

Abstract
Site-specific delivery of drugs while minimizing unwanted distribution has been one of the pursued goals in cancer therapy. In this endeavor, we have developed targeted polymeric nanoparticles called amphiphilic urethane acrylate nonionomer (UAN) for encapsulation of diverse water-insoluble drugs and diagnostic agents, as well as for simple and reproducible surface conjugation of targeting ligands. Using monoclonal antibodies or lymphocyte function-associated antigen-1 (LFA-1) I domain engineered for varying affinities to intercellular adhesion molecule (ICAM)-1, we were able to deliver UAN nanoparticles to human cancer cells with the efficiency dependent on the strength of the molecular interactions and the degree of ICAM-1 expression on cell surface. Compared to non-specific uptake of free drugs, targeted delivery of UAN nanoparticles carrying equal amount of drugs produced more potent cytotoxicity. Notably, without the targeting ligands attached, UAN nanoparticles were largely precluded from non-specific uptake by the cells, resulting in much lower toxicity. The versatility of our UAN nanoparticles in both payload encapsulation and presentation of targeting ligands may facilitate developing a robust platform for evaluating various combinations of cancer drugs and molecular interactions toward developing effective cancer therapy formulations.
Keywords: self-assembled nanoparticle, targeted nanoplatform, adhesion molecule, tumor targeting, drug delivery, payload encapsulation
Introduction
Nanoparticles are emerging as a powerful platform for delivery of imaging [1] and therapeutic entities [2], particularly due to their potential applications in cancer therapy for early detection, enhanced therapeutic potency, and reduced side effects [3]. For example, microspheres, liposomes, nanoshells, dendrimers, and biodegradable polymers have been utilized as specific drug delivery systems [4]. Compared to the conventional methods of delivering therapeutic agents directly via intravenous or enteral routes, or as molecular conjugates, nanoparticle-mediated delivery may protect the therapeutic agents from chemical and enzymatic degradation and circumvent the need for chemical modification of therapeutic agents, which often leads to reduced potency and stability of drugs [5, 6]. Yet, a successful targeted delivery of nanoparticles needs to address the challenges in delivering hydrophobic payloads, controlling drug release, and accomplishing specific delivery while minimizing adverse immune response [7, 8].
Recently, polymeric nanoparticles have been reported to exhibit characteristics of sustained release, effective solubilization of hydrophobic drugs, and reduced interactions with the reticuloendothelial system [6, 9, 10]. In order to achieve selective targeting by nanoparticles, targeting ligands are often attached to the surface of the particles. The high surface-to-volume ratio in these nanoparticles increases the surface density of ligands, leading to a multivalent, high affinity binding effect [11]. Specific targeting is additionally improved by the enhanced permeation and retention effect in tumor environments due to a leaky vasculature structure, allowing appropriately sized nanoparticles to extravasate [12]. Nanoparticles of certain size range (10-100 nm) have been shown to accumulate more readily in tumors through the retention effect compared to the larger ones, making them optimal for tumor penetration.
To take advantage of these unique characteristics of polymeric nanoparticles, we have developed a versatile nanoparticle platform using amphiphilic urethane acrylate nonionomer (UAN) [13] with two functional layers. The hydrophilic outer shell is coated with a high density of Nickel-nitrilotriacetic acid (Ni-NTA) for simple conjugation of tumor-specific targeting molecules [14] and the hydrophobic core has a large capacity for water-insoluble chemotherapy drugs and imaging agents, where the encapsulation process occurs through the diffusion of hydrophobic molecules from water into the hydrophobic core [14, 15]. Through this approach, we demonstrate efficient encapsulation of water-insoluble fluorescent dyes, 9,10-diphenylanthracene (DPA) and fluorescein isothiocyanate (FITC), and cytotoxic drugs, such as camptothecin, a topoisomerase inhibitor [16], and celastrol, a proteasome inhibitor [17].
Some of the current approaches in targeting tumors include ligands specific to Her2 [18], transferrin receptor [19], epidermal growth factor receptor [20], folate receptor [21], integrin αVβ3 [22], hyaluronan receptor [23], and chondroitin sulfate [24]. As the target molecule in the present study, we have chosen intercellular adhesion molecule (ICAM)-1. ICAM-1 is present in low concentrations in the membranes of endothelial cells, and epithelial cells, and a subset of immune cells but is greatly up-regulated in response to inflammatory signals [25, 26]. In addition, constitutive over-expression of ICAM-1 has also been found in many carcinomas, such as breast, colon, non-small cell lung, renal-cell, pancreas, and gastric carcinomas compared to the respective normal epithelial cells [27-32] as well as in the tumor microenvironments, wherein an inflammatory milieu promotes angiogenesis and tumor growth [33].
Though most molecular targeting approaches have been based on antibodies [34] and small peptides [35], herein, we demonstrate ICAM-1 targeting with a native ligand, called the inserted (I) domain derived from lymphocyte function-associated antigen (LFA)-1. LFA-1 is a member of the integrin family, expressed on all leukocytes [36], that mediates leukocyte adhesion to endothelial cells and transmigration into inflamed tissues [37]. We used the I domains that have previously been engineered by various activating mutations (F292A, F292G, F265S/F292G) to increase the affinity to ICAM-1 in a step-wise manner [38]. Using ICAM-1 expressing HeLa cells [39] as a model cancer cell line, we demonstrate specific delivery of UAN encapsulating hydrophobic dyes and therapeutic agents to the cells, in a manner dependent on the affinity between the LFA-1 I domains and ICAM-1, as well as the expression level of ICAM-1.
Materials and Methods
Synthesis and characterization of NTA-coupled UAN nanoparticles
The synthesis of UAN was previously described [40]. In brief, glycerol propoxylate (n∼5, MW ∼ 1000, Sigma Aldrich) was mixed with toluene diisocyanate (TDI, Sigma Aldrich) to react the hydroxyl groups of glycerol propoxylate and the isocyanate (-N=C=O) groups of TDI (Fig. 1A). The remaining isocyanate groups in TDI were reacted with the hydroxyl groups of 2-hydroxyethyl methacrylate (2-HEMA, n∼34, Sigma Aldrich) and polyethylene glycol (PEG, MW = 1500, Sigma Aldrich). The final product, urethane acrylate nonionomer, consisted of a 1:3:2:1 molar ratio of glycerol propoxylate:TDI:HEMA:PEG. 50 mg of the UAN monomers were then cross-linked by the reaction between the vinyl groups (-CH=CH2) of HEMA. This polymerization was accomplished by 2 mg of azobisisobutyronitrile (AIBN, Sigma Aldrich), which creates free radical initiators when heated. The cross-linking step was carried out in 10 ml of dimethyl sulfoxide (DMSO, Sigma Aldrich) at 65°C overnight with vigorous stirring (Fig. 1B). The average molecular weight of the synthesized UAN chains was 6,700 (MALDI Micro MX, Waters) with a polydispersity of 2.0 measured via ambient temperature gel permeation chromatography (GPC, Waters). To create the NTA moiety on the hydroxyl end of the PEG groups in polymerized UAN, 10 mg of Nα,Nα-Bis(carboxymethyl)-L-lysine hydrate (NTA analog, Sigma Aldrich) in 0.1 ml DMSO was mixed with 6.4 mg of TDI (molar ratio of NTA to TDI is 1:1) for 2.5 hours at room temperature to prepare NCO-terminated NTA-TDI complexes. Then NTA-TDI was mixed with 50 mg of cross-linked UAN overnight at room temperature to covalently link NCO of NTA-TDI to OH groups of UAN chains. In order to form nanoparticles from these monomers, the UAN chains dissolved in DMSO were diluted in distilled water (dH2O) at a volume ratio of 1:10 (DMSO:dH2O). The payloads such as FITC, camptothecin, and celastrol were added to UAN chains in DMSO at 0-20% of UAN weight prior to dilution with water.
Figure 1. Synthesis and characterization of UAN.
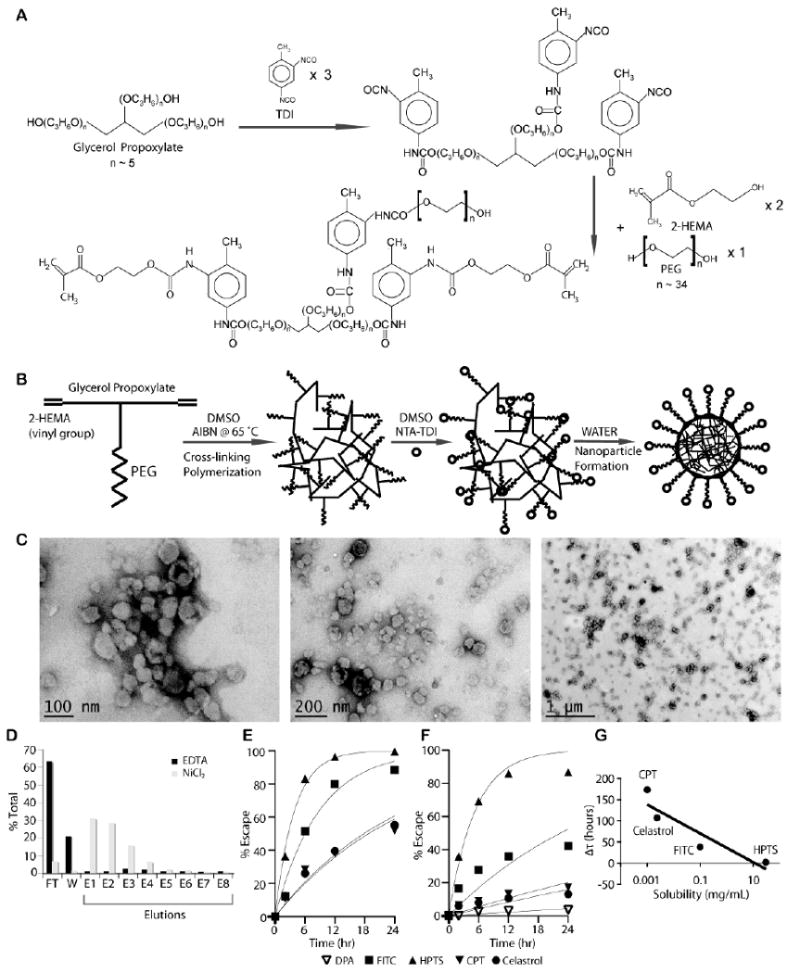
(A) UAN monomer is synthesized by covalently linking 2-HEMA, PEG, and glycerol propoxylate at 2:1:1 molar ratio. (B) NTA-UAN is formed by cross-linking UAN monomers with AIBN and conjugation of NTA to PEG. NTA-UAN forms nanoparticles when suspended in aqueous solution. (C) TEM images of UAN nanoparticles after staining with uranyl acetate. Scale bar is shown. (D) The function and specificity of Ni-NTA on UAN nanoparticles were confirmed by their binding to a His-peptide column. (E-F) The release kinetics of hydrophobic dyes and drugs from UAN were inferred by measuring the rate of escape through dialysis tubes without (E) or with (F) UAN. The fit of the first-order kinetics to the data is shown as a line. (G) The delay in the rate of escape (τ_UAN-τ) of the payloads (HPTS, FITC, celastrol, CPT) is plotted against their respective solubility values in water on a semi-log plot.
Dynamic light scattering (DLS, Malvern Instruments) was used to measure the average size of the UAN nanoparticles after synthesis, after NTA conjugation, and after payload encapsulation. Transmission electron microscopy (TEM; FEI Tecnai™) images of the UAN particles were taken after staining with 1% uranyl acetate. 5 μl of the sample was placed on a glow-discharge grid for two minutes, after which excess solution was removed with filter paper. 5 μl of 1% uranyl acetate was then placed on the grid for one minute and removed with filter paper. Images were taken after air-drying the grid for 30 minutes.
His-peptide column and Ni-NTA functionality test
His-peptide column was made by covalently attaching 2 mg of His-tag peptide (NH2-Cys-Gly4-Trp-Ser-His6-COOH; subscripts denote the number of repeats) to 1 ml of aldehyde-activated agarose resin (AminoLink Coupling Resin and Kit, Pierce). The Schiff base formed between the amine and aldehyde groups was then reduced by cyanoborohydride according to the manufacturer's protocol.
‘Escape’ Kinetics Assay
The release kinetics of the payloads encapsulated in UAN were indirectly measured using dialysis tubes. A highly water-soluble dye, 8-Hydroxypyrene-1,3,6-trisulfonic acid trisodium salt (HPTS, Anaspec) was used as a comparison with hydrophobic agents such as FITC (Pierce), celastrol (Cayman Chemical), and camptothecin (MP Biomedicals) to demonstrate UAN's ability to delay the ‘escape’ or the rate of diffusion of hydrophobic molecules through dialysis tubes. 1 ml of each UAN(payload) sample was placed in separate dialysis tubes (MWCO 7,000 Da, Fisher) and subsequently placed in separate 2 liter water-baths with stirring. After overnight dialysis, the concentrations of the payloads encapsulated within UAN were measured and the samples were subsequently placed in separate fresh dialysis tubes. 2 μl of each of the samples was taken out from the dialysis tubes and the absorbance levels were measured at different time points using a spectrophotometer (Nanodrop™ 2000) set at appropriate wave-lengths (HPTS: λmax = 454 nm, FITC: λmax = 495 nm, celastrol: λmax = 424 nm, camptothecin: λmax = 365 nm). Since DPA (Sigma Aldrich) was completely water-insoluble, DPA precipitated out of solution did not diffuse through the dialysis membrane. Therefore, UAN(DPA) was placed in a 1.7 ml microcentrifuge tube, and the supernatant after removing the DPA pellet that was released from UAN was measured for absorbance (λmax = 413 nm). The extinction coefficient and water solubility of each of the molecules are as follows: HPTS: 20,800 M-1cm-1 and highly water soluble, FITC: 70,000 M-1cm-1 and 100 μg/ml, celastrol: 10,063 M-1cm-1 and 2.33 μg/ml, camptothecin: 42,282 M-1cm-1 and 1 μg/ml, and DPA: 14,000 M-1cm-1 and water-insoluble. The same procedure was carried out to evaluate the escape kinetics of free materials with the initial concentrations of the payloads set to be equal to those encapsulated in UAN. The percent escape was plotted as 100% [C0-C(t)]/C0, where C(t) and C0 refer to the measured payload concentration inside the dialysis tube at time = t and at time = 0 h, respectively. The rate of escape from dialysis tubes (τ) was then calculated by assuming the first order diffusion model, C(t) = C0 exp(-t/τ) and curve-fitting the resulting equation, 100% [1- exp(-t/τ)], to the percent escape data.
Production of I domains, protein A, and R6.5 antibody
LFA-1 I domains and immunoglobulin G-binding recombinant protein A (17 kDa fragment) with a His tag at the C-terminal were produced in Escherichia coli BL21(DE3) (Invitrogen) using pET20b and pET28a vectors, respectively. To produce protein A, a 1 liter bacteria culture with OD600 of 0.4-0.5 was induced with 1 mM isopropyl-b-D-thiogalactoside (IPTG, Gold Biotechnology) for 4 hours, after which it was centrifuged (3000 rpm, 4°C, 10 minutes) to pellet the cells. The soluble fraction of protein A was extracted by sonication and was purified using a Ni-NTA column (Novagen). Elutions from the column were further purified by size exclusion in a liquid chromatography column (Akta, GE Healthcare). Recombinant LFA-1 I domains (Asn-129 to Tyr-307) were produced as described previously [38]. Briefly, after protein induction, inclusion bodies were resuspended in 10 ml of the washing buffer (50 mM Tris (pH 8.0), 23% w/v sucrose, 0.5% w/v Triton X-100, 1 mM ethylenediaminetetraacetic acid (EDTA)), sonicated, and centrifuged again to wash the inclusion body. This process was repeated three times. Then, the inclusion bodies were solubilized with 20 ml of the solubilization buffer (50 mM Tris (pH 8.0), 6 M Guanidine-HCl) for 1.5 hours at 4°C with stirring. This solution was diluted with 2 liter of refolding buffer (50 mM Tris (pH 8.0), 10% glycerol, 1 mM MgCl2) and stirred slowly at 4°C overnight to initiate protein refolding. Finally, the refolded solution was concentrated down to 1-5 ml by centrifugation (Amicon 5 kDa MWCO Millipore), filtered through 0.45 μm, and purified by gel filtration chromatography. Monoclonal antibody (mAb) R6.5 was produced from hybridoma (ATCC), and purified by a Protein A column (Pierce) followed by size exclusion.
Cell culture and labeling with UAN
HeLa cells were grown in Advanced DMEM (Gibco) containing 10% FBS (Atlanta Biologicals) and 2mM L-glutamine (GlutaMAX™, Gibco) at 37°C in a 5% CO2 humidified incubator. HeLa cells in 96 well plates (∼90 % confluency) were washed twice with 100 μl of the labeling buffer (phosphate buffer saline (PBS), pH 7.4, 5% bovine serum albumin (BSA), 5 mM MgCl2) and incubated with UAN (250 μg/ml) conjugated to I domain in 100 μl of labeling buffer at room temperature for 30 minutes. UAN(payload) nanoparticles were kept in a dialysis tube (MWCO 7,000 Da, Fisher) until they were used for cell labeling, with a minimum dialysis time of 48 hours to remove unencapsulated payloads. The unbound UAN particles were aspirated out and the cells were washed three times with the labeling buffer. The cells were then washed with the culture media and kept at 37°C, while being observed at different time points. Human breast adenocarcinomas (MDA-MB-231) were cultured in αMEM containing 10% FBS and 2mM L-glutamine, human thyroid cancer cell line (KTC-1) [41] in RPMI 1640 with 10% FBS and 2mM L-glutamine, and human microvascular endothelial cell (HMEC)-1 in MCDB 131 media (Gibco) containing 10% fetal bovine serum, 2mM L-glutamine, 10 ng/ml human epidermal growth factor, 1 μg/ml hydrocortisone, and 1% penicillin-streptomycin at 37°C in a 5% CO2 humidified incubator. Human acute monocytic leukemia cell line, THP-1 (ATCC), were cultured in RPMI 1640 media with 10% fetal bovine serum, and 0.05 mM 2-mercaptoethanol at 37°C in a 5% CO2 humidified incubator. In order to induce upregulation of ICAM-1, the cells were treated with 1 μg/ml of lipopolysaccharide (LPS) in the appropriate culture media for 24 hours.
Immunofluorescence Flow Cytometry
HeLa, KTC-1, MDA-MB-231, and HMEC-1 cells (∼200,000 cells per labeling) were trypsinized and washed with the labeling buffer. Since THP-1 is a suspension cell line, it was centrifuged (700 rpm, 4°C) and washed with the labeling buffer. 250 μg/ml of UAN coated with I domain and containing FITC (I domain-UAN(FITC)) in 100 μl was added to the cells. After 15 minutes of incubation, cells were washed and re-suspended in 100 μl of the labeling buffer for flow cytometry (COULTER® EPICS® XL-MCL™ Flow Cytometer, Beckman Coulter). In evaluating the expression level of ICAM-1 in different cell lines, cells were labeled with 10 μg/ml of mAb R6.5 or LB2 (Santa Cruz Biotech), washed, and subsequently labeled with goat-anti-mouse-PE secondary antibody (Santa Cruz Biotech).
Quantification of Cell Viability
To quantify the number of viable cells after drug delivery, cells were fixed with 10% formalin for 5 minutes and stained with 0.05% crystal violet (Fisher Scientific) in distilled water for 30 minutes. After washing out the dye outside of the cells, methanol was added to release the crystal violet from the cells, which was quantified by detecting the absorbance level at 540 nm.
Statistical Analysis
Data were expressed as mean ± standard deviation of at least triplicate samples. One-way ANOVA was performed on the four groups of HeLa cells, each treated with UAN(drug) conjugated to different I domain mutants (WT, F292A, F292G, F265S/F292G). The means of the number of viable cells from each of the groups were compared. Post hoc Tukey's HSD test was carried out in order to assess which groups significantly differed from others. All of the statistical analyses were performed using GraphPad Prism5 (GraphPad Software).
Results
Synthesis and characterization of targeted UAN nanoparticles
When NTA-coupled UAN (NTA-UAN) dissolved in DMSO is diluted in water, polypropylene oxide-based segments associate with each other to form micelle-like nanoparticles with hydrophobic interior. Being covalently bound to the OH groups of hydrophilic polyethylene oxide, NTA groups are more likely to be exposed on the surface of the UAN nanoparticles (as depicted in Fig. 1B). Similar to the surfactant micelles, UAN chains can absorb small hydrophobic payloads within their hydrophobic core. Thus, the hydrophobic agents can be added to NTA-UAN in DMSO (FITC, celastrol, camptothecin) or in water after dilution (DPA), which would cause the agents to be encapsulated in the core of the nanoparticles through hydrophobic interactions. Dynamic light scattering was used to measure the size distribution (hydrodynamic radius) of the following formulations: bare UAN (32.6 nm ± 2.7), NTA-UAN (42.98 nm ± 4.12), and NTA-UAN after encapsulation of various hydrophobic agents (NTA-UAN(DPA): 47.72 nm ± 5.56, NTA-UAN(FITC): 44.29 nm ± 2.19, NTA-UAN(camptothecin): 52.22 nm ± 5.3, NTA-UAN(celastrol): 43.7 nm ± 1.82). TEM images revealed that bare UAN nanoparticles were spherical, uniformly sized, and well dispersed (Fig. 1C).
The presence of NTA on the surface of UAN was studied using a His-peptide column (Fig. 1D). NTA-UAN nanoparticles were first charged with nickel ions by incubation with 10 mM NiCl2 in PBS (pH7.4) and loaded to the His-peptide column, after which the amount of flow through and samples eluted with 50 mM EDTA were determined by the absorbance levels at 280 nm. Approximately 90% of NTA-UAN nanoparticles were bound to the column, and eluted entirely with 50 mM EDTA. On the other hand, NTA-UAN pretreated with EDTA to remove nickel ions from UAN nanoparticles did not bind to the His-peptide column, indicating the binding of NTA-UAN nanoparticles to the column was solely dependent on Ni-NTA interaction with His-peptide.
The amounts of the payloads encapsulated in 1 mg of NTA-UAN nanoparticles after overnight dialysis were 110 μg of FITC, 87.8 μg of celastrol, 4 μg of DPA, and 28.6 μg of camptothecin. To study the release kinetics of the payloads from UAN nanoparticles, we measured the rate of diffusion or the escape rate of the payloads through a dialysis membrane with the appropriate molecular weight cutoff to contain UAN nanoparticles but allow free payloads to diffuse through. We found that the time constants (τ) for the first-order escape kinetics of the payloads from dialysis tubes without UAN were in correlation with their hydrophobicity: the 95% confidence interval for τ values of HPTS, FITC, celastrol, and camptothecin were, respectively, 3.9 ± 0.7, 9.4 ± 2.6, 27.5 ± 7.0, 30.3 ± 9.9 hours (Fig. 1E). The rate of escape of the payloads after encapsulation into UAN (τ_UAN) was delayed in proportion to the payloads’ hydrophobicity: the τ_UAN values of HPTS, FITC, celastrol, and camptothecin were, respectively, 6.1 ± 2.3, 47.7 ± 27.0, 203.7 ± 115.5, 134.4 ± 61.8 hours (Fig. 1F, G). The release rate of DPA from UAN was measured at 757.1 ± 417.4 hour.
Affinity Dependent Cell Labeling Assay
LFA-1 I domain exhibits multiple conformations that are linked to differential affinity to ICAM-1 (Fig. 2A, B). Using a directed evolution approach, we have previously isolated a number of point mutations that led to an increase in the affinity of the I domain to ICAM-1 [38]. The equilibrium dissociation constants (KD) for WT, F292A, F292G, and F265S/F292G were determined, respectively, to be 1500 ± 200 μM, 20 ± 0.8 μM, 0.119 ± 0.017 μM, and 0.006 ± 0.0002 μM. (Fig. 2D). Compared to the more commonly used targeting approaches based on antibodies or short peptides, the I domain is a native, small (∼20 kDa), and highly specific ligand to the ICAM family molecules, and can be produced in bacteria in a large-scale. The I domain was produced with a His-tag for non-covalent conjugation to NTA-UAN nanoparticles. To estimate a maximum coating capacity of UAN, LFA-1 I domain was added in excess to UAN-NTA, after which the unbound I domain was removed by gel filtration. By the Lowry method (DC Protein Assay Kit, BioRad), the maximum capacity of UAN in binding the I domain was determined to be ∼100 μg of I domain/mg of UAN. In order to obviate the need for removing unbound proteins, the weight ratio of I domain to UAN for conjugation was used at 1:25 throughout this study. This corresponds to a molar ratio of 136:1 (I domain :UAN nanoparticle), given the molecular weight of the I domain of ∼20 kDa, the measured value of UAN density of 1 mg/ml, and the molecular weight of 68 MDa estimated for UAN with a 60 nm diameter.
Figure 2. LFA-1 I domains engineered for high affinity for targeting ICAM-1.
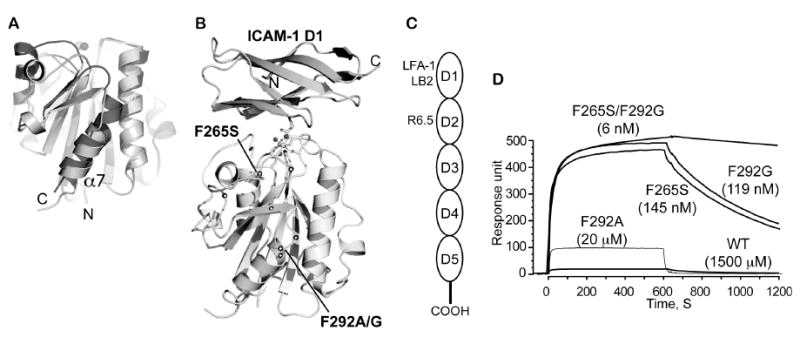
(A) Structural diagrams of low (inactive) and high affinity (active) conformations of LFA-1 I domains. Structurally conserved regions between the two states are in light gray, while the regions that differ more than 1 Å in their Cα positions are in dark gray (inactive) and in medium gray (active). The metal ions in the metal ion-dependent adhesion site are shown as spheres. (B) The structure of the I domain in complex with ICAM-1 domain 1 (D1). Allosteric activation sites found in our previous study [38] are displayed in spheres along the peptide backbone. The metal ion and three oxygen atoms of water molecules are depicted as spheres, while the residues that coordinate to the metal ion are drawn with the sticks. (A) and (B) are adapted from Fig. 5 of Xuebo et al [51]. (C) Schematic of ICAM-1 domains. Domain 1 binds to LFA-1 I domain. The mAbs LB2 and R6.5 bind domain 1 and domain 2, respectively. (D) Surface plasmon resonance data for binding of I domains (100 μM of WT (wild-type) and F292A and 1 μM of F265S, F292G, and F265S/F292G) to ICAM-1 immobilized (adapted from Fig. 2 of Jin et al [38]). Compared to the wild-type, each mutation of I domain (F292A, F265S, F292G, F265S/F292G) increases the binding affinity of I domain to ICAM-1 in a step-wise manner. The numbers in parentheses denote the equilibrium dissociation constants, KD.
To determine how the nature of different I domain mutants coating UAN would influence their interactions with ICAM-1, we examined the binding and uptake of nanoparticles by living cells. Cervical cancer cells (HeLa) were used as a model for the study, representing a group of cancer cells expressing high levels of ICAM-1. HeLa cells were labeled with I domain-UAN(FITC), washed with the culture media, and maintained at 37°C, while being observed at different time points (images were taken 2 hours after incubation) (Fig. 3A). The fluorescence intensity exhibited by the cells reflected LFA-1 I domain/ICAM-1 affinity dependent labeling: while F292A-UAN(FITC) showed only slightly higher cell labeling than the wild-type, F292G-UAN and F265S/F292G-UAN led to greatly enhanced fluorescence. The observed affinity dependent binding of UAN(FITC) was confirmed in a separate experiment, in which trypsinized HeLa cells were labeled with I domain-UAN(FITC) and the fluorescence intensity was measured by flow cytometry (Fig. 3B).
Figure 3. Affinity- and expression-dependent delivery of I domain-UAN(FITC) to ICAM-1 expressing cells.
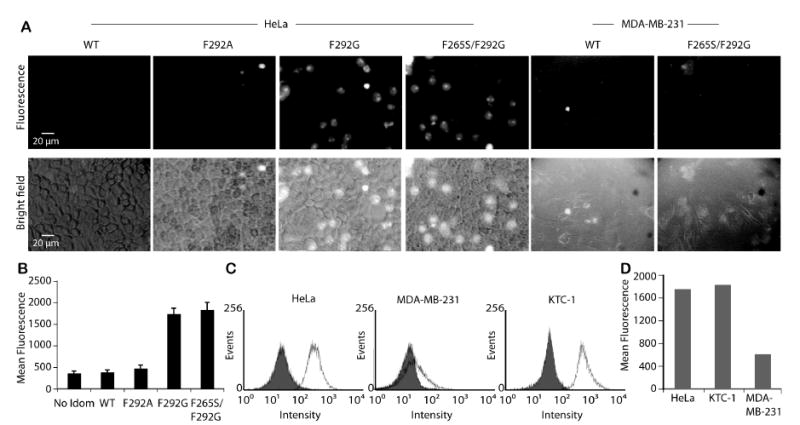
(A) UAN nanoparticles encapsulating FITC and conjugated to different I domains were delivered to HeLa and MDA-MB-231 cells. (B) Immunofluorescence flow cytometry measurement of the delivery into HeLa of UAN(FITC) with no I domain, WT, or I domain variants (n = 3). (C) The levels of ICAM-1 expression in HeLa, MDA-MB-231, and KTC-1 cells were measured by mAb R6.5 (open histogram) or with secondary antibody alone as a control (filled histogram). (D) Immunofluorescence flow cytometry measurements of F265S/F292G-UAN(FITC) binding to HeLa, MDA-MB-231, and KTC-1 cells.
In order to examine if the expression level of surface antigen influences the efficiency of nanoparticle delivery, the level of I domain-UAN(FITC) binding to HeLa cells was compared to the association with other cancer cell lines, KTC-1 and MDA-MB-231, which express high and low levels of ICAM-1, respectively (Fig. 3C). As expected, the levels of I domain-UAN(FITC) binding to KTC-1 and MDA-MB-231 were in good correlation with the levels of ICAM-1 expression (Fig. 3D). The dependence of UAN targeting on the levels of antigen expression will be an important facet in achieving preferential delivery into cells over-expressing tumor-specific antigens.
Affinity Dependent Cellular Cytotoxicity Assay
We examined the cellular cytotoxicity effects of UAN nanoparticles encapsulating celastrol, a quinone methide triterpene derived from the Chinese plant Tripterygium wilfordii [17, 42], which is known to inhibit the function of proteasomes[42]. Celastrol has been shown to inhibit the proliferation of a variety of tumor cells, including those of leukemia, gliomas, prostate cancer [43], and melanomas [44]. After incubation with the UAN conjugated to I domains of different affinity (WT, F292A, F292G, F265S/F292G) for 30 minutes, HeLa cells were washed and maintained in culture media for 24 hours before assaying for cell viability. UAN encapsulating high (125 μM) and low (20 μM) concentrations of celastrol caused I domain affinity dependent cytotoxicity to cells (Fig. 4). In the case of UAN encapsulating 20 μM celastrol, more than 80% cell death was detected with F292G-UAN and F265S/F292G-UAN, and a much lower cytotoxic effect with WT-UAN and F292A-UAN (Fig. 4A, D). Though a similar pattern was detected with UAN encapsulating 125 μM celastrol, a significant number of cells had died and lifted off from the culture flask when targeted with WT-UAN as well (Fig. 4B, E). Monomeric interaction between the wild-type I domain and ICAM-1 is low (1500 ± 200 μM) [45], but the high density of I domains attached to UAN would have increased the effective affinity to ICAM-1 via multivalency or avidity effect, resulting in a sufficient amount of celastrol delivered into the cells. The discrepancy between the minimal cell labeling by WT-UAN(FITC) and the significant cell death caused by WT-UAN(celastrol) can be attributed to the higher sensitivity with which cellular cytotoxicity can be detected compared to the sensitivity of our imaging system in detecting fluorescence. To further explore the efficacy of these nanoparticles in delivering chemotherapeutic agents, we replaced celastrol with camptothecin. Camptothecin loaded at 2 μM produced cytotoxic effect comparable to celastrol loaded at 20 μM, producing a potent cell death when targeted by F265S/F292G and negligible cytotoxicity with the wild-type I domain (Fig. 4C, F). Cytotoxicity caused by UAN(camptothecin/celastrol) conjugated to different I domains indicated that the differences in the cell viability were in fact due to the different affinities to ICAM-1 (Fig. 4G; p<0.0001, one-way ANOVA). Cell viability due to WT-UAN vs. F292G-UAN and WT-UAN vs. F265S/F292G-UAN was also significantly different (p<0.001 by Tukey's HSD Post Hoc Test). The observed cell death was not due to the UAN polymer, as the F265S/F292G-UAN without drug encapsulation at up to 500 μg/ml led to negligible cytotoxic effect (data not shown).
Figure 4. Affinity-dependent cytotoxicity by I domain-UAN(celastrol or CPT).
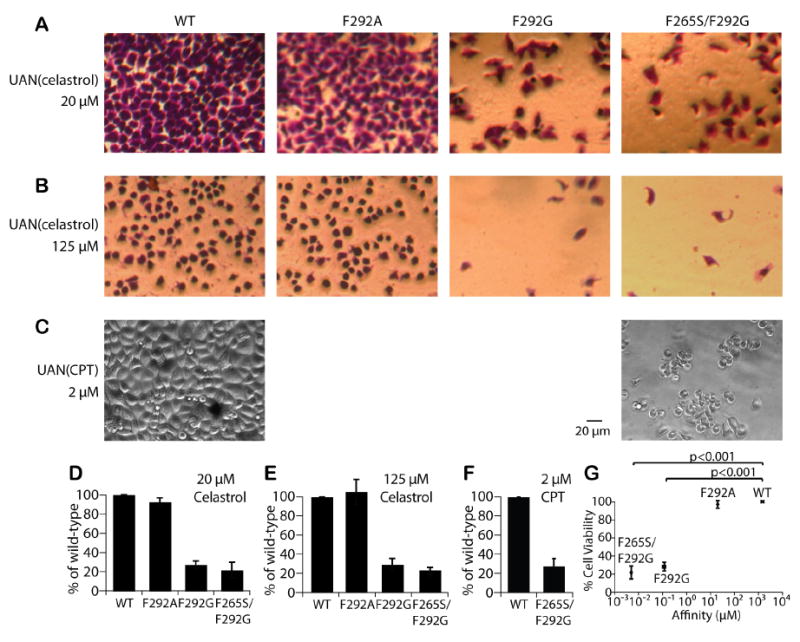
I domain-UAN nanoparticles encapsulating celastrol (A, B) or CPT (C) were delivered to HeLa cells. The cells treated with celastrol were stained with crystal violet for better visualization. Cell viability was then quantified by measuring the amount of crystal violet after cell lysis and shown as bar graphs (D-F), normalized to the levels with WT-UAN (n = 3). (G) The percentage of viable cells after HeLa cells treated with UAN(celastrol or CPT) was plotted as a function of I domain affinity to ICAM-1 (n=8). Values were normalized to that of the wild-type. The significance of the differences in cell viability due to I domain-UAN was analyzed by one-way ANOVA and Tukey's HSD post hoc test statistics.
Free celastrol vs. UAN(celastrol)
In order to demonstrate that targeted delivery of drugs carried by UAN is more potent than non-specific uptake of free drugs, we treated HeLa cells with UAN(celastrol) with no I domain, F265S/F292G-UAN(celastrol), and celastrol alone, each with 100 μM of celastrol (Fig. 5). After 30 minutes of incubation, cells were washed with the culture media and kept at 37°C for 28 hours. The percent cell viability was measured using the crystal violet assay. Though celastrol alone was able to cause substantial amount of cell death (78%), F265S/F292G-UAN(celastrol) induced significantly higher cytotoxicity (∼90%) (Fig. 5B). Notably, UAN(celastrol) without the I domain caused negligible amount of cell death, providing strong evidence that cytotoxicity induced by F265S/F292G-UAN was due to the specific uptake of UAN(celastrol) into the cells. Additionally, UAN nanoparticles were effectively precluded from non-specific uptake by the cells, and the cytotoxicity caused by celastrol diffusing out of UAN during the 30 min incubation was insignificant.
Figure 5. Comparison of cytotoxicity due to free celastrol, UAN(celastrol) without targeting, and F265S/F292G-UAN(celastrol).
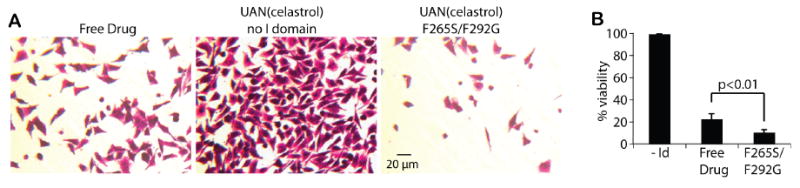
(A) Cytotoxicity of HeLa due to the treatment with celastrol, UAN(celastrol), or F265S/F292G-UAN(celastrol) was analyzed. HeLa cells were stained with a crystal violet for better visualization (images were taken 28 hours after the treatment) and subsequently lysed for quantification of cell viability. Each condition contained 100 μM of celastrol. (B) The measured cell viability was shown in bar graphs, normalized to that of ‘No I domain Control’ (n = 3).
Exploiting the Versatility of UAN Nanoparticle Platform
The versatility of our nanoparticle platform comes not only from its ability to encapsulate many different naturally available hydrophobic therapeutic and contrasting agents, but also from the fact that it can be conjugated to any targeting molecules with a His-tag. To demonstrate such versatility, we used a recombinant His-tagged protein A as the linker between our nanoparticle and R6.5, a mAb against ICAM-1. We also encapsulated a different dye, DPA, for cell labeling, and camptothecin for cellular cytotoxicity assay (Fig. 6). For encapsulation of DPA, a ‘soaking’ method [15] of pre-formulated particles was used. DPA was dissolved in dichloromethane at 25 mg/ml and 25 μl of this DPA solution was added to UAN nanoparticles (500 μg/ml) in 10 ml water. The mixture was stirred for 24 hours to evaporate off dichloromethane, during which DPA was encapsulated into the core of UAN. Unencapsulated DPA was precipitated out of solution and removed by a 0.2 μm filter. Protein A was conjugated to NTA-UAN nanoparticle at a weight ratio of 1:25 (protein A:UAN) for 30 minutes at 4°C, which was subsequently incubated with R6.5 at a molar ratio of 1:1 (R6.5:protein A) under the same conditions. Three additional conditions were included as control samples: bare NTA-UAN particles, NTA-UAN mixed with R6.5, and NTA-UAN with protein A. These conditions showed no detectable labeling or cytotoxicity. Only with both protein A and R6.5 conjugated onto the surface of UAN nanoparticles, an efficient delivery was observed resulting in effective cell labeling and cell death. Cell viability was quantified using the crystal violet assay, which showed an approximately 80% decrease in the number of viable cells.
Figure 6. Antibody-mediated targeting of ICAM-1.
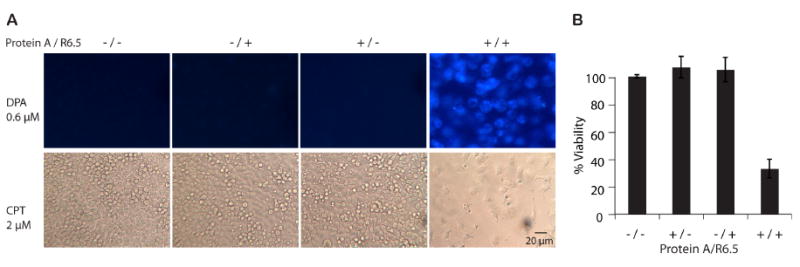
(A) In order to demonstrate the versatility of UAN platform, UAN nanoparticles were encapsulated with DPA or CPT, while the surface of UAN was coated with four different combinations of protein A and mAb R6.5. HeLa cells were imaged 2 hours after incubation with UAN(DPA) and 28 hours with UAN(CPT). (B) The bar graphs were normalized to that of no protein A and no R6.5 control (‘-/-’, n=3).
I domain-UAN Shows Minimal Binding to Cells Expressing Basal Levels of ICAM-1
ICAM-1 is ubiquitously present in lymphocytes, macrophages, and endothelial cells at low levels but is greatly upregulated under inflammatory conditions. To examine the possibility of UAN nanoparticle in targeting normal cells with low levels of ICAM-1, we used endothelial (HMEC-1) and monocytic (THP-1) cell lines, and measured the delivery efficiency of UAN into the cells with and without the treatment with LPS, a potent inducer of inflammation and ICAM-1 expression (Fig. 7). The increase in ICAM-1 expression in HMEC-1 and THP-1 cells after 24 hours of LPS treatment was confirmed by immunofluorescence flow cytometry with the mAb, LB2 (Fig. 7A). When these cells were labeled with F265S/F292G-UAN prior to LPS induction, little binding was measured (Fig. 7B). However, with LPS treatment, a 4-5 fold increase in the levels of UAN binding was observed (Fig. 7B, C), implying a requirement for a threshold or minimum concentration of ICAM-1 on cell surface to be targeted by a sufficient dose of UAN nanoparticles to produce cytotoxic effects.
Figure 7. Minimal binding of I domain-UAN to cells with basal expression of ICAM-1.
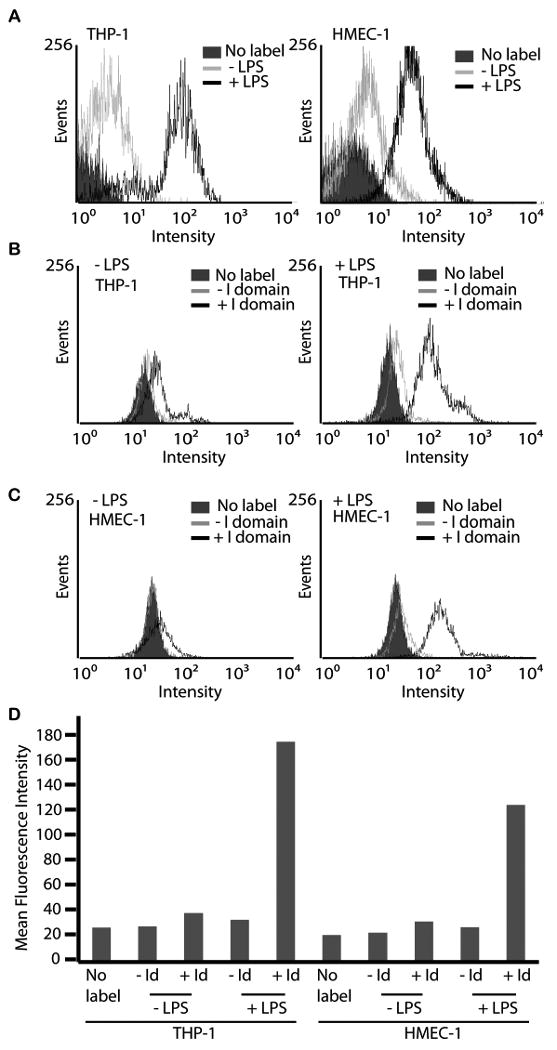
(A) Immunofluorescence flow cytometry measurement of mAb LB2 binding to HMEC-1 and THP-1 cells with (black open) or without (gray open) LPS treatment, or without labeling (filled histograms) to show the induction of ICAM-1 expression level. (B, C) Immunofluorescence flow cytometry measurement of F265S/F292G-UAN(FITC) (black open) binding to HMEC-1 and THP-1 cells with or without LPS treatment. UAN(FITC) without the I domain (gray open histograms) was used to measure non-specific binding to cells. Cells without labeling is shown in filled histograms. (D) Immunofluorescence flow cytometry results expressed as a bar graph.
Discussion
The delivery of drugs to the target cells and tissues with minimal distribution into unwanted targets will be critical in maximizing payload concentration at the sites of interest while preventing side effects. Thus far, no molecular target has been identified that is present only in tumors. Enhancing the specificity of drug carriers by controlling the affinity and avidity of molecular interactions will, therefore, aid to minimize binding to normal cells with basal levels of the molecular target. In this study, we have demonstrated that UAN nanoparticles are highly versatile and robust in terms of the methods used for conjugation, selection of optimal surface density of targeting molecules, and encapsulation of hydrophobic imaging and therapeutic agents. Using LFA-1 I domain mutants as tools to validate our UAN platform, we demonstrated affinity dependent delivery of both therapeutic and diagnostic agents to ICAM-1 expressing HeLa cells. We have also found that the level of ICAM-1 on the surface of target cells contributed to a great degree in specific binding of UAN nanoparticles. This finding is evidenced by the fact that despite ubiquitous expression of ICAM-1 in many different types of cells such as endothelium, epithelium, and a subset of leukocytes, our UAN nanoparticles exhibited little binding to them under resting conditions, and significant binding to endothelium and leukocytes only after induction of ICAM-1 by inflammatory molecules. ICAM-1 expression dependent delivery of UAN was also seen with two other cancer cell lines, KTC-1 and MDA-MB 231, which demonstrated a correlation between the level of UAN binding and ICAM-1 expression.
A large proportion of chemotherapeutic drugs are derived from plants [46] and contain hydrophobic aromatic groups and a chain of hydrocarbon, making them water insoluble [6]. Though much work has been done to modify these drugs to yield better analogues for solubility and bioavailability, these efforts have often resulted in drugs with reduced potency or stability [6]. Studies have shown, however, that chemotherapeutic drugs in their native forms can be encapsulated in the hydrophobic core of polymeric nanoparticles and protected from degradation, leading to an increased potency [5, 47]. The ability of UAN nanoparticles to retain hydrophobic materials was confirmed in the release kinetics study, which showed an increase in retention in correlation with their degree of hydrophobicity. Although the exact mechanism by which the payloads diffuse out of UAN after internalization into the cells is uncertain, the rate of diffusion may change once the UAN nanoparticles are exposed to a lower pH and lipid components in the endosomes.
Due to the presence of Ni-NTA, the versatility of UAN extends to its ability to be linked to any molecule with a His-tag, expanding the repertoire of targeting molecules that can be utilized. Recombinant engineering of His-tagged proteins is a common practice in molecular biology and these proteins can be produced in a variety of expression systems. Thus, a large library of recombinant His-tagged proteins is available for immobilization onto our nanoparticles [14]. Rather than applying different surface chemistry for conjugation of different targeting molecules, Ni-NTA/His-tag interaction provides a universal and reproducible conjugation process. This non-covalent but high affinity conjugation method obviates the problems associated with the conjugation via chemical activation (e.g., carbodiimide), such as the lack of control over the degree of conjugation and orientation of the molecules, and frequent aggregation of nanoparticles and proteins. By using a ratio of proteins to nanoparticle that does not fully saturate the NTA moieties on UAN while ensuring a coating density sufficiently high for tight binding, we eliminated the purification step of removing unbound proteins from the final formulation of UAN nanoparticles. The fact that encapsulation of drugs and conjugation of targeting molecules to UAN nanoparticles do not require any chemical conjugation and special purification methods will be of significant advantage in the potential therapeutic use of UAN in clinics.
An important facet of targeted delivery of nanoparticles is the prospect of achieving specific and more potent treatment than the conventional therapy methods. Here, we have demonstrated that UAN encapsulating celastrol was much more potent than celastrol alone, which is currently being evaluated in preclinical studies for cancer therapy [44], and that UAN itself causes no toxicity. Furthermore, UAN(celastrol) without the I domain was shown to cause much less cytotoxicity, indicating that non-specific uptake of UAN nanoparticles by the cells was much lower than that of free drugs. This characteristic will be of significant importance since the administration of chemotherapeutic drugs alone lack specificity and cause significant damage to non-cancerous tissues, leading to serious, unwanted side effects in bone marrow, gut epithelial cells, reticuloendothelial system, and hair loss. Owing to the ability to encapsulate hydrophobic materials and achieve consistent surface modification via the Ni-NTA/His-tag interaction as presented, UAN provides a promising platform for effective drug delivery in numerous disease models.
Conclusion
In this study, we have demonstrated that UAN nanoparticles possess ideal properties to achieve effective encapsulation of hydrophobic agents as well as specific delivery of imaging and cytotoxic agents to the target cells in a manner dependent on the levels of target receptor expression. The versatility of UAN nanoparticles allows for reproducible and controlled conjugation of targeting ligands via the Ni-NTA/His-tag interaction for specific targeting of diverse disease markers, while facilitating encapsulation of various hydrophobic materials in the core. Though we only demonstrated this ability using DPA, FITC, camptothecin and celastrol, the encapsulation method should be applicable to a wide array of hydrophobic materials. A drug carrier system targeting molecules over-expressed in tumor or inflammation, such as ICAM-1, can be effective in not only targeting tumors over-expressing ICAM-1 but also tumor microenvironments, which have been shown to display characteristics of inflammation sites [48-50]. Finally, the use of physiological ligands such as LFA-1 I domain used in this study may be superior to antibodies or short peptides in ensuring target specificity and preventing adverse immune responses associated with foreign antigens.
Acknowledgments
This work was supported, in whole or in part, by an American Heart Association Scientist Development Grant, NIH R01 GM090320, and Business for International Cooperative R&D between Industry, Academy, and Research Institute (Korea Small and Medium Business Administration).
Footnotes
Author Contributions: S.P., R.Z., M.M.J., and K.J-Y. designed research, performed research, and wrote the paper; S.K., A.V and Y.V. performed research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weissleder R. Molecular imaging in cancer. Science. 2006;312:1168–71. doi: 10.1126/science.1125949. [DOI] [PubMed] [Google Scholar]
- 2.Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AK, Han MS, Mirkin CA. Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science. 2006;312:1027–30. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- 3.Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–22. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 4.Mainardes RM, Silva LP. Drug delivery systems: past, present, and future. Curr Drug Targets. 2004;5:449–55. doi: 10.2174/1389450043345407. [DOI] [PubMed] [Google Scholar]
- 5.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–82. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 6.Srivastava V, Negi AS, Kumar JK, Gupta MM, Khanuja SP. Plant-based anticancer molecules: a chemical and biological profile of some important leads. Bioorg Med Chem. 2005;13:5892–908. doi: 10.1016/j.bmc.2005.05.066. [DOI] [PubMed] [Google Scholar]
- 7.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161–71. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 8.Langer R. Drug delivery. Drugs on target Science. 2001;293:58–9. doi: 10.1126/science.1063273. [DOI] [PubMed] [Google Scholar]
- 9.Zentner GM, Rathi R, Shih C, McRea JC, Seo MH, Oh H, et al. Biodegradable block copolymers for delivery of proteins and water-insoluble drugs. J Control Release. 2001;72:203–15. doi: 10.1016/s0168-3659(01)00276-0. [DOI] [PubMed] [Google Scholar]
- 10.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103:6315–20. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker JR, Jr, Banaszak Holl MM. The binding avidity of a nanoparticle-based multivalent targeted drug delivery platform. Chem Biol. 2007;14:107–15. doi: 10.1016/j.chembiol.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 12.Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:15549–54. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim JY, Shim SB, Shim JK. Effect of amphiphilic polyurethane nanoparticles on sorption-desorption of phenanthrene in aquifer material. J Hazard Mater. 2003;98:145–60. doi: 10.1016/s0304-3894(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 14.Fischer NO, Blanchette CD, Chromy BA, Kuhn EA, Segelke BW, Corzett M, et al. Immobilization of His-tagged proteins on nickel-chelating nanolipoprotein particles. Bioconjug Chem. 2009;20:460–5. doi: 10.1021/bc8003155. [DOI] [PubMed] [Google Scholar]
- 15.Zhu H, McShane MJ. Loading of hydrophobic materials into polymer particles: implications for fluorescent nanosensors and drug delivery. J Am Chem Soc. 2005;127:13448–9. doi: 10.1021/ja052188y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hertzberg RP, Caranfa MJ, Holden KG, Jakas DR, Gallagher G, Mattern MR, et al. Modification of the hydroxy lactone ring of camptothecin: inhibition of mammalian topoisomerase I and biological activity. J Med Chem. 1989;32:715–20. doi: 10.1021/jm00123a038. [DOI] [PubMed] [Google Scholar]
- 17.Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–65. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- 18.Park JW, Kirpotin DB, Hong K, Shalaby R, Shao Y, Nielsen UB, et al. Tumor targeting using anti-her2 immunoliposomes. J Control Release. 2001;74:95–113. doi: 10.1016/s0168-3659(01)00315-7. [DOI] [PubMed] [Google Scholar]
- 19.Ishida O, Maruyama K, Tanahashi H, Iwatsuru M, Sasaki K, Eriguchi M, et al. Liposomes bearing polyethyleneglycol-coupled transferrin with intracellular targeting property to the solid tumors in vivo. Pharm Res. 2001;18:1042–8. doi: 10.1023/a:1010960900254. [DOI] [PubMed] [Google Scholar]
- 20.Mamot C, Drummond DC, Greiser U, Hong K, Kirpotin DB, Marks JD, et al. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR- and EGFRvIII-overexpressing tumor cells. Cancer Res. 2003;63:3154–61. [PubMed] [Google Scholar]
- 21.Leamon CP, Weigl D, Hendren RW. Folate copolymer-mediated transfection of cultured cells. Bioconjug Chem. 1999;10:947–57. doi: 10.1021/bc990066n. [DOI] [PubMed] [Google Scholar]
- 22.Montet X, Montet-Abou K, Reynolds F, Weissleder R, Josephson L. Nanoparticle imaging of integrins on tumor cells. Neoplasia. 2006;8:214–22. doi: 10.1593/neo.05769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peer D, Margalit R. Loading mitomycin C inside long circulating hyaluronan targeted nano-liposomes increases its antitumor activity in three mice tumor models. Int J Cancer. 2004;108:780–9. doi: 10.1002/ijc.11615. [DOI] [PubMed] [Google Scholar]
- 24.Lee CM, Tanaka T, Murai T, Kondo M, Kimura J, Su W, et al. Novel chondroitin sulfate-binding cationic liposomes loaded with cisplatin efficiently suppress the local growth and liver metastasis of tumor cells in vivo. Cancer Res. 2002;62:4282–8. [PubMed] [Google Scholar]
- 25.Dustin ML, Rothlein R, Bhan AK, Dinarello CA, Springer TA. Induction by IL 1 and interferon-gamma: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1) J Immunol. 1986;137:245–54. [PubMed] [Google Scholar]
- 26.Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1) Cell. 1987;51:813–9. doi: 10.1016/0092-8674(87)90104-8. [DOI] [PubMed] [Google Scholar]
- 27.Ogawa Y, Hirakawa K, Nakata B, Fujihara T, Sawada T, Kato Y, et al. Expression of intercellular adhesion molecule-1 in invasive breast cancer reflects low growth potential, negative lymph node involvement, and good prognosis. Clin Cancer Res. 1998;4:31–6. [PubMed] [Google Scholar]
- 28.Kelly CP, O'Keane JC, Orellana J, Schroy PC, 3rd, Yang S, LaMont JT, et al. Human colon cancer cells express ICAM-1 in vivo and support LFA-1-dependent lymphocyte adhesion in vitro. Am J Physiol. 1992;263:G864–70. doi: 10.1152/ajpgi.1992.263.6.G864. [DOI] [PubMed] [Google Scholar]
- 29.Passlick B, Izbicki JR, Simmel S, Kubuschok B, Karg O, Habekost M, et al. Expression of major histocompatibility class I and class II antigens and intercellular adhesion molecule-1 on operable non-small cell lung carcinomas: frequency and prognostic significance. Eur J Cancer. 1994;30A:376–81. doi: 10.1016/0959-8049(94)90259-3. [DOI] [PubMed] [Google Scholar]
- 30.Tomita Y, Nishiyama T, Watanabe H, Fujiwara M, Sato S. Expression of intercellular adhesion molecule-1 (ICAM-1) on renal-cell cancer: possible significance in host immune responses. Int J Cancer. 1990;46:1001–6. doi: 10.1002/ijc.2910460609. [DOI] [PubMed] [Google Scholar]
- 31.Koyama S, Ebihara T, Fukao K. Expression of intercellular adhesion molecule 1 (ICAM-1) during the development of invasion and/or metastasis of gastric carcinoma. J Cancer Res Clin Oncol. 1992;118:609–14. doi: 10.1007/BF01211806. [DOI] [PubMed] [Google Scholar]
- 32.Shimoyama S, Gansauge F, Gansauge S, Widmaier U, Oohara T, Beger HG. Overexpression of intercellular adhesion molecule-1 (ICAM-1) in pancreatic adenocarcinoma in comparison with normal pancreas. Pancreas. 1997;14:181–6. doi: 10.1097/00006676-199703000-00011. [DOI] [PubMed] [Google Scholar]
- 33.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsumura Y, Gotoh M, Muro K, Yamada Y, Shirao K, Shimada Y, et al. Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Ann Oncol. 2004;15:517–25. doi: 10.1093/annonc/mdh092. [DOI] [PubMed] [Google Scholar]
- 35.Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000;60:722–7. [PMC free article] [PubMed] [Google Scholar]
- 36.Kurzinger K, Reynolds T, Germain RN, Davignon D, Martz E, Springer TA. A novel lymphocyte function-associated antigen (LFA-1): cellular distribution, quantitative expression, and structure. J Immunol. 1981;127:596–602. [PubMed] [Google Scholar]
- 37.Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106:584–92. doi: 10.1182/blood-2004-12-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin M, Song G, Carman CV, Kim YS, Astrof NS, Shimaoka M, et al. Directed evolution to probe protein allostery and integrin I domains of 200,000-fold higher affinity. Proc Natl Acad Sci U S A. 2006;103:5758–63. doi: 10.1073/pnas.0601164103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grunert HP, Wolf KU, Langner KD, Sawitzky D, Habermehl KO, Zeichhardt H. Internalization of human rhinovirus 14 into HeLa and ICAM-1-transfected BHK cells. Med Microbiol Immunol (Berl) 1997;186:1–9. doi: 10.1007/s004300050039. [DOI] [PubMed] [Google Scholar]
- 40.Kim JY, Kim HM, Shin DH, Ihn KJ. Synthesis of CdS nanoparticles dispersed within poly(urethane acrylate-co-styrene) films using an amphiphilic urethane acrylate nonionomer. Macromolecular Chemistry and Physics. 2006;207:925–32. [Google Scholar]
- 41.Kurebayashi J, Okubo S, Yamamoto Y, Ikeda M, Tanaka K, Otsuki T, et al. Additive antitumor effects of gefitinib and imatinib on anaplastic thyroid cancer cells. Cancer Chemother Pharmacol. 2006;58:460–70. doi: 10.1007/s00280-006-0185-x. [DOI] [PubMed] [Google Scholar]
- 42.Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR, 3rd, Segatori L, et al. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–81. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sethi G, Ahn KS, Pandey MK, Aggarwal BB. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB activation. Blood. 2007;109:2727–35. doi: 10.1182/blood-2006-10-050807. [DOI] [PubMed] [Google Scholar]
- 44.Abbas S, Bhoumik A, Dahl R, Vasile S, Krajewski S, Cosford ND, et al. Preclinical studies of celastrol and acetyl isogambogic acid in melanoma. Clin Cancer Res. 2007;13:6769–78. doi: 10.1158/1078-0432.CCR-07-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimaoka M, Xiao T, Liu JH, Yang Y, Dong Y, Jun CD, et al. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell. 2003;112:99–111. doi: 10.1016/s0092-8674(02)01257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balunas MJ, Kinghorn AD. Drug discovery from medicinal plants. Life Sci. 2005;78:431–41. doi: 10.1016/j.lfs.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 47.Kawano K, Watanabe M, Yamamoto T, Yokoyama M, Opanasopit P, Okano T, et al. Enhanced antitumor effect of camptothecin loaded in long-circulating polymeric micelles. J Control Release. 2006;112:329–32. doi: 10.1016/j.jconrel.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 48.Nakashima Y, Raines EW, Plump AS, Breslow JL, Ross R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler Thromb Vasc Biol. 1998;18:842–51. doi: 10.1161/01.atv.18.5.842. [DOI] [PubMed] [Google Scholar]
- 49.Davies ME, Sharma H, Pigott R. ICAM-1 expression on chondrocytes in rheumatoid arthritis: induction by synovial cytokines. Mediators Inflamm. 1992;1:71–4. doi: 10.1155/S0962935192000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hofer S, Bopp C, Hoerner C, Plaschke K, Faden RM, Martin E, et al. Injury of the blood brain barrier and up-regulation of icam-1 in polymicrobial sepsis. J Surg Res. 2008;146:276–81. doi: 10.1016/j.jss.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 51.Hu X, Kang S, Chen X, Shoemaker CB, Jin MM. Yeast Surface Two-hybrid for Quantitative in Vivo Detection of Protein-Protein Interactions via the Secretory Pathway. J Biol Chem. 2009;284:16369–76. doi: 10.1074/jbc.M109.001743. [DOI] [PMC free article] [PubMed] [Google Scholar]