

Abstract
Brain edema in acute liver failure (ALF) remains lethal. The role of vasogenic mechanisms of brain edema has not been explored. We previously demonstrated that matrix metalloproteinase-9 (MMP-9) contributes to the pathogenesis of brain edema. Here, we show that MMP-9 mediates disruptions in tight junction proteins in vitro and in brains of mice with ALF. We transfected murine brain endothelial cells with MMP-9 cDNA using pc DNA3.1 (+)/Myc-His A expression vector. Tissue inhibitor of matrix metalloproteinases (TIMP-1) cDNA transfection or GM6001 was used to inhibit MMP-9. ALF was induced in mice with azoxymethane. Endogenous overexpression of MMP-9 in brain endothelial cells resulted in significant degradation of tight junction proteins occludin and claudin-5. The alterations in tight junction proteins correlated with increased permeability to FITC-dextran molecules. The degradation of tight junction proteins and the increased permeability were reversed by TIMP-1 and GM6001. Similar results were found when MMP-9 was exogenously added to brain EC. We also found that tight junction proteins degradation was reversed with GM6001 in brains of mice with ALF.
Conclusions
Tight junction proteins are significantly perturbed in brains of mice with ALF. These data corroborate the important role of MMP-9 in the vasogenic mechanism of brain edema in ALF.
Keywords: acute liver failure, blood brain barrier permeability
Brain capillary endothelial cells and their tight junctions (TJ) form the blood-brain barrier (BBB) that regulates what goes in and out the brain. The paracellular passage of small molecules, such as water and solutes, is highly regulated by the TJ proteins, including occludin, claudin-5, and the associated proteins zona occluden (ZO)-1 and -2 (1).
In acute liver failure (ALF), brain edema and sepsis are the two leading causes of death (2). ALF occurs when there is a sudden loss of hepatic function in a person without preexisting liver disease. The mechanisms responsible for the development of brain edema in ALF remain inadequately characterized. Although the cytotoxic mechanism of brain edema is commonly known (3), the role of a vasogenic mechanism in ALF is not known.
Others have reported that vasogenic BBB failure in ALF results in increased selective permeability to small molecules(4–6). However, light and electron microscopy have shown that in subjects with ALF the BBB and its TJ are grossly intact(4–8). These findings are consistent with the emerging concept that a vasogenic brain edema results from subtle modifications of TJ proteins without an obvious disruption of the BBB(9). Consistent with a leaky BBB that lacks obvious structural breakdown, a perturbation in the extracellular loops of occludin can disturb the barrier function(10–12). Similarly, a deletion in claudin-5 results in increased BBB permeability to molecules <800 Da despite an intact BBB ultrastructure(13).
MMPs play significant roles in highly complex processes, including regulating cell behavior, cell-cell communication, and tumor progression(14, 15). MMPs mediate many different proteolytic reactions involving cellular surface elements, including adhesion molecules, receptors and intercellular junction proteins. We have shown that matrix metalloproteinase-9 (MMP-9) contributes to the pathogenesis of brain edema in ALF(16, 17). Moreover, in rodents with induced ALF we demonstrated that MMP-9 upregulation is coupled with downregulation of tissue inhibitor of MMP (TIMP)-1 and that blocking MMP-9 with specific monoclonal antibodies or a synthetic inhibitor (GM6001) successfully attenuated brain extravasation and edema(16, 17). These results suggest that a vasogenic mechanism contributes to the pathogenesis of brain edema in ALF.
MMP-9 has been shown to affect the TJ proteins, leading to increased BBB permeability. MMP-9 has also been implicated in BBB alterations in brain ischemia and trauma(18–20). Recently, MMP-9 was demonstrated to degrade occludin and claudin-5 in focal cerebral ischemia(21). Similarly, MMP-9 has been shown to alter occludin, claudin-5, and ZO-1 and -2 in early diabetic retinopathy(22–24) and in experimental dry eye(25). We have thus hypothesized that MMP-9 plays a critical role in BBB failure in ALF by directly affecting TJ proteins, particularly occludin and claudin-5.
In this study, we demonstrate that MMP-9 alters the TJ proteins. We examined the effects of MMP-9 on TJ integrity and BBB permeability when MMP-9 is endogenously overexpressed and when it is exogenously added to brain endothelial cells in vitro. We also determined the pattern of TJ alterations in brains of mice with induced ALF that were treated with the MMP-9 inhibitor GM6001 versus vehicle(16).
METHODS
An MMP-9 cDNA clone in pBluescript II KS+ vector was obtained from Dr. Gregory I. Goldberg at Washington University (St. Louis, MO). The insert was cleaved with Xba I, and the MMP-9 cDNA was amplified with forward primer 5′-CTCCGGGTACCATGTTCCAAACCTTTGAGGGCGAC-3′ and reverse primer 5′-GGTCAAGAATTCGTCCTCAGGGC-3′(26). The MMP-9 cDNA was subcloned to a pc DNA3.1 (+)/Myc-His A expression vector between the EcoR I and Kpn I digestion sites. MMP-9 was detected using anti-MMP-9 antibody or anti tag-His antibody. Sequence was confirmed by DNA Sequencing Core Laboratory at Mayo Clinic Rochester. TIMP-1 cDNA clone was purchased (MC202082, OriGene, Rockville, MD). MMP-9 antibody was purchased from Santa Cruz Biotechnology (sc-13520, Santa Cruz, CA); rabbit anti-claudin (Zy-34-1600), anti-occludin (Zy-71-1500), and anti-ZO-1 (Zy-40-2300) from Invitrogen-Zymed Laboratories (Carlsbad, CA); and mouse ZO-2 antibody from BD Transduction Laboratories (San Jose, CA).
Overexpressing MMP-9 and TIMP-1 in mouse brain endothelial (bEnd3) cells in vitro
The murine brain endothelial cell line bEnd3 was purchased from ATCC (CRL-2299, Manassas, VA). The bEnd3 cells were grown on 100-cm2 tissue culture plates (Falcon) until confluence in DMEM with 4.5 g/L glucose, 3.7 g/L sodium bicarbonate, 4 mM glutamine, 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were transfected with MMP-9 and TIMP-1 plasmid DNAs by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Cells that were transfected with pc DNA expression vector served as controls.
Exogenous MMP-9 added to bEnd3 cells
The bEnd3 cells were grown to confluence, and 100 ng/mL of human recombinant active MMP-9 (Calbiochem, LaJolla, CA) was added for 18 hours with and without 100 nM of the potent broad-spectrum MMP-9 inhibitor GM6001 (Calbiochem).
Immunofluorescent microscopy
The bEnd3 cells were grown to confluence. After the studies, the culture plates were fixed in 3% paraformaldehyde for 30 min, permeabilized in 0.2% Triton X-100, and blocked for 30 min with PBS containing 3% (w/v) BSA. Immunostaining for claudin-5 (1:200), occludin (1:200), and ZO-1 (1:200) was performed with primary antibodies for 1 hour at room temperature. Normal rabbit/mouse IgG (Invitrogen) was used for the controls. Slides were washed with PBS/glycine and incubated with Alexa Fluor 488 dye-conjugated goat anti-mouse and goat anti-rabbit Alexa 596 at 1:600 (Invitrogen). The fluorescent microscope (DM5000B; Leica) with a 63×/1.4 HCX planApo oil objective (Leica) was used. Photos were acquired with the FX4000 program (Leica) using a charge-coupled device camera (DFC350FX, Leica).
Gelatin zymography
Gelatinase activities including MMP-9 were measured by gelatin zymography(16). Protein levels were determined by BCA (Pierce Biotechnology, Rockford, IL). Twenty-microgram samples were electrophoresed at 4°C in 10% SDS-PAGE containing 1 mg/mL of gelatin (BioRad, Hercules, CA). MMP-9 and MMP-2 (Chemicon, Billerica, MA) were used as standards. The gels were processed, incubated at 37°C for 40 h, fixed, and stained with 0.5% Coomassie Blue R-250. Gelatinase activity presented as clear bands against a blue background and was quantitated using ImageQuant.
Western blotting for tight junction protein analysis
Triton X-100-soluble and -insoluble fractions were prepared for analysis of TJ proteins(12, 27). The cells were washed with PBS, scraped, and rinsed into 1 mL of lysate buffer (50 mM Tris-HCl pH 7.3, 150 mM NaCl, 3 mM MgCl, 1 mM dithiothreitol [DTT], 1 mM EDTA, 1 mM EGTA, 1.0 % Triton X-100), supplemented with protease and phosphatase inhibitors. Supernatant was collected as a Triton X-100-soluble fraction. The remaining residue was extracted by heat (95°C) with lysis buffer containing 1% SDS (including 50 mM Tris-HCl pH 7.3, 150 mM NaCl, 3 mM MgCl, 1 mM DTT, 1 mM EDTA, 1 mM EGTA) as a Triton X-100-insoluble fraction. Equal amounts of protein (30 μg) from each sample were resolved on 10–16% SDS-PAGE gels, transferred to nitrocellulose membrane, and immunoblotted. Rabbit polyclonal anti-claudin-5, anti-occludin, and anti-ZO-1 and mouse anti-ZO-2 antibodies were used at 1:1000 to detect TJ protein expression. MMP-9 and TIMP-1 were detected with anti-MMP-9 antibody and anti-TIMP-1 antibody (Calbiochem), respectively.
Paracellular permeability assay
In vitro permeability was assayed using FITC-dextran fluorescein. The bEnd3 cells were seeded onto 6-well collagen-coated Transwell inserts (0.4 μM pore size) (Corning Costar Corp., Cambridge, MA) at 5×105 per well and were cultured in BD Endothelial Culture Medium (BD Biocoat™ Endothelial Cell Growth Environment, BD Bioscience, San Jose, CA). The medium was replaced every second day with 1.5 mL to the outer and 0.5 mL to the inner chambers. When the cells reached confluence at day 5, they were transfected with MMP-9 and TIMP-1 plasmid DNA. After 18 hours, 50 μL of 5 mg/mL FITC-dextran 40 (Sigma-Aldrich, St. Louis, MO), was added to the inner chamber with final volume of 0.5 mL. In the studies examining the exogenous MMP-9 and GM-6001, confluent bEnd3 cells were treated with 100 ng/mL recombinant MMP-9 with and without 50 nM GM6001 for 18 hours.
To determine the permeability of the bEnd3 cellular barrier, aliquots of 100 μL from the outer chamber were taken after 3 hours. The extravasated FITC-dextran 40 was measured with excitation and emission wavelengths at 485 and 535 nm using a SpectraMax spectrophotometer, and results were analyzed using Softmax.
Brains from mice with ALF
ALF was induced according to our previous description (16). Control mice were injected with saline and vehicle alone. ALF was induced with intraperitoneal injection with azoxymethane (AOM) (Sigma-Aldrich) at 50 μg/g. Twelve hours later, the study mice were treated intraperitoneally with either GM6001 at 2 mg/mouse or vehicle every 12 h for three doses. At the comatose stages of ALF, the study mice were killed, and their brains were removed for analysis. Hemibrains were homogenized at 150 mg tissue/mL of extraction buffer (50 mM Tris-HCl pH 7.3, 150 mM NaCl, 3 mM MgCl, 1 mM DTT, 1 mM EDTA, 1 mM EGTA, 1.0% Triton X-100). Homogenates were centrifuged at 10,000 g for 10 minutes. Supernatants were collected as a Triton X-100-soluble fraction. The remaining residues were extracted by heat (95°C) with 1% SDS buffer (including 50 mM Tris-HCl pH 7.3, 150 mM NaCl, 3 mM MgCl, 1 mM DTT, 1 mM EDTA, 1 mM EGTA) as a Triton X-100-insoluble fraction. Three separate experiments were performed.
Data analysis
The results were expressed as mean ± standard deviation. Student t-test and ANOVA were used for statistical analysis. A p value < 0.05 was considered statistically significant.
RESULTS
Overexpression of MMP-9 and TIMP-1 in bEnd3 cells
In brain ischemia, MMP-9 is upregulated in brain capillary endothelial cells, astrocytes, and neurons(28, 29). We investigated the local effect of MMP-9 on TJ composition by transfecting bEnd3 cells with MMP-9 cDNA with and without TIMP-1. The bEnd3 cells were derived from mouse brains and have BBB characteristics including the TJ proteins occludin, claudin-5, and ZO-1 and ZO-2(30, 31). Transfection with MMP-9 alone resulted in 5-fold upregulation of MMP-9 activity and concentration compared with control cells (Figure 1A,B). Cotransfection of TIMP-1 suppressed the MMP-9 activity (Figure 1A,B). TIMP-1 was mildly upregulated in MMP-9-transfected cells (Figure 1D). In contrast, cotransfection with both TIMP-1 and MMP-9 resulted in 300% increase in TIMP-1 (Figure 1D) and in 35% reduction in both amount and activity of MMP-9 (Figure 1C,D). In contrast, MMP-2, which is the closely related gelatinase of MMP-9, was not affected by MMP-9 and TIMP-1 overexpression.
Figure 1.
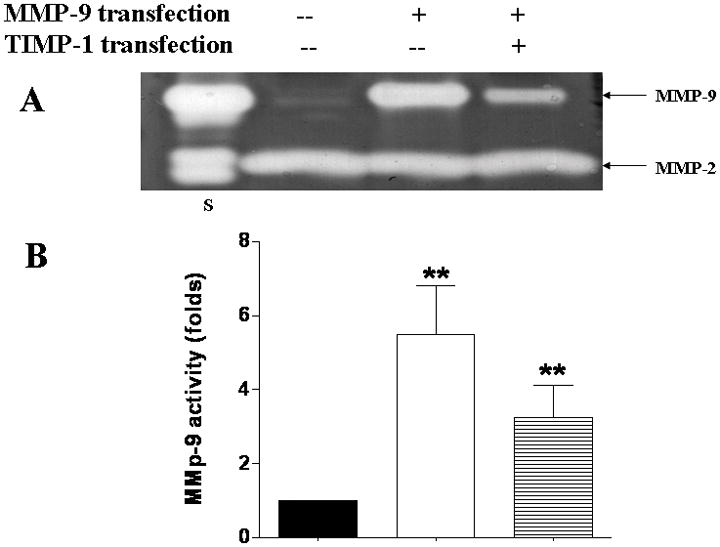
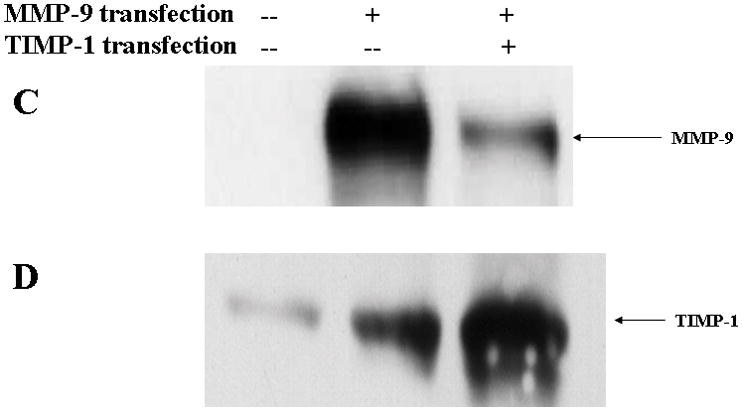
Endogenous overexpression of MMP-9 and TIMP-1 in bEnd3 cells. (A & B) Gelatin zymography and densitometric analysis of conditioned medium containing bEnd3 cells transfected with vector alone, with MMP-9, and with both MMP-9 and TIMP-1; Human MMP-9 and -2 are used as standards (lane S). Values are expressed as mean ± SD based on three individual tests (**p<0.01). (C & D) Western blot analysis of MMP-9 and TIMP-1 expression.
MMP-9 degrades occludin and claudin-5 in bEnd3 cells
The bEnd3 cells that were treated with vector alone, MMP-9 transfection, or both MMP-9 and TIMP-1 cotransfection were analyzed for the composition of TJ proteins. Triton X-100-soluble and -insoluble fractions were prepared(12, 27). The Triton X-100-soluble fraction represented the cytoplasmic and extracellular-associated forms of TJ proteins, and the Triton X-100-insoluble fraction was associated with the transmembrane portion of the TJ complex. Upregulation of MMP-9 induced a significant degradation of occludin in Triton X-100-soluble fractions. Both isoforms of occludin, i.e., the β (65 kDa) and a (63 kDa) forms, were equally susceptible to the degradation to 50-kDa form. The ratio of the 50-kDa to the 63- and 65-kDa isoforms of occludin markedly increased in MMP-9-transfected cells. The occludin degradation and 50/65–63 ratio were reversed by TIMP-1 (Figure. 2A,B,C). We observed similar alterations of occludin in Triton X-100-insoluble fractions (Figure. 2D,E).
Figure 2.
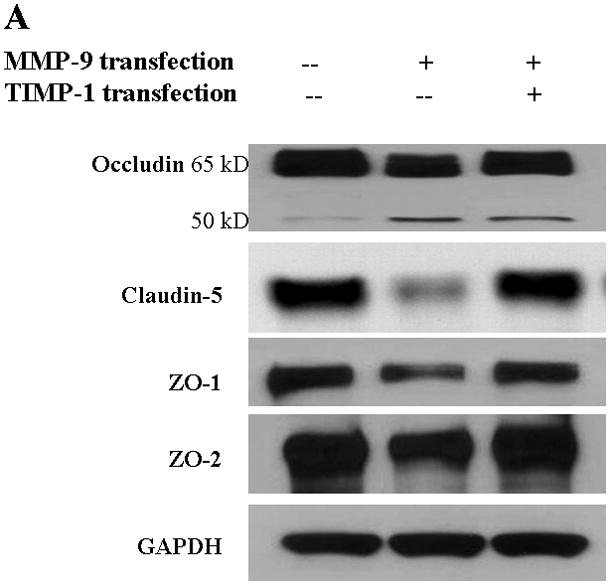
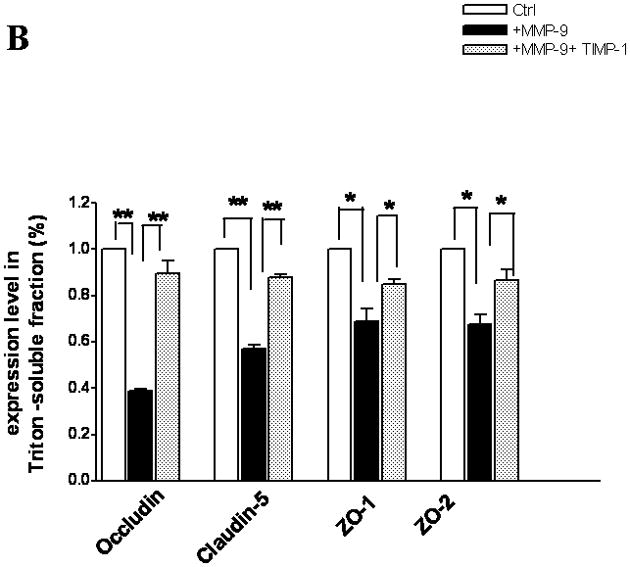
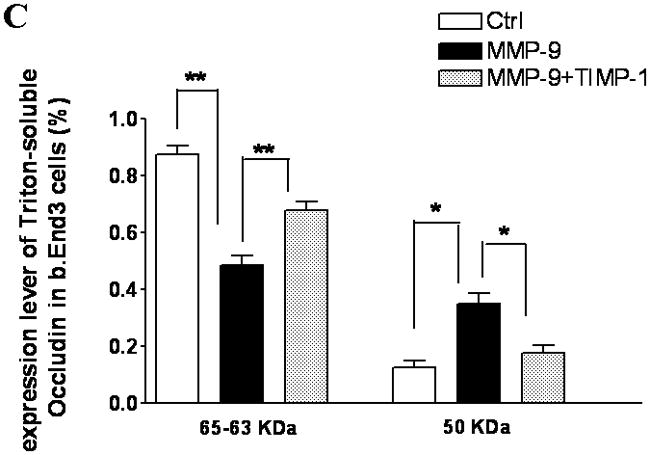
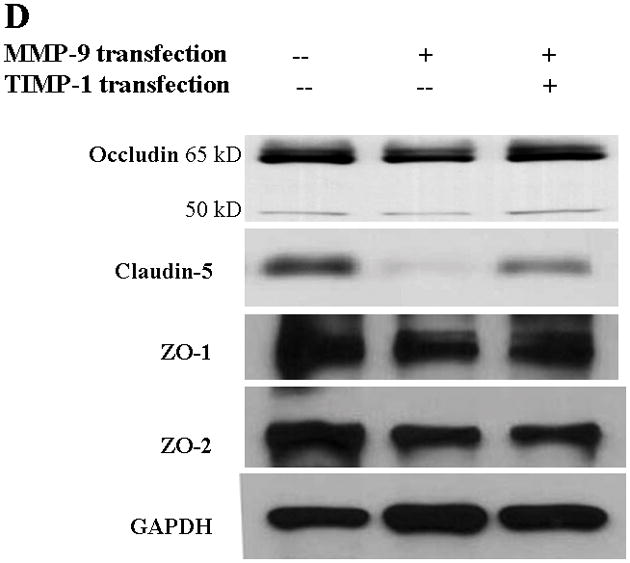
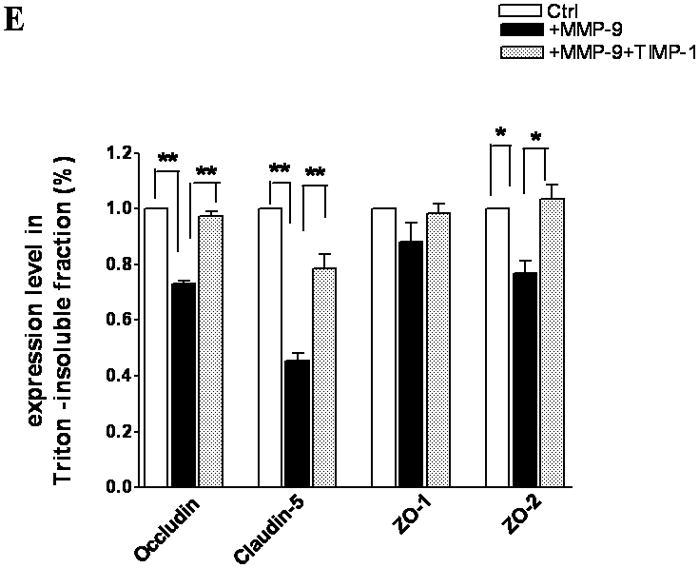
TJ protein expression in bEnd3 cells endogenously expressing MMP-9 and TIMP-1. (A & B) Immunoblot of occludin, claudin-5, ZO-1, and ZO-2 expression levels in Triton X-100-soluble fractions and densitometric analyses of bEnd3 cells transfected with vector alone, with MMP-9, and with both MMP-9 and TIMP-1. (C) The distribution levels of 65-kD and 50-kD isoforms of occludin in Triton X-100-soluble fractions in the three groups. (D & E) Immunoblot of TJ proteins in Triton-X-100-insoluble fractions and densitometric analyses of bEnd3 cells in the three groups. GAPDH expression was used as loading control. Values are expressed as mean percentage ± SD (*p<0.05, **p<0.01).
Similarly, MMP-9 upregulation induced significant degradation of claudin-5 in both Triton X-100-soluble and -insoluble fractions. Claudin-5 degradations were attenuated by upregulation of TIMP-1 (Figure 2B,E). At the same time, both ZO-1 and ZO-2 accessory proteins of TJ were significantly affected by MMP-9 in Triton X-100-soluble fractions (Figure 2B). Only ZO-2 was significantly affected in the Triton X-100-insoluble fractions (Figure 2A).
Exogenous MMP-9 degrades TJ proteins in bEnd3 cells
Because the brain is an innocent bystander with secondary injury in ALF, we sought to determine whether MMP-9, when present in a luminal fluid on the surface of the endothelial cell, would influence the TJ composition. Hence, we added exogenous MMP-9 to the confluent bEnd3 monolayer. We found that occludin, ZO-1, and ZO-2 were significantly decreased in Triton X-100-soluble fractions compared with the controls and that GM6001, a broad-spectrum MMP inhibitor, partly reversed the loss of the TJ proteins (Figure 3A,B). ZO-1 and ZO-2 were similarly altered. Although claudin-5 was affected negatively, its degradation was not reversed by GM6001 (Figure 3A,B). In contrast, there was no change in TJ protein expression in Triton X-100-insoluble fractions (Figure 3C,D).
Figure 3.
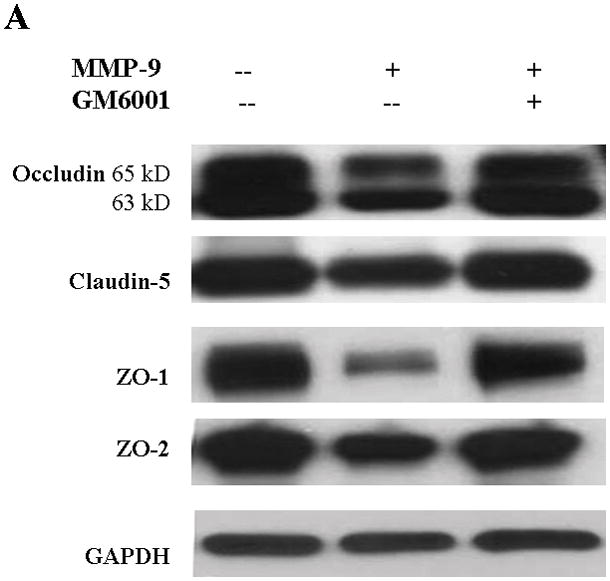
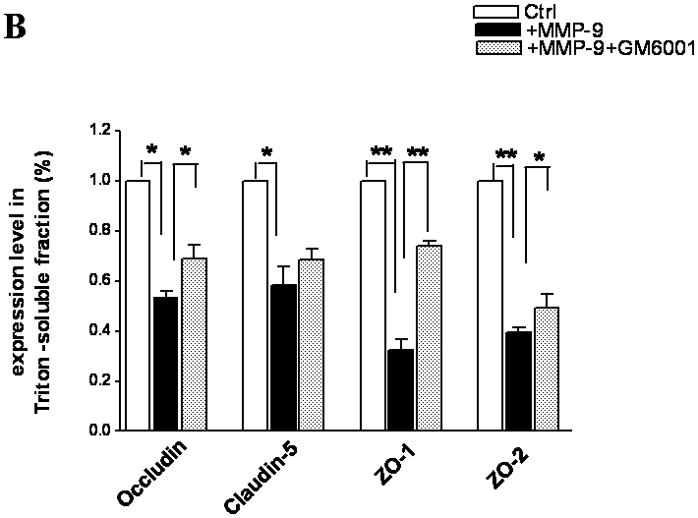
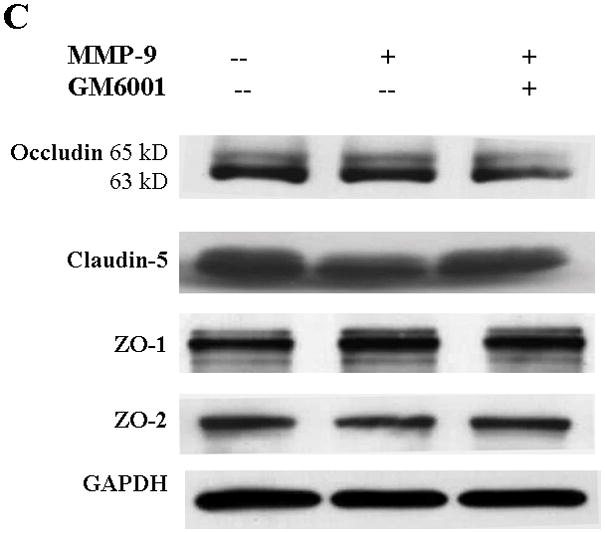
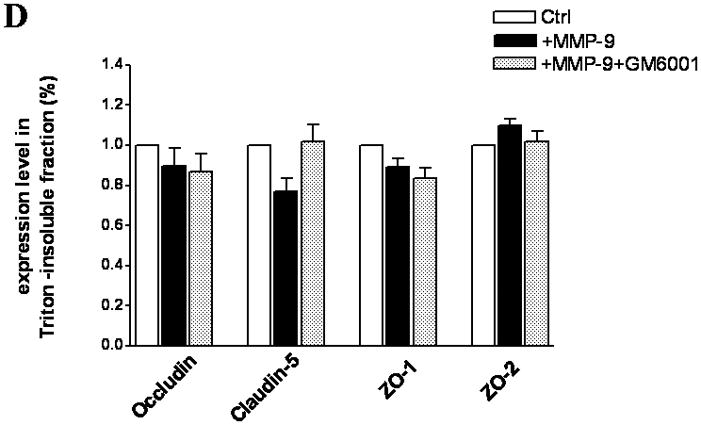
TJ protein expression in bEnd3 cells treated with exogenous MMP-9 and GM6001. (A & B) Immunoblot of occludin, claudin-5, ZO-1, and ZO-2 expression levels in Triton X-100-soluble fractions and densitometric analyses of bEnd3 cells incubated with culture medium alone, with 100 ng/mL of MMP-9, and with MMP-9 and 100 nM of GM6001. (C & D) Immunoblot of TJ proteins in Triton X-100-insoluble fractions and densitometric analyses of bEnd3 cells in the three groups. GAPDH expression was used as loading control. Values are expressed as mean percentage ± SD (*p<0.05, **p<0.01).
MMP-9 altered TJ protein distributions in bEnd3 cells
Under fluorescent microscopy, we observed a normal distribution of occludin, claudin-5, ZO-1, and ZO-2 forming a fine near-continuous line at the junction of cell-cell contact among the bEnd3 cells that were transfected with the vector alone (Control column in Figure 4). The bEnd3 cells that were transfected with MMP-9 showed marked irregularity and discontinuity in the distribution of occludin, claudin-5, ZO-1, and ZO-2 (MMP-9 column in Figure 4). The bEnd3 cells that were cotransfected with TIMP-1 and MMP-9 had significantly less perturbed distribution of the TJ components (MMP-9 + TIMP-1 column in Figure 4). Although not shown, a similar pattern of TJ alterations, to a lesser extent, was observed when MMP-9 was added to the bEnd3 monolayer and the alterations were attenuated with GM6001.
Figure 4.
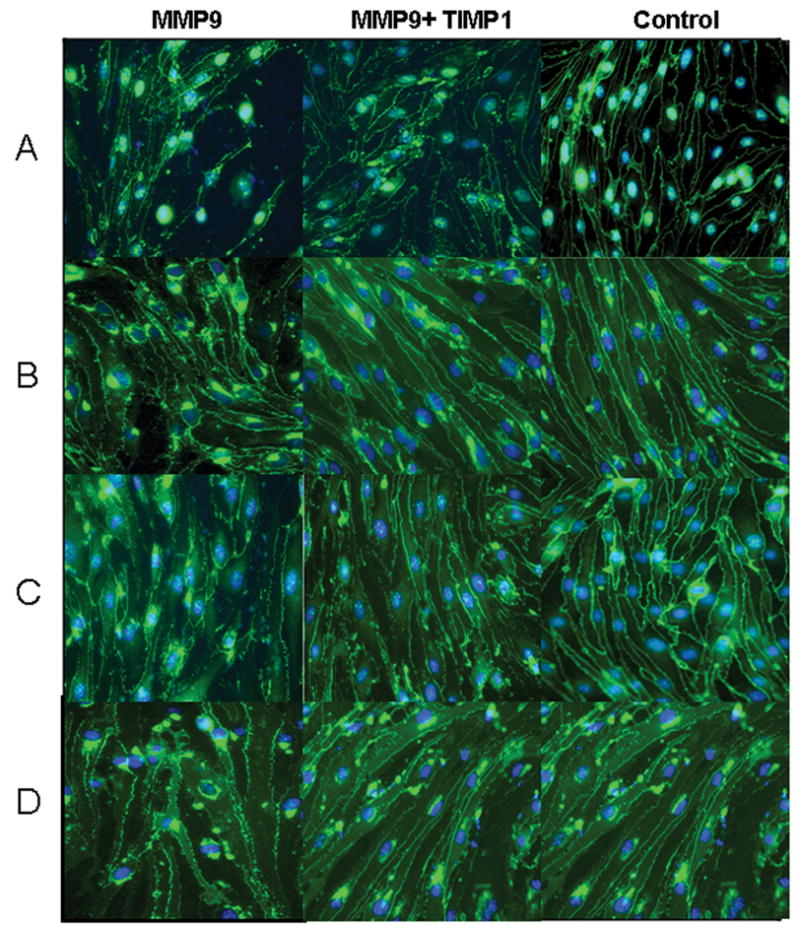
TJ protein distribution in monolayers of bEnd3 cells assessed by immunofluorescent microscopy. The bEnd3 cells were transfected with vector alone (control column), with MMP-9 alone (MMP-9 column), or with both MMP-9 and TIMP-1 (MMP-9 + TIMP-1 column). TJ proteins were immunohistologically targeted for occludin (row A), claudin-5 (row B), ZO-1 (row C), and ZO-2 (row D). (Magnifications at 400X and scale bar = 50 μm).
MMP-9 increases bEnd3 barrier permeability
We next determined the effect of MMP-9 on BBB permeability. The bEnd3 cells in a confluent monolayer possess characteristics of BBB, including TJ composition(30), and can be used to simulate the BBB in vitro(30, 31). The permeability across the BBB was assessed using FITC-dextran in a Transwell chamber. The bEnd3 cells were seeded on a collagen-coated Transwell filter for 5 days until the cells covered the entire surface of the insert and cell-free regions were virtually absent. FITC-dextran 40 was added to the upper compartment. After 3 hours, the extravasation of the FITC-dextran across the monocellular bEnd3 was significantly increased with MMP-9-transfected cells; the increased permeability was significantly attenuated with TIMP-1 cotransfection (Figure 5). Similarly, exogenous MMP-9 administration induced increased permeability of FITC-dextran, which was reduced with GM6001 (not shown).
Figure 5.
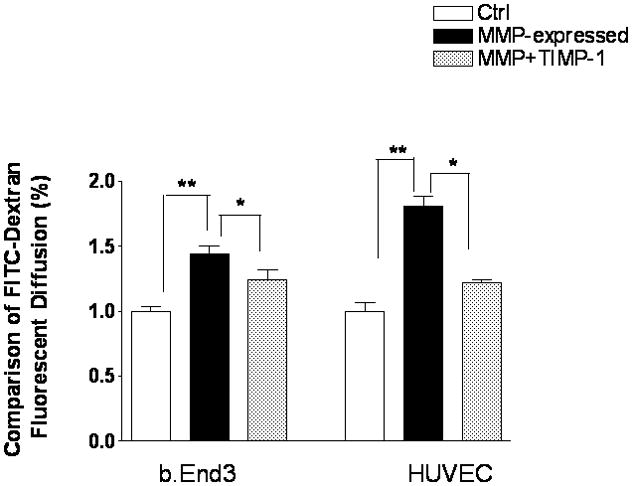
Effect of endogenously overexpressed MMP-9 on paracellular permeability in bEnd3 cells. bEnd3 cells were plated to confluence in collagen-coated 6-well Transwell inserts. Active MMP-9 was transfected with TIMP-1 plasmid DNA. For comparison, human umbilical vein endothelial cells (HUVEC) were similarly treated. Paracellular permeability assay was assessed using FITC-dextran fluorescein. Data are expressed as mean ± SD (*p<0.05, **p<0.01).
HUVEC have been used frequently for monocellular BBB permeability, so we corroborated the permeability study using HUVEC(32, 33). The HUVEC cells were transfected with MMP-9 and TIMP-1 in the same manner as the bEnd3 cells. We showed that endogenous MMP-9 expression resulted in increased BBB permeability with HUVEC that was significantly ameliorated with TIMP-1 (Figure 5).
Occludin and claudin-5 are degraded in brains of mice with ALF
We have previously demonstrated that blockage of MMP-9 with specific MMP-9 mAb or with broad-spectrum synthetic inhibitor GM6001 attenuated brain extravasation and edema in mice with AOM-induced ALF(16). The brains from normal control mice, ALF mice treated with vehicle, and ALF mice treated with GM6001 were examined with immunoblotting for the TJ proteins occludin, claudin-5, ZO-1, and ZO-2. We found a significant reduction of 65- and 63-kDa isoforms of occludin with a concomitant increase in the 50-kDa form of occludin in mice with ALF (Figure 6A,B). The decrease in 65- and 63-kD isoforms of occludin was significantly attenuated with GM6001 treatment (Figure 6A,B). The ratio of 50/60–63 occludin was reversed toward normal when ALF mice were treated with GM6001 (Figure 6C).
Figure 6.
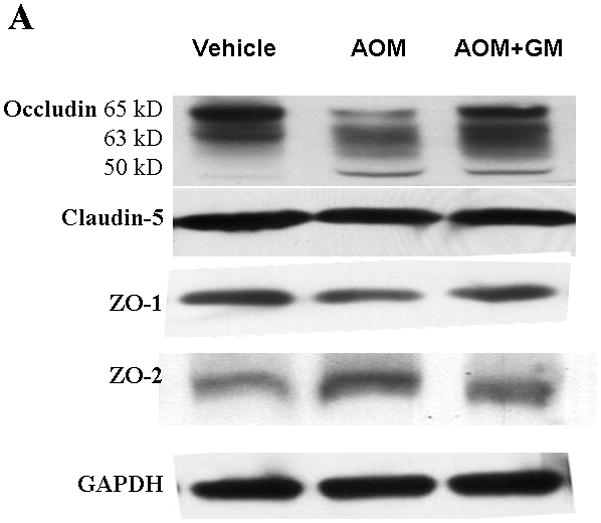
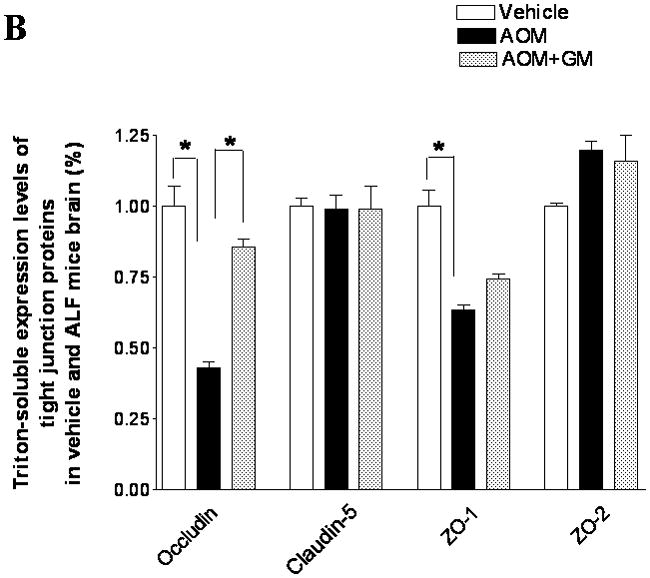
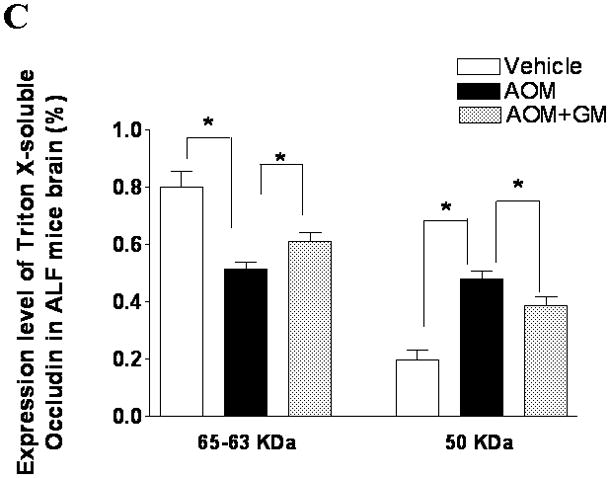
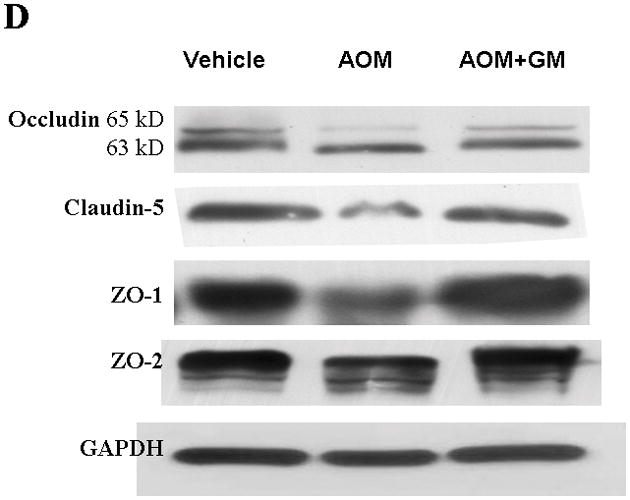
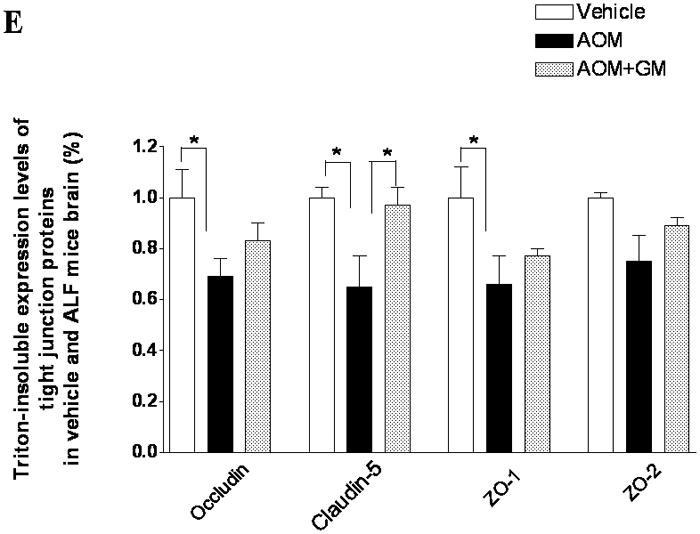
Alterations in TJ proteins in brains of mice with AOM-induced ALF. (A & B) Immunoblot of occludin, claudin-5, ZO-1, and ZO-2 expression levels in Triton X-100-soluble fractions and densitometric analyses of brains from control mice, mice with AOM-induced ALF, and mice with AOM-induced ALF treated with GM6001. (C) Distribution levels of 65-kD and 50-kD isoforms of occludin in Triton X-100-soluble fractions in the three groups. (D & E) Immunoblot of TJ proteins in Triton X-100-insoluble fractions and densitometric analyses of brains from mice in the three groups. GAPDH expression was used as loading control. Values are expressed as mean percentage ± SD (*p<0.05, n=8 per group).
Triton X-100-insoluble claudin-5 expression was also downregulated in brains of the ALF mice, and GM6001 prevented the effects of the downregulation. In contrast, the occludin and ZO-1 decreases were not reversed with GM6001 (Figure 6D, E).
These data corroborate the results observed with brain endothelial cells in vitro, providing support for the major role of MMP-9 in the proteolysis of occludin and other TJ proteins in ALF.
DISCUSSION
Since the early report of brain edema as a complication of ALF(34), the role of a vasogenic mechanism has not been examined. In this study, we demonstrated that the TJ proteins occludin and claudin-5 are significantly perturbed in brains of mice with experimentally induced ALF. Similar alterations in the TJ proteins were observed in vitro when brain endothelial cells were subjected to MMP-9. These alterations were reversed by MMP-9 inhibition with TIMP-1 or with the broad synthetic inhibitor GM6001. These findings support our hypothesis that vasogenic mechanism contributes to brain edema in ALF.
To determine whether MMP-9 influences BBB integrity, we examined TJ proteins in a murine brain endothelial cell line, bEnd3, which possesses characteristics of the BBB, including TJ proteins occludin, claudin-5, and ZO-1 and ZO-2.(30, 31) When MMP-9 was expressed by or added to bEnd3, we observed significant degradation of Triton X-100-soluble, i.e., extracellular, occludin and claudin-5 and their associated cytoplasmic accessories ZO-1 and ZO-2. These findings suggested that MMP-9 can directly modify the extracellular components of the TJ proteins. The upregulation of TIMP-1 resulted in a robust reversal of the degradation of the TJ proteins. In contrast, GM6001 was less successful at inhibiting the degradation of occludin, ZO-1, and ZO-2. Moreover, GM6001 failed to reverse the claudin-5 loss. The difference might be related to the much stronger and more specific inhibition capacity of TIMP-1 to MMP-9 than GM6001. In parallel, when we examined the Triton X-100-insoluble fractions derived from brain endothelial cells with endogenously expressed MMP-9, we observed significant perturbations in occludin, claudin-5, and ZO-2, which were mitigated with TIMP-1. However, no significant alterations in the TJ components were seen with the Triton X-100-insoluble fractions derived from brain endothelial cells with exogenously added MMP-9. The perturbations of the TJ proteins by MMP-9 correlated with in vitro BBB permeability. We observed increased BBB permeability in vitro when the bEnd3 cells endogenously expressed MMP-9 or when MMP-9 was added to their surface. Inhibiting MMP-9, particularly with TIMP-1, attenuated the BBB permeability.
The analysis of TJ proteins from mice with AOM-induced ALF reveals a pattern of TJ degradations and perturbations that resembles the in vitro findings, with some differences. The AOM-induced ALF models the liver failure that is seen in humans. Specifically, in the comatose stages, mice with ALF had increased brain extravasation and edema that recapitulates the clinical course that is observed in humans(16). In this study, we examined the composition of the tight junction in brains of comatose mice with ALF that were treated with GM6001 versus vehicle alone. The control mice were treated with saline without AOM. In mice with ALF, the Triton X-100-soluble occludin and ZO-1 were decreased, but claudin-5 and ZO-2 were not. Treatment with GM6001 attenuated loss of occludin but not ZO-1. Conversely, in the Triton X-100-insoluble fractions, although occludin, claudin-5, ZO-1, and ZO-2 were all decreased, only claudin-5 loss was reversed with GM6001. Importantly, the specific degradation of the 65- and 63-kD isoforms of occludin to the truncated 50-kD isoform was consistently observed in the Triton X-100-soluble fractions from brains of mice with ALF. This specific degradation of occludin was attenuated by GM6001. This observation is consistent with our in vitro results in which bEnd3 cells were transfected with MMP-9 and TIMP-1. The specific degradation of occludin is consistent with previous reports(21, 23–25, 35). Our results thus corroborate that MMP-9 directly modifies TJ proteins, particularly occludin.
Our observations that MMP-9 can degrade TJ proteins are consistent with previous reports of brain ischemic and traumatic injuries. MMP-9 mediates the BBB opening in brain ischemia(21, 36, 37). MMP-9 has recently also been implicated in the subtle alterations in the blood-retinal barrier in early diabetic retinopathy(22) and in experimental dry eyes(25). Interestingly, the pattern of occludin degradation of the 65–63 kDa isoforms versus the 50-kDa isoform suggests that MMP-9 plays a central role in regulating occludin. In contrast, MMP-2, a closely related relative of MMP-9, does not play a part in brain ischemic and traumatic injury(19). Furthermore, it has been shown that MMP-2 produces a different pattern in the degradation of occludin(35). The consistent profile of occludin degradation in vivo as well as in vitro strengthens the likelihood that MMP-9 has a role in altering TJ composition in ALF.
Thus, it appears that injury to the TJ in mice with ALF could result from either luminal exposure to circulating MMP-9 or from MMP-9 being produced by the endothelial cell itself. Our previous study suggested that brain cellular elements do not participate in producing injury to the TJ per se because we did not see an upregulation of MMP-9 in brains of mice with ALF(16). However, the possibility of inadequacy of our assays to detect a tiny concentration of MMP-9 cannot be excluded, because endothelial cells and other immediately surrounding cells could produce MMP-9(28, 29) in response to stress and circulating cytokines. In addition, the role of cerebral inflammation due to the circulating inflammatory cytokines in ALF has recently been implicated(38, 39). Although we observed microglial activation in the brains of the ALF mice (data not shown), we consistently did not find an upregulation of MMP-9 in the brains of these mice(16). Therefore, it would be unlikely that the degradation of the TJ proteins in this model of ALF was due to microglial cell activation.
It has been well recognized that MMP-9 and its proform are upregulated in the liver in response to injury(16, 40, 41). Within a few hours of onset of experimentally induced ALF, MMP-9 is continuously released into the systemic circulation(16, 42). Therefore, we speculated that MMP-9 might directly affect the integrity of the BBB in ALF. We demonstrated that a specific monoclonal antibody against MMP-9 effectively attenuated the brain extravasation and edema in ALF mice(16). Moreover, we showed that TIMP-1 is significantly decreased in the systemic circulation of animals with ALF and that GM6001, a broad-spectrum inhibitor of MMP-9, successfully reduced brain extravasation and edema in animals with ALF(17). In the present study, we demonstrated that the TJ protein occludin is degraded, thereby generating a 50-kDa isoform in the brains of mice with ALF and in vitro when MMP-9 is either endogenously induced in or exogenously administered to the brain endothelial cells. The alteration in occludin is ameliorated by TIMP-1 and GM6001. These findings suggest that MMP-9 targets the extracellular loop of occludin somewhere between amino acid 106 and 120 from the N-terminus(35). However, the exact nature of injury to occludin and other TJ proteins in ALF requires further investigation. A recent paper showed that occludin was decreased in the brain of comatose rats with surgically-induced ALF(43). Coupled with our data, the evidences support the concept that BBB dysfunction occurs as a consequence of occludin alterations. However, different mechanisms appear to contribute to the disease process. In addition, the division into Triton X-100-soluble versus -insoluble fractions is not rigorously adequate to distinguish the extracellular and transmembrane components of the TJ elements. Moreover, potential contributions of intracellular signals that are transduced by the cleavage of occludin have not been assessed(44, 45).
In conclusion, we demonstrated that in ALF, occludin and other TJ proteins are significantly disrupted, resulting in increased paracellular permeability across the BBB. These findings corroborate an important role for MMP-9 and its inhibitors in the pathogenesis and potential therapy of brain edema in ALF. Further understanding of the BBB and TJ regulation will permit more effective treatment of brain edema.
Acknowledgments
The work was supported (to JHN) by R01NS051646-01A2, AHA 0655589B, Deason Foundation, and Sandra and W. Eugene Davenport. The authors wish to acknowledge Diane Morell, Kathleen Norton and Lisa Maroski for their editorial assistance, and Dr. Michael B. Wallace for his critical review of the paper.
Abbreviations used in this article
- ALF
acute liver failure
- AOM
azoxymethane
- bEnd3
mouse brain endothelial cell
- BBB
blood-brain barrier
- MMP-9
matrix metalloproteinase-9
- TIMP-1
tissue inhibitor of MMP
- TJ
tight junction
- ZO
zona occluden
References
- 1.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Lee WM, Squires RH, Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: Summary of a workshop. Hepatology. 2008;47:1401–1415. doi: 10.1002/hep.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ede RJ, Williams RW. Hepatic encephalopathy and cerebral edema. Semin Liver Dis. 1986;6:107–118. doi: 10.1055/s-2008-1040594. [DOI] [PubMed] [Google Scholar]
- 4.Livingstone AS, Potvin M, Goresky CA, Finlayson MH, Hinchey EJ. Changes in the blood-brain barrier in hepatic coma after hepatectomy in the rat. Gastroenterology. 1977;73:697–704. [PubMed] [Google Scholar]
- 5.Gove CD, Hughes RD, Ede RJ, Williams R. Regional cerebral edema and chloride space in galactosamine-induced liver failure in rats. Hepatology. 1997;25:295–301. doi: 10.1002/hep.510250207. [DOI] [PubMed] [Google Scholar]
- 6.Scorticati C, Prestifilippo JP, Eizayaga FX, Castro JL, Romay S, Fernandez MA, Lemberg A, et al. Hyperammonemia, brain edema and blood-brain barrier alterations in prehepatic portal hypertensive rats and paracetamol intoxication. World J Gastroenterol. 2004;10:1321–1324. doi: 10.3748/wjg.v10.i9.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato M, Hughes RD, Keays RT, Williams R. Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology. 1992;15:1060–1066. doi: 10.1002/hep.1840150615. [DOI] [PubMed] [Google Scholar]
- 8.Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology. 1992;15:449–453. doi: 10.1002/hep.1840150316. [DOI] [PubMed] [Google Scholar]
- 9.Kimelberg HK. Water homeostasis in the brain: basic concepts. Neuroscience. 2004;129:851–860. doi: 10.1016/j.neuroscience.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 10.Wong V, Gumbiner BM. A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol. 1997;136:399–409. doi: 10.1083/jcb.136.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tavelin S, Hashimoto K, Malkinson J, Lazorova L, Toth I, Artursson P. A new principle for tight junction modulation based on occludin peptides. Mol Pharmacol. 2003;64:1530–1540. doi: 10.1124/mol.64.6.1530. [DOI] [PubMed] [Google Scholar]
- 12.Nusrat A, Parkos CA, Verkade P, Foley CS, Liang TW, Innis-Whitehouse W, Eastburn KK, et al. Tight junctions are membrane microdomains. Journal of Cell Science. 2000;113:1771–1781. doi: 10.1242/jcs.113.10.1771. [DOI] [PubMed] [Google Scholar]
- 13.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cauwe B, Van den Steen PE, Opdenakker G. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit Rev Biochem Mol Biol. 2007;42:113–185. doi: 10.1080/10409230701340019. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen JH, Yamamoto S, Steers J, Sevlever D, Lin W, Shimojima N, Castanedes-Casey M, et al. Matrix metalloproteinase-9 contributes to brain extravasation and edema in fulminant hepatic failure mice. J Hepatol. 2006;44:1105–1114. doi: 10.1016/j.jhep.2005.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamamoto S, Nguyen JH. TIMP-1/MMP-9 imbalance in brain edema in rats with fulminant hepatic failure. J Surg Res. 2006;134:307–314. doi: 10.1016/j.jss.2005.11.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gasche Y, Fujimura M, Morita-Fujimura Y, Copin JC, Kawase M, Massengale J, Chan PH. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 1999;19:1020–1028. doi: 10.1097/00004647-199909000-00010. [DOI] [PubMed] [Google Scholar]
- 19.Asahi M, Sumii T, Fini ME, Itohara S, Lo EH. Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. Neuroreport. 2001;12:3003–3007. doi: 10.1097/00001756-200109170-00050. [DOI] [PubMed] [Google Scholar]
- 20.Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401–6408. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- 22.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacological Reviews. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 23.Giebel SJ, Menicucci G, McGuire PG, Das A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Invest. 2005;85:597–607. doi: 10.1038/labinvest.3700251. [DOI] [PubMed] [Google Scholar]
- 24.Hawkins BT, Lundeen TF, Norwood KM, Brooks HL, Egleton RD. Increased blood-brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. 2007;50:202–211. doi: 10.1007/s00125-006-0485-z. [DOI] [PubMed] [Google Scholar]
- 25.Pflugfelder SC, Farley W, Luo L, Chen LZ, de Paiva CS, Olmos LC, Li DQ, et al. Matrix metalloproteinase-9 knockout confers resistance to corneal epithelial barrier disruption in experimental dry eye. American Journal of Pathology. 2005;166:61–71. doi: 10.1016/S0002-9440(10)62232-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogata Y, Enghild JJ, Nagase H. Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J Biol Chem. 1992;267:3581–3584. [PubMed] [Google Scholar]
- 27.Yoo J, Nichols A, Mammen J, Calvo I, Song JC, Worrell RT, Matlin K, et al. Bryostatin-1 enhances barrier function in T84 epithelia through PKC-dependent regulation of tight junction proteins. Am J Physiol Cell Physiol. 2003;285:C300–309. doi: 10.1152/ajpcell.00267.2002. [DOI] [PubMed] [Google Scholar]
- 28.Planas AM, Sole S, Justicia C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis. 2001;8:834–846. doi: 10.1006/nbdi.2001.0435. [DOI] [PubMed] [Google Scholar]
- 29.Rosenberg GA, Cunningham LA, Wallace J, Alexander S, Estrada EY, Grossetete M, Razhagi A, et al. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res. 2001;893:104–112. doi: 10.1016/s0006-8993(00)03294-7. [DOI] [PubMed] [Google Scholar]
- 30.Song L, Pachter JS. Culture of murine brain microvascular endothelial cells that maintain expression and cytoskeletal association of tight junction-associated proteins. In Vitro Cell Dev Biol Anim. 2003;39:313–320. doi: 10.1290/1543-706X(2003)039<0313:COMBME>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 31.Brown RC, Morris AP, O’Neil RG. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res. 2007;1130:17–30. doi: 10.1016/j.brainres.2006.10.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fontijn RD, Rohlena J, van Marle J, Pannekoek H, Horrevoets AJ. Limited contribution of claudin-5-dependent tight junction strands to endothelial barrier function. Eur J Cell Biol. 2006;85:1131–1144. doi: 10.1016/j.ejcb.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 33.Villasante A, Pacheco A, Ruiz A, Pellicer A, Garcia-Velasco JA. Vascular endothelial cadherin regulates vascular permeability: Implications for ovarian hyperstimulation syndrome. J Clin Endocrinol Metab. 2007;92:314–321. doi: 10.1210/jc.2006-1231. [DOI] [PubMed] [Google Scholar]
- 34.Ware AJ, D’Agostino AN, Combes B. Cerebral edema: a major complication of massive hepatic necrosis. Gastroenterology. 1971;61:877–884. [PubMed] [Google Scholar]
- 35.Bojarski C, Weiske J, Schoneberg T, Schroder W, Mankertz J, Schulzke JD, Florian P, et al. The specific fates of tight junction proteins in apoptotic epithelial cells. J Cell Sci. 2004;117:2097–2107. doi: 10.1242/jcs.01071. [DOI] [PubMed] [Google Scholar]
- 36.Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100. doi: 10.1016/s0006-8993(99)01843-0. [DOI] [PubMed] [Google Scholar]
- 37.Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- 38.Jalan R, Williams R. The inflammatory basis of intracranial hypertension in acute liver failure. J Hepatol. 2001;34:940–942. doi: 10.1016/s0168-8278(01)00038-1. [DOI] [PubMed] [Google Scholar]
- 39.Jiang W, Desjardins P, Butterworth RF. Direct evidence for central proinflammatory mechanisms in rats with experimental acute liver failure: protective effect of hypothermia. J Cereb Blood Flow Metab. 2009;29:944–952. doi: 10.1038/jcbfm.2009.18. [DOI] [PubMed] [Google Scholar]
- 40.Kim TH, Mars WM, Stolz DB, Michalopoulos GK. Expression and activation of pro-MMP-2 and pro-MMP-9 during rat liver regeneration. Hepatology. 2000;31:75–82. doi: 10.1002/hep.510310114. [DOI] [PubMed] [Google Scholar]
- 41.Knittel T, Mehde M, Grundmann A, Saile B, Scharf JG, Ramadori G. Expression of matrix metalloproteinases and their inhibitors during hepatic tissue repair in the rat. Histochem Cell Biol. 2000;113:443–453. doi: 10.1007/s004180000150. [DOI] [PubMed] [Google Scholar]
- 42.Wielockx B, Lannoy K, Shapiro SD, Itoh T, Itohara S, Vandekerckhove J, Libert C. Inhibition of matrix metalloproteinases blocks lethal hepatitis and apoptosis induced by tumor necrosis factor and allows safe antitumor therapy. Nat Med. 2001;7:1202–1208. doi: 10.1038/nm1101-1202. [DOI] [PubMed] [Google Scholar]
- 43.Sawara K, Desjardins P, Chatauret N, Kato A, Suzuki K, Butterworth RF. Alterations in expression of genes coding for proteins of the neurovascular unit in ischemic liver failure. Neurochem Int. 2009;55:119–123. doi: 10.1016/j.neuint.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 44.Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–934. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- 45.Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta. 2008;1778:794–809. doi: 10.1016/j.bbamem.2007.09.003. [DOI] [PubMed] [Google Scholar]