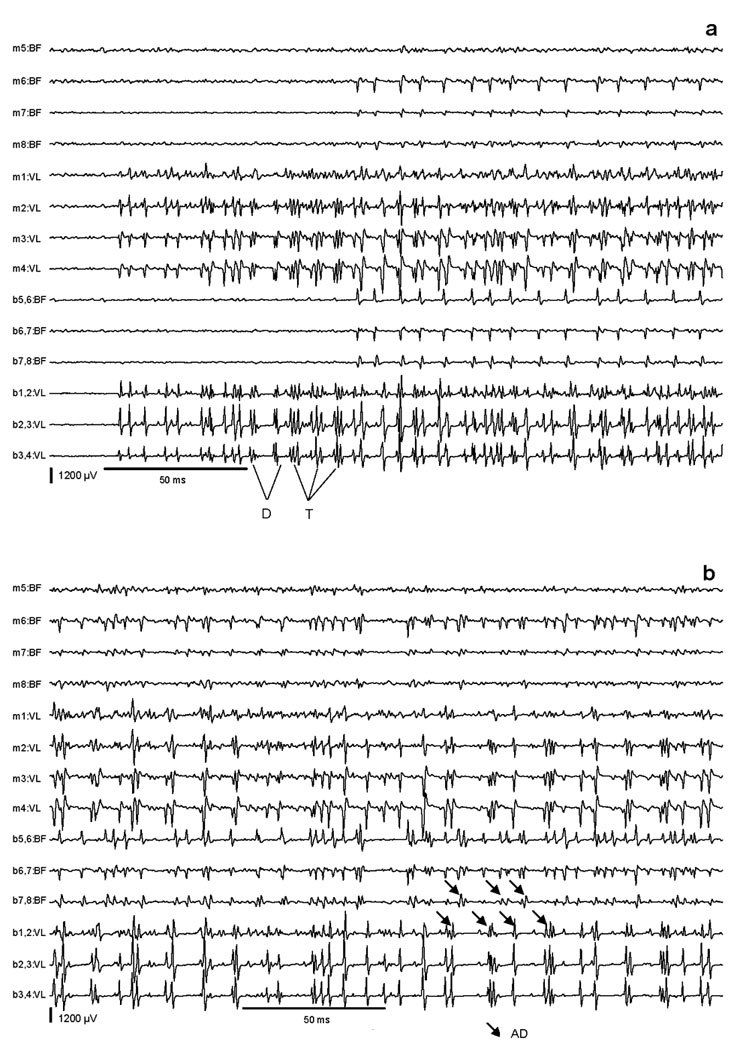
High resolution EMG recordings during a paroxysmal dystonic attack. The early phase (a) shows simultaneous EMG activities in BF and VL with high-amplitude bursts in VL partly appearing as doublets (D) and triplets (T). The late phase (b) shows simultaneous EMG activities in VL and BF, partly with interposed sequences of alternating short discharges (AD).
Figure 5.
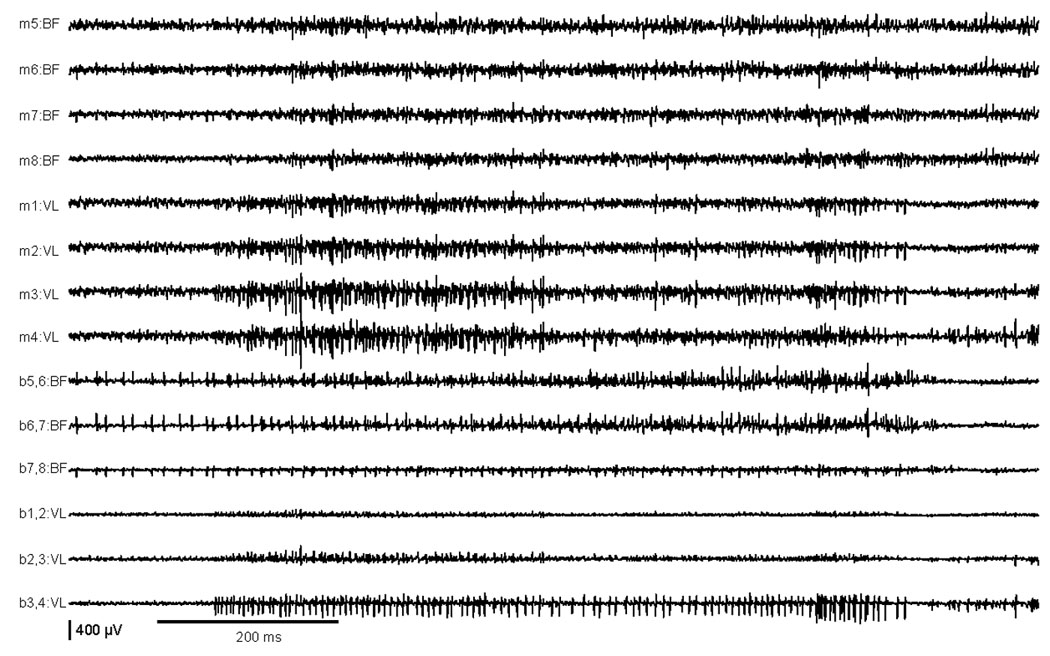
EMG recordings during the peak of an attack of paroxysmal dystonia in a tottering mutant, showing simultaneous prolonged semi-rhythmic EMG discharges of VL and BF associated with lengthy co-contraction of these normally antagonistic muscles.
Discussion
These studies provide several useful insights into the utility of video kinematics and multi-channel EMG for characterizing complex central motor syndromes of mice. The merits and limitations of these methods for assessing different types of motor disorders and comparing results with similar disorders of humans are addressed below.
Video kinematics
Video kinematics can be valuable for characterizing complex movement disorders by breaking them down into component parts that can be examined separately. Standard videos are collected at 25–30 frames/sec, but higher frame rates are needed for capturing faster movements of small animals. Mice take multiple steps per second with average stance and swing times of only 120 and 72 msec respectively.9 At standard frame rates of 25–30 frames/sec, each frame spans epochs of 33–40 msec, capturing fewer than 4 frames to define limb positions during stance phase and only 2 frames during swing phase. In comparison, our frame rate of 400 Hz spans epochs of 2.5 msec, capturing 48 positions during stance and 28 positions during swing. The higher frame rate allows for a more precise definition of the temporal and spatial position of the foot in relation to the step cycle for kinematic analyses. The high sampling rate also permits time-normalization of step cycles of different durations, for statistical comparisons across different walking speeds.
The video kinematics of tottering mice exhibiting their typical baseline ataxic gait during treadmill locomotion revealed increased intra- and inter-individual variation of stance and swing times, excessive leg movements with occasional vertical hip displacements, and slower spontaneous walking velocities. These abnormalities are similar to those described for ataxia associated with Purkinje cell degeneration in lurcher mutant mice, where the salient features included abnormal inter-limb coordination, increased step variability, vertical hip displacement, and exaggerated hind limb flexion.15 Contrary to tottering mice, lurchers also showed shortened steps. This last difference could be explained by differences in the expression of ataxia of the two mouse mutants, or to non-ataxic contributions to their gaits.
A quantitative kinematic analysis of the gait on treadmill was not feasible during the intermittent attacks of more severely disabling dystonic movements in tottering mutants, because the animals could not walk during the spells. Qualitatively, the videotapes revealed sustained twisting movements and odd postures characteristic of human dystonia. Similar movements have been reported for dystonia in the dtsz hamster16, 27 and the dt rat.27, 28 In these other models, kinematic analyses of the gait and other repetitive movements also have not been possible. These observations highlight one of the limitations of classical kinematic analyses. This technique cannot be used in animals wherein the motor disorder is sufficiently severe to prevent recording of repetitive limb movements, such as walking.
Several additional studies would be valuable for improving the utility of video kinematics for animals with movement disorders. The first involves examining a broader group of other ataxic mice to delineate the core features of ataxia from features that may be idiosyncratic to specific models. Studies of other ataxic mice also would be valuable for parsing different types of ataxia into different subtypes based on specific physiological parameters. Once such a database of information regarding ataxia is developed, it could then be useful for characterizing motor syndromes that are too subtle to recognize by eye, and for discriminating components of a potentially mixed motor disorder.
Multichannel EMG in unrestrained animals
As noted in the introduction, EMG has proved useful for characterizing disorders of nerve and muscle in rodents. It has received less attention for characterizing disorders of central motor control, partly due to added technical demands. For central disorders, animals must be studied awake, since the disorder often abates during sleep. They also must be studied when they are free to move, to avoid artifacts related to struggling in restraints. Most importantly, multiple simultaneous EMG channels must be employed, to reveal abnormal coordination among different muscles, such uncoupling of normal relationships during ataxia or dystonia.
In tottering mice displaying their typical baseline ataxic gait during treadmill locomotion, the EMG showed reduced mean amplitudes in both BF and VL, increased variance of EMG amplitudes during stance phases, and poorly coordinated activities between BF and VL during the step cycles with occasional co-activation or late onset of VL activity. The ataxia of Lurcher mice was associated with similar EMG characteristics of poorly coordinated activity, late and variable EMG onset relative to foot contact, and premature activation of the flexor muscle.
During episodes of paroxysmal dystonia in tottering mice, the EMG showed a very different picture. There were sustained and simultaneous EMG bursts of both the BF and VL muscles, often with a semi-rhythmic pattern of doublets and triplets. These abnormalities associated with dystonic attacks were quite distinct from those seen during baseline periods of ataxic movement in the tottering mice. Similar EMG results were associated with dystonia in the dtsz hamster.16 In these studies, freely moving dtsz hamsters demonstrated nearly continuous activity in the tibialis cranialis with superimposed phasic bursts simultaneously in the gastrocnemius of the hind limbs. In another study, the hamstring muscles of lightly restrained normal mice showed high levels of resting muscle activity with exaggerated and prolonged discharges associated with dystonic provoked by ±Bay K 8644.29 An increase in EMG activity also has been associated with exaggerated blinking in a rat model for blepharospasm.30 A recent study showed similar results in the Gunn rat model of dystonia associated with kernicterus.17
The main limitation of EMG is that it is technically demanding, and not readily amenable for evaluation of large numbers of animals. Further development of this method might include some of the same studies suggested above for video kinematics such as evaluation of groups of mice with defined motor disorders to discriminate essential features from idiosyncratic ones, to parse different disorders into different subtypes based on specific criteria, and to aid in the diagnosis of mice with particularly subtle or unusual manifestations that defy simpler approaches for diagnosis.
Comparisons with human movement disorders
The clinical features of ataxia in humans are well known to most clinicians. The key features can be recorded via video kinematics and include abnormal timing of movements among the limbs and across different joints within a limb, increased variation in the speed and distance of movements, and abnormal truncal displacements during walking.31 Movements often are slowed too. The EMG often shows relatively low amplitudes compared to normal movements, increased variability for repetitious movements, and poor timing of both synergistic and antagonistic muscles.
Paroxysmal dystonia is typically labeled paroxysmal dyskinesia in humans to acknowledge a combination of dystonia with other abnormal movements.32, 33 To our knowledge, there are no detailed kinematic studies of human paroxysmal dyskinesias. There are no EMG studies specifically for the paroxysmal dyskinesias, but there are several EMG studies of its primary component, dystonia. These studies typically show exaggerated firing rates and amplitudes relative to normal movements, occasional repetitive discharges, and sometimes co-contraction of antagonistic muscles.34–38 The kinematic features of dystonia in humans include slowing with twisting movements and odd postures, often with a repetitive or stereotyped pattern.34, 39–41 Co-contraction of antagonistic muscles is sometimes described as a diagnostic feature of dystonia, but it is neither specific to dystonia nor consistently found in humans with dystonia.34, 42 Co-contraction of antagonists may occur during voluntary movements or struggling in restraints, and in ataxia during poor timing of antagonistic muscles.
These studies in humans provide a useful framework for interpreting the results from animals. The kinematic and EMG characteristics of ataxic humans are similar to those for tottering mutants during baseline ataxic gait, as well as other rodents with ataxia summarized above. The EMG characteristics of humans with dystonia also are quite similar to those for tottering mutants during attacks of paroxysmal dystonia, as well as other rodents with dystonia described above. Of course, comparisons across species must be done with appropriate caution. For example, ataxia in biped humans is much more disabling than ataxia in quadriped rodents, since a poorly placed step for one of two legs is more likely to cause staggering and falling than a poorly placed step of one of four legs. However, the similarities are more striking than the differences.
In summary, we have characterized the motor behavior of tottering mutant mice with kinematic and EMG parameters. The results demonstrate some strengths and limitations of both methods for different motor syndromes, and how the findings may be compared with similar movement disorders in humans. Further studies of these methods in other mouse models with defined motor disorders would be valuable for delineating essential features of different motor syndromes, for discriminating core features from idiosyncratic ones, and for defining various subgroups of each disorder that reflect a different pathogenesis. These methods also might provide useful adjunctive tools for confirming provisional diagnoses made by visual inspection of rodents, especially when the motor syndromes are subtle or otherwise difficult to characterize.
Acknowledgements
We thank Dr. Harald Schubert and Mrs. Petra Dobermann, Institute of Laboratory Animal Science of University Hospital Jena, and Günter Ditze and his co-workers, Scientific Workshop of University Hospital Jena, for technical support to facilitate the surgical intervention in animals. Supported in part by NIH grant NS40470 and NS33592.
References
- 1.Jinnah HA, Hess EJ. The assessment of movement disorders in mice. In: Le Doux MS, editor. Animal models of movement disorders. San Diego: Elsevier Academic Press; 2005. pp. 55–71. [Google Scholar]
- 2.Carter GT, Longley KJ, Entrikin RK. Electromyographic and nerve conduction studies in the mdx mouse. Am J Phys Med Rehabil. 1992 Feb. 571 doi: 10.1097/00002060-199202000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Chandran AP, Oda K, Shibasaki H, Kikuchi T, Pisharodi M. Stimulus induced repetitive muscle potentials in the gracile axonal dystrophy (GAD) mouse. Electromygr Clinc Neurophysiol. 1995;35:225–230. [PubMed] [Google Scholar]
- 4.Entrikin RK, Abresch RT, Sharman RB, Larson DB, Levine NA. Contractile and EMG studies of murine myotonia (mto) and muscular dystrophy (dy/dy) Muscle Nerve. 1987;10:293–298. doi: 10.1002/mus.880100403. [DOI] [PubMed] [Google Scholar]
- 5.Shirakawa T, Sakai K, Kitagawa Y, Hori A, Hirose G. A novel murine myotonia congenita without molecular defects in the CLC-1 and the SCN4A. Neurology. 2002;59:1091–1094. doi: 10.1212/wnl.59.7.1091. [DOI] [PubMed] [Google Scholar]
- 6.Zielasek J, Martini R, Suter U, Toyka KV. Neuromyotonia in mice with hereditary myelinopathies. Muscle Nerve. 2000;23:696–701. doi: 10.1002/(sici)1097-4598(200005)23:5<696::aid-mus5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 7.Biedermann F, Schumann NP, Fischer MS, Scholle HC. Surface EMG recordings using a miniaturised matrix electrode: a new technique for small animals. J Neurosci Meth. 2000;97:69–75. doi: 10.1016/s0165-0270(00)00170-9. [DOI] [PubMed] [Google Scholar]
- 8.Whelan PJ. Electromyogram recordings from freely moving animals. Methods. 2003;30:127–141. doi: 10.1016/s1046-2023(03)00074-4. [DOI] [PubMed] [Google Scholar]
- 9.Schumann NP, Biedermann FH, Arnold D, et al. Treadmill locomotion in normal mice: Step related multi-channel EMG profiles of thigh muscles. Pathophysiology. 2006;13:245–255. doi: 10.1016/j.pathophys.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Scholle HC, Biedermann F, Arnold D, Jinnah HA, Grassme R, Schumann NP. A surface EMG multi-electrode technique for characterizing muscle activation patterns in mice during treadmill locomotion. J Neurosci Meth. 2005;146:174–182. doi: 10.1016/j.jneumeth.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Schumann NP, Biedermann FHW, Kleine BU, et al. Multi-channel EMG of the M. triceps brachii in rats during treadmill locomotion. Clin Neurophysiol. 2002;113:1142–1151. doi: 10.1016/s1388-2457(02)00143-8. [DOI] [PubMed] [Google Scholar]
- 12.Scholle HC, Schumann NP, Biedermann F, et al. Spatiotemporal surface EMG characteristics from rat triceps brachii muscle during treadmill locomotion indicate selective recruitment of functionally distinct muscle regions. Exp Brain Res. 2001;138:26–36. doi: 10.1007/s002210100685. [DOI] [PubMed] [Google Scholar]
- 13.Leblond H, L'Esperance M, Orsal D, Rossignol S. Treadmill locomotion in the intact and spinal mouse. J Neurosci. 2003;23:11411–11419. doi: 10.1523/JNEUROSCI.23-36-11411.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milner TE, Cadoret G, Lessard L, Smith AM. EMG analysis of harmaline-induced tremor in normal and three strains of mutant mice with Purkinje cell degeneration and the role of the inferior olive. J Neurophysiol. 1995;73:2568–2577. doi: 10.1152/jn.1995.73.6.2568. [DOI] [PubMed] [Google Scholar]
- 15.Fortier PA, Smith AM, Rossignol S. Locomotor deficits in the mutant mouse, Lurcher. Exp Brain Res. 1987;66:271–286. doi: 10.1007/BF00243304. [DOI] [PubMed] [Google Scholar]
- 16.Loscher W, Fisher JE, Schmidt D, Fredow G, Honack D, Iturrian WB. The sz mutant hamster: a genetic model of epilepsy or of paroxysmal dystonia? Mov Disord. 1989;4:219–232. doi: 10.1002/mds.870040304. [DOI] [PubMed] [Google Scholar]
- 17.Chaniary K, Baron M, Rice A, Wetzel P, Shapiro S. Electromyographic characterization in an animal model of dystonia. Mov Disord. 2008;23:1122–1129. doi: 10.1002/mds.22040. [DOI] [PubMed] [Google Scholar]
- 18.Clarke KA, Still J. Gait analysis in the mouse. Physiol Behav. 1999;66:723–729. doi: 10.1016/s0031-9384(98)00343-6. [DOI] [PubMed] [Google Scholar]
- 19.Clarke KA, Still J. Development and consistency of gait in the mouse. Physiol Behav. 2001;73:159–164. doi: 10.1016/s0031-9384(01)00444-9. [DOI] [PubMed] [Google Scholar]
- 20.Ganor I, Golani I. Coordination and integration in the hindleg step cycle of the rat: kinematic synergies. Brain Res. 1980;195:57–67. doi: 10.1016/0006-8993(80)90866-5. [DOI] [PubMed] [Google Scholar]
- 21.Neychev V, Fan X, Mitev VI, Hess EJ, Jinnah HA. The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain. 2008;131:2499–2509. doi: 10.1093/brain/awn168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shirley TL, Rao LM, Hess EJ, Jinnah HA. Paroxysmal dyskinesias in mice. Mov Disord. 2008;23:259–264. doi: 10.1002/mds.21829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hess EJ, Jinnah HA. Mouse models of dystonia. In: Le Doux MS, editor. Animal models of movement disorders. San Diego: Elsevier Academic Press; 2005. pp. 265–277. [Google Scholar]
- 24.Fureman BE, Jinnah HA, Hess EJ. Triggers of paroxysmal dyskinesias in the calcium channel mouse mutant tottering. Pharmacol Biochem Behav. 2002;73:631–637. doi: 10.1016/s0091-3057(02)00854-7. [DOI] [PubMed] [Google Scholar]
- 25.Campbell DB, North JB, Hess EJ. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp Neurol. 1999;160:268–278. doi: 10.1006/exnr.1999.7171. [DOI] [PubMed] [Google Scholar]
- 26.Campbell DB, Hess EJ. L-type calcium channels contribute to the tottering mouse dystonic episodes. Mol Pharmacol. 1999;55:23–31. doi: 10.1124/mol.55.1.23. [DOI] [PubMed] [Google Scholar]
- 27.Lorden JF. Animal models of dystonia. In: Tsui JKC, Calne DB, editors. Handbook of dystonia. New York: Marcel Dekker; 1995. May, p. 42. [Google Scholar]
- 28.Lorden JF, McKeon TW, Baker HJ, Cox N, Walkley SU. Characterization of the rat mutant dystonic (dt): a new animal model of dystonia musculorum deformans. J Neurosci. 1984;4:1925–1932. doi: 10.1523/JNEUROSCI.04-08-01925.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jinnah HA, Sepkuty JP, Ho T, et al. Calcium channel agonists and dystonia in the mouse. Mov Disord. 2000;15:542–551. doi: 10.1002/1531-8257(200005)15:3<542::AID-MDS1019>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 30.Schicatano EJ, Basso MA, Evinger C. Animal model explains the origins of the cranial dystonia benign essential blepharospasm. J Neurophysiol. 1997;77:2842–2846. doi: 10.1152/jn.1997.77.5.2842. [DOI] [PubMed] [Google Scholar]
- 31.Bastian AJ, Martin TA, Keating JG, Thach WT. Cerebellar ataxia: abnormal control of interaction torques across multiple joints. J Neurophysiol. 1996;76:492–509. doi: 10.1152/jn.1996.76.1.492. [DOI] [PubMed] [Google Scholar]
- 32.Demirkiran M, Jankovic J. Paroxysmal dyskinesias: clinical features and classification. Ann Neurol. 1995;38:571–579. doi: 10.1002/ana.410380405. [DOI] [PubMed] [Google Scholar]
- 33.Fahn S, Marsden CD. The paroxysmal dyskinesias. In: Marsden CD, Fahn S, editors. Movement disorders 3. Oxford: Butterworth-Heinemann; 1994. pp. 310–347. [Google Scholar]
- 34.Malfait N, T.D. S. Does dystonia always include co-contraction? A study of unconstrained reaching in children with primary and secondary dystonia. Exp Brain Res. 2006 doi: 10.1007/s00221-006-0606-4. [DOI] [PubMed] [Google Scholar]
- 35.Grosse P, Edwards M, Tijssen MA, et al. Patterns of EMG-EMG coherence in limb dystonia. Mov Disord. 2004;19:758–769. doi: 10.1002/mds.20075. [DOI] [PubMed] [Google Scholar]
- 36.Tijssen MA, Munchau A, Marsden JF, Lees A, Bhatia KP, Brown P. Descending control of muscles in patients with cervical dystonia. Mov Disord. 2002;17:493–500. doi: 10.1002/mds.10121. [DOI] [PubMed] [Google Scholar]
- 37.Tijssen MA, Marsden JF, Brown P. Frequency analysis of EMG activity in patients with idiopathic torticollis. Brain. 2000;123:677–686. doi: 10.1093/brain/123.4.677. [DOI] [PubMed] [Google Scholar]
- 38.Farmer SF, Sheean GL, Mayston MJ, et al. Abnormal motor unit synchronization of antagonist muscles underlies pathological co-contraction in upper limb dystonia. Brain. 1998;121:801–814. doi: 10.1093/brain/121.5.801. [DOI] [PubMed] [Google Scholar]
- 39.Jenkins ME, Mink JW. Kinematic analysis of a reach-grasp-drink task in children with primary dystonia and age-matched controls. Mov Disord. 2004;19:S89. [Google Scholar]
- 40.Inzelberg R, Flash T, Schechtman E, Korczyn AD. Kinematic properties of upper limb trajectories in idiopathic torsion dystonia. J Neurol Neurosurg Psychiatry. 1995;58:312–319. doi: 10.1136/jnnp.58.3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beuter A, Legros A, Cif L, Coubes P. Quantifying motion in dystonic syndromes: the bare essentials. J Clin Neurophysiol. 2004;21:209–214. doi: 10.1097/00004691-200405000-00008. [DOI] [PubMed] [Google Scholar]
- 42.MacKinnon CD, Velickovic M, Drafta C, hesquijarosa A, Brin MF. Corticospinal excitability accompanying ballistic wrist movements in primary dystonia. Mov Disord. 2004;19:273–284. doi: 10.1002/mds.20017. [DOI] [PubMed] [Google Scholar]