

Abstract
Nanoparticle (NP) size has been shown to significantly effect the biodistribution of targeted and non-targeted NPs in an organ specific manner. Herein we have developed NPs from carboxy-terminated poly (d,l-lactide-co-glycolide)-block-poly(ethylene glycol) (PLGA-b-PEG-COOH) polymer and studied the effects of altering the following formulation parameters on the size of NPs, including: 1) polymer concentration, 2) drug loading, 3) water miscibility of solvent, and 4) the ratio of water to solvent. We found that NP mean volumetric size correlates linearly with polymer concentration for NPs between 70 and 250 nm in diameter (linear coefficient = 0.99 for NPs formulated with solvents studied). NPs with desirable size, drug loading, and polydispersity were conjugated to the A10 RNA aptamer (Apt) that binds to the Prostate Specific Membrane Antigen (PSMA), and NP and NP-Apt biodistribution was evaluated in a LNCaP (PSMA+) xenograft mouse model of PCa. The surface functionalization of NPs with the A10 PSMA aptamer significantly enhanced delivery of NPs to tumors vs. equivalent NPs lacking the A10 PSMA aptamer (a 3.77-fold increase at 24 hrs; NP-Apt 0.83% ± 0.21% vs. NP 0.22% ± 0.07% of injected dose per gram of tissue; mean ± s.d., n = 4, p = 0.002). The ability to control NP size together with targeted delivery may result in favorable biodistribution and development of clinically relevant targeted therapies.
Keywords: Drug delivery, Nanoparticle, PLGA, Prostate cancer, Targeting, Aptamer
1. Introduction
Several decades of biomaterials research has led to a progressively heightened interest in the use of biodegradable polymeric nanoparticles (NPs) for drug delivery applications [1-5]. NPs can accumulate in tumors after systemic administration, and their biodistribution is largely determined by their physical and biochemical properties, such as particle size, nature of the polymer and drug, and surface biochemical properties [6]. The effect of size on NP biodistribution has been shown to be organ specific and non-linear [7]. This is in part due to organ-specific physical and physiological barriers that systemically administered NPs encounter [8]. Studies on liposomes have shown that splenic sequestration of particles decreases linearly with decreases in particle size [9,10]. For the liver, the dependence on size is non-linear. While larger NPs are sequestered in the liver consistent with the observations in the spleen, the very small NPs (less than 70nm) can pass through the sinusoidal fenestrations in the liver and be entrapped by underlying parenchymal cells [11]. In the case of bone marrow, larger particles are generally excluded, but smaller particles that escape spleen and liver sequestration can partition more effectively in the bone marrow depending on their surface characteristics [12]. The effect of NP size on biodistribution is therefore non-linear and varies from organ to organ, underscoring the importance of tuning NP size for each distinct in vivo application.
Recently, we developed ∼250 nm NP-aptamer (NP-Apt) bioconjugates using poly(d,l-lactide)-block-poly(ethylene glycol) (PLA-b-PEG) copolymer and the A10 RNA aptamer [13] that can bind the extracellular domain of the Prostate Specific Membrane Antigen (PSMA), and demonstrated their capability for active binding and uptake by the targeted cancer cells in vitro [14]. We also developed ∼180 nm docetaxel-encapsulated NP-Apt bioconjugates using poly(d,l-lactide-co-glycolide)-block-poly(ethylene glycol) (PLGA-b-PEG) copolymer that showed remarkable antitumor efficacy in vivo after a single intratumoral administration to subcutaneous xenograft mouse models of prostate cancer [15]. Although intratumoral drug delivery is suitable for localized prostate cancer disease, patients with advanced prostate cancer will require drugs to be administered systemically for the treatment of disseminated tumors [16]. Systemic delivery of targeted NPs presents other challenges for developing an effective NP drug delivery vehicle with desirable pharmacological properties capable of extended circulation in blood and targeted drug delivery.
Herein we report formulation strategies to control the size of PLGA-b-PEG NPs during both synthesis and post-synthesis processing. The ability to control NP size may be broadly important to their use in various clinical applications, allowing for optimization of NP delivery vehicles for systemic administration. For our technology, we chose one NP-Apt bioconjugate formulation that has size and drug loading characteristics that are potentially suitable for systemic administration and tested its tumor targeting efficiency and biodistribution when administered systemically to a xenograft mouse model of prostate cancer.
2. Materials and Methods
2.1 Materials
Docetaxel and 14C-paclitaxel were purchased from Sigma-Aldrich (St. Louis, MO). Poly(d,l-lactide-co-glycolide) (50/50) with terminal carboxylate groups (PLGA, inherent viscosity 0.20 dl/g in hexafluoroisopropanol, Mw ∼ 17 kDa) was obtained from Absorbable Polymers International (Pelham, AL). NH2-PEG-COOH (MW 3400) was purchased from Nektar Therapeutics (San Carlos, CA). All reagents were analytical grade or above and used as received, unless otherwise stated. Molecular biology buffers were purchased from Boston BioProducts (Worcester, MA). Tissue culture reagents and the LNCaP cell line were obtained from American Type Culture Collection (Manassas, VA). RNA aptamer (sequence: 5′-NH2-spacer-GGG/AGG/ACG/AUG/CGG/AUC/AGC/CAU/GUU/UAC/GUC/ACU/CCU/UGU/CAA/UCC/UCA/UCG/GCiT-3′ with 2′-fluoro pyrimidines, a 5′-amino group attached by a hexaethyleneglycol spacer and a 3′-inverted T cap) was custom synthesized by RNA-TEC (Leuven, Belgium) at a purity above 90%.
2.2 Methods
2.2.1 Synthesis of PLGA-b-PEG
Carboxylate-functionalized copolymer PLGA-b-PEG was synthesized by the conjugation of COOH-PEG-NH2 to PLGA-COOH. PLGA-COOH (5 g, 0.28 mmol) in methylene chloride (10 mL) was converted to PLGA-NHS with excess N-hydroxysuccinimide (NHS, 135 mg, 1.1 mmol) in the presence of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC, 230 mg, 1.2 mmol). PLGA-NHS was precipitated with ethyl ether (5 mL), and repeatedly washed in an ice-cold mixture of ethyl ether and methanol to remove residual NHS. After drying under vacuum, PLGA-NHS (1 g, 0.059 mmol) was dissolved in chloroform (4 mL) followed by addition of NH2-PEG-COOH (250 mg, 0.074 mmol) and N,N-diisopropylethylamine (28 mg, 0.22 mmol). The co-polymer was precipitated with cold methanol after 12h and washed with the same solvent (3 × 5 mL) to remove unreacted PEG. The resulting PLGA-PEG block co-polymer was dried under vacuum and used for NP preparation without further treatment. 1H NMR (CDCl3 at 300Hz) δ 5.2 (m, ((OCH(CH3)C(O)OCH2C(O))n-(CH2CH2O)m), 4.8 (m, ((OCH(CH3)C(O)OCH2C(O))n-(CH2CH2O)m), 3.7 (s, ((OCH(CH3)C(O)OCH2C(O))n-(CH2CH2O)m), 1.6 (d, ((OCH(CH3)C(O)OCH2C(O))n-(CH2CH2O)m).
2.2.2 Formulation of taxane drug-loaded PLGA-b-PEG NPs
The nanoprecipitation method was employed for the formation of drug-encapsulated carboxylated PLGA-b-PEG NPs, similarly to previously described [15,17]. Briefly, docetaxel (or 14C-paclitaxel) was dissolved in various organic solvents that are miscible with water. Polymer was likewise dissolved and mixed with the drug. NPs were formed by adding the drug-polymer solution to water, a non-solvent. The resulting NP suspension was allowed to stir uncovered for 6 h at room temperature. NPs were purified by centrifugation (10 min, 10000 × g) or by ultrafiltration (15 min, 3000 × g, Amicon Ultra, Ultracel membrane with 100,000 NMWL, Millipore, Billerica, MA). The PLGA-b-PEG NPs were re-suspended, washed with water, and collected likewise.
Parameters controlling formation of the NPs were systematically varied in this study. Generally, the starting formulation was as follows: PLGA-b-PEG (10 mg/mL) and docetaxel (0.1 mg/mL) were dissolved in acetonitrile. The mixture was added dropwise to a 2× volume of stirring water. The NPs were produced with the nanoprecipitation method in four solvents: N,N-dimethylformamide (DMF), acetone, acetonitrile, and tetrahydrofuran (THF). The effects of the various solvents were assayed on the overall size of the NPs. For each solvent, the ratio of solvent to water was varied from 0.1 to 1 (using 10 mg/mL polymer for each). Further, a range of polymer concentrations in the organic phase from 5 mg/mL to 50 mg/mL was used for formation of NPs in a 2 × volume of water. The NPs were processed as above in triplicate, noting trends in formulation parameters. In another study, NPs containing variable amounts of docetaxel were synthesized by adjusting docetaxel drug loading from 0% to 10% by weight of the added polymer, formulating the NPs from 10mg/ml polymer in acetonitrile and a 2 × volume of water.
NP post-formulation stability was studied for both the purification and bioconjugation steps, and through the storage in solid-state after freeze-drying. NPs were also flash-frozen in liquid nitrogen prior to lyopholization for freeze-drying.
2.2.3 Determination of particle sizes and polydispersities
The particle size distributions were measured by the dynamic light scattering (Brookhaven Instruments Corporation 90 plus Particle Sizer, 676 nm laser) at 25 °C and at a scattering angle of 90 ° at a concentration of approximately 1mg NP/mL water. The intensity-weighted mean value was recorded as the average of three measurements.
2.2.4 Determination of drug content
NPs were dissolved in acetonitrile and measured by HPLC in triplicates to determine docetaxel content. The Agilent 1100 HPLC (Palo Alto, CA) was equipped with a UV detector and a reverse-phase pentafluorophenyl column (Curosil-PFP, 250 × 4.6 mm, 5 μ, Phenomenex, Torrance, CA) with a non-gradient mobile phase of water and acetonitrile (v/v 50/50) at a constant flow rate 1 mL/min. The docetaxel peak was measured at a wavelength of 227 nm and quantitatively determined by comparing with a standard curve.
2.2.5 Conjugation of aptamer to the PLGA-b-PEG-COOH NPs
PLGA-b-PEG NPs (10 μg/μL) were suspended in water and were incubated with EDC (400 mM) and NHS (200 mM) for 20 min. NPs were then repeatedly washed in DNase-, RNase-free water (3 × 15 mL) followed by ultrafiltration. The NHS-activated NPs were reacted with 5′-amino-RNA aptamer (1 μg/μL). The resulting NP-Apt bioconjugates were washed with ultrapure water (15 mL) by ultrafiltration, and the surface-bound aptamers were denatured at 90 °C and allowed to assume binding conformation during snap-cooling on ice. The NP suspensions were kept at 4 °C until use.
NP-Apt bioconjugation was confirmed on 10% TBE-Urea PAGE. NPs were incubated as above with (+EDC) and without (-EDC) the crosslinker to confirm covalent conjugation. Aptamer, NP, NP+Apt (+EDC), NP+Apt (-EDC), washed NP+Apt (+EDC), and washed NP+Apt (-EDC) were separated by PAGE. The molecular weight (MW) DNA marker and free aptamer served as standards for a 57 base pair band on the gel.
2.2.6 In vivo tumor targeting and biodistribution of NP-Apt bioconjugates
All animal studies were carried out under the supervision of MIT's Division of Comparative Medicine and in compliance with NIH's Principles of Laboratory Animal Care. Human xenograft prostate cancer tumors were induced in 8-week old balb/c nude mice (Charles River Laboratories, Wilmington, MA). Mice were injected subcutaneously in the right flank with 3×106 LNCaP cells suspended in a 1:1 mixture of media and matrigel (BD Biosciences, Franklin Lakes, NJ). Prior to use in tumor induction, LNCaP cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 100 units/mL penicillin G, and 100 μg/mL streptomycin.
Tumor targeting studies were carried out after the mice developed ∼100 mg tumors. Mice were divided into groups of four, minimizing tumor size variations between groups. Mice were anesthesized by intraperitoneal injection of avertin (200 mg/kg body weight), and dosed with NPs or NP-Apt bioconjugates by retro-orbital injection. The NPs were traced by encapsulating 14C-paclitaxel and suspended in 200 μL PBS (1 ×) prior to administration. Different groups were euthanized at 2, 6 or 24 hours, and 200 μL of blood was drawn by cardiac puncture from each mouse. The tumor, heart, lungs, liver, spleen and kidneys were harvested from each animal. The 14C content of tissues was assayed in a Packard Tri-Carb Scintillation Analyser (Downers Grove, IL). The tissues were solubilized in Solvable (Packard), and activity was counted in Hionic-Fluor scintillation cocktail (PerkinElmer, Boston, MA). The liver from each mouse was homogenized due to its large size, and ∼100 mg of tissue was placed in a scintillation vial for analysis. The other organs were placed directly in scintillation vials. Each organ was solubilized in 2 mL Solvable for ∼12 hours at 60 °C, and the resulting solution was de-colored with 200 μL hydrogen peroxide for 1 hour at 60 °C. For the blood, 400 μL Solvable was added, and the vials were otherwise treated similarly to the tissues. To determine 100% dose, vials of the formulated NPs were counted along with the tissues. Data is presented as percent injected dose per gram of tissue.
2.2.7 Statistical Analysis
The statistical analysis of the samples was undertaken using a student's t-test, and p-values < 0.05 were considered statistically significant. All data reported are means +/- standard deviations, unless otherwise noted.
3. Results and Discussion
3.1 Synthesis of PLGA-b-PEG-COOH Copolymer
We synthesized carboxyl-functionalized PLGA-b-PEG copolymer by direct conjugation of PLGA-COOH with NH2-PEG-COOH, both having fixed block length, to generate PLGA-b-PEG-COOH (Scheme 1). The carboxyl group in the copolymer is at the terminal end of the hydrophilic PEG block; therefore, upon NP formulation the PEG should facilitate the presentation of the carboxyl groups on the NP surface making it available for surface chemistry. The RNA aptamers are synthesized with 5′-amino groups that can be conjugated to the carboxyl groups on the NP surface using carbodiimide coupling chemistry (Scheme 1). After preparing the polymer, the efficiency of the coupling reaction was determined by 1H NMR, which revealed that approximately 83% of PLGA was conjugated with the PEG segment.
Scheme 1.

Synthesis of PLGA-b-PEG-COOH NP, and conjugation of aptamer to NP. Docetaxel was encapsulated within PLGA-b-PEG-COOH NP using the nanoprecipitation method. The PLGA-PEG NP/Docetaxel was covalently conjugated to amine-terminated A10 PSMA aptamer (Apt) in the presence of EDC.
3.2 Effects of varying formulation parameters to control NP size
As a starting point for controlling the NP size distribution, we first studied the effect of varying the type of organic solvent used to solubilize the drug and polymer. Previous studies have suggested that the miscibility of the organic solvent in water can impact NP size for a given solvent:water system [18,19]. Generally, the miscibility can be quantitatively expressed by comparing the solubility parameters (δ) of both the solvent and water [20]. As solvents become more miscible, the difference in solubility parameters between the solvents (Δδ) is minimized. We chose to investigate the relationship of NP size and solvent miscibility with water using four organic solvents and observed a dependence of NP size on the solubility parameters. As shown in Figure 1, the sizes of PLGA-b-PEG NPs and the water-miscibility of the four organic solvents used in this study were generally correlated; an increase of water miscibility (decrease in Δδ, as indicated by the arrow shown in Figure 1) led to a decrease in the mean NP size, with all other formulation parameters held constant. NPs prepared in DMF, the most water miscible solvent tested, resulted in the smallest particles, which is presumably due to more efficient solvent diffusion and polymer dispersion into water.
Figure 1.

Effect of varying formulation parameters on NP size. (A) Varying the solvent:water ratio (1:1, 1:2, 1:5, 1:10) while keeping the PLGA-b-PEG polymer constant at 10 mg/ml, and (B) varying the polymer concentrations in organic phase (5, 10, 20 or 50 mg/ml) while keeping the solvent:water ratio constant at 1:2.
In conjunction with our investigation of the effect of solvent-water miscibility, we studied the effect of altering the solvent:water ratio during NP formulation. When solvent:water ratios were varied for a fixed polymer concentration (10 mg/mL) as shown in Figure 1A, no clear correlation of particle size with solvent-to-water ratio was observed. Most of the NP sizes remained relatively unchanged when the ratio was in a range of 0.1 - 0.5. In acetone, for example, NP sizes increased from 115.3 ± 5.1 nm to 120.9 ± 6.9 nm as the Vsolvent/Vwater ratio increased from 0.1 to 0.5, respectively (mean ± s.d., n = 3 for each formulation; p > 0.05). For THF, the size remained consistent as the ratio was varied from 0.1 to 0.5, with respective sizes of 130 ± 0.5 nm and 129 ± 15.5 nm. At the solvent:water ratio of 1, a large increase in particle size was observed presumably due to poor phase separation—NPs formulated in acetonitrile and THF were sized greater than 200 nm (p < 0.05, comparing sizes for ratio of 1 vs. 0.5).
When polymer concentrations were varied during NP formulation at a fixed solvent:water ratio (Figure 1B), we observed a trend of increasing NP size with increasing polymer concentration. For example, NP sizes increased from 69.0 nm to 165.0 nm in DMF as the polymer concentration increased 10 times from 5 mg/mL to 50 mg/mL. Similar trends were observed in all other solvents investigated. Interestingly, interpreting the data in terms of changes in volumetric size showed linear agreement between size and polymer concentration. The R2 values for the plot of mean NP volume and polymer concentration (Figure 2) were 0.997, 0.985, 0.998, and 0.997 for DMF, acetone, THF, and acetonitrile, respectively. For our polymer system, using the linear correlation of the NP volumetric size and polymer concentration allows for formulation of NP with predefined, desired sizes.
Figure 2.

Correlation of NP volumetric sizes with polymer concentrations at constant solvent:water ratio.
3.3 NP Polydispersity at different drug loadings
For our PLGA-b-PEG system, we studied the effect of docetaxel loading on resulting NP size distributions, comparing NPs loaded with 1%, 5% and 10% docetaxel. For a given NP formulation (150 nm NPs), the polydispersity of the particle preparations increased with docetaxel concentration as follows: from 0.154 for the 1% loading to 0.203 for the 5% loading and 0.212 for the 10% loading. The size distribution of the NPs exhibited a biphasic trend with a smaller diameter particle distribution accompanied by a distribution of larger diameter particles (Figure 3). The distribution corresponding to the smaller particles did not shift with the increase of drug concentration. The larger diameter locus of the two size distributions shifted higher as the drug loading increased (the size increasing from approximately 300 nm to 1200 nm, Figure 3). Since the only difference between these formulations is the amount of drug loading, a significant amount of the NPs formed may be due to aggregation of unencapsulated docetaxel, due to its poor water solubility.
Figure 3.

Effect of the docetaxel loading on PLGA-b-PEG NP polydispersity.
Previous studies have shown that in order to release taxane drugs at a sustainable rate from PLGA NP, the drug loading concentrations should be limited [21], especially in the case of pegylated NPs [22]. Some studies have suggested that drug loading can be increased [23] or that release can be sustained [24] by using high molecular weight PLGA. In our polymer system, we found that drug loadings of 1% minimized NP polydispersity, and thus these NPs may behave most predictably in vivo.
3.4 Control of NP size during post-formulation treatment
NPs formed by nanoprecipitation generally do not require surfactant; however, the lack of surfactant can cause NP aggregation after formulation. High-speed centrifugation, for example, can substantially increase particle size due to aggregation upon pelleting. After NPs (∼80 nm) were centrifuged at 10,000 × g for 10 minutes, an increase in diameter of approximately 20-30% was observed for each of the centrifugation steps (Figure 4A). However, the mechanical force that causes aggregation can be substantially avoided by low-speed ultrafiltration (Figure 4A). Use of a commercially available centrifuge filtration device reproducibly controls the particle size during multiple washing steps.
Figure 4.

PLGA-b-PEG NP size stability. (A) Effect of centrifugation vs ultrafiltration on NP size (12000 ×g, 15 min vs. 3000 ×g, 15min); (B) effect of sucrose prior to lyopholization on NP size, after storage and resuspension.
For translation to clinical use of any biodegradable formulation, stability upon storage is a concern. Freeze-drying the NPs and storing frozen in solid-state is a common approach, and sugars like sucrose can act as a lyoprotectant during the process [25,26]. Addition of 10% sucrose to an aqueous NP suspension (10 mg/mlL) allows recovery of NPs of very similar size as originally formulated (Figure 4B). Without sucrose as a lyoprotectant, the NPs aggregated to a few micrometers in size and were not useful upon reconstitution for in vivo systemic delivery (Figure 4B). Further investigation is necessary to determine whether other properties of the NPs, such as drug release profile, are unchanged.
3.5 Conjugation of aptamer to NP
PAGE was utilized to examine the conjugation of the NPs to aptamers, and to demonstrate successful removal of unconjugated aptamers after the reaction. The mixing of aptamer and NP without the addition of the coupling agent (-EDC, Figure 5) did not show any band of conjugated aptamer, indicating a lack of non-specific interaction between the aptamer and NP. Conjugation with the addition of EDC (+EDC, Figure 5) leads to RNA bands consistent with RNA covalently bound to the NPs and unable to run on the gel, both before and after washing. After repetitive washing by ultrafiltration, the unconjugated aptamer was no longer detectable (Figure 5).
Figure 5.
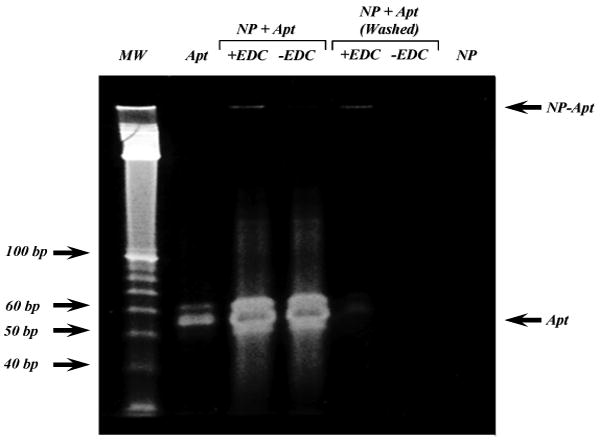
Confirmation of NP-Apt conjugation. The A10 PSMA aptamer (Apt) was incubated with PLGA-b-PEG NP in the absence (-) or presence (+) of EDC and the reactions were resolved on a 10% TBE-Urea PAGE directly, or after washing to remove any unconjugated Apt, The bands corresponding to the A10 PSMA Apt and NP-Apt are indicated by arrows. Nucleic acid molecular weight marker (MW) is shown on left,
3.6 In Vivo tumor targeting and biodistribution of NP-Apt bioconjugates
As a result of our investigations of formulation parameters and their effects on NP size, an optimal NP formulation in terms of size and drug loading was chosen for in vivo study. For the study, 14C-paclitaxel (serving as a tracing agent) was encapsulated at a drug loading of 1% into the PLGA-b-PEG NPs. Paclitaxel is a taxane drug related to docetaxel and is available commercially as a radiochemical. The resulting NPs were sized at 156.8 +/- 3.9 nm, and after bioconjugation with the aptamers, the final size of the NP-Apt bioconjugates was measured to be 188.1 ± 4.0 nm. At all three time-points, the 14C-paclitaxel dose recovered in the tumor was higher for the NP-Apt targeted groups compared to the control NP groups (Figure 6). The values in % injected dose per gram of tissue at 2, 6, and 24 hours for the NP-Apt group were 1.49 ± 0.92, 1.98 ± 1.72, and 0.83 ± 0.21, respectively (mean ± S.D., n = 4). For the NP control group, the respective values at 2, 6, and 24 hours were 1.10 ± 0.20, 0.96 ± 0.44, and 0.22 ± 0.07. At the 24 hour endpoint of the study, the level in the tumor was 3.77-fold higher for the NP-Apt group (p = 0.002, n = 4). At the 2 and 6 hour time-points, the level of NP-Apt in the tumor was 1.35-fold and 2.06-fold higher than the control, respectively, but this difference was not statistically significant. Interestingly in both 2 and 6 hr groups the intra-tumoral concentrations of the NP-Apt increased as compared to the NP control, while the levels in most other tissues decreased in parallel to less NPs in circulation. We postulated that the concentration of the recovered drug in the tumor over time shows both the EPR effect and the effect of targeting. The ability of the NP-Apt bioconjugates to maintain a significantly higher concentration in the tumor at 24 hours is possibly due to uptake by the targeted LNCaP cells, while the NP group without targeting ligand diffused away from the tumor over time in the absence of cell uptake. Similar strong binding of the NP-Apt bioconjugates to LNCaP cells was observed in vitro [14]. Additionally, the concentration in the tumor for both groups may decline from the 6 and 24 h time-points due to the burst effect of the NPs, which can release a large percentage of the drug during this time [17]. Drug released at the tumor site, if not internalized by the cells, can diffuse away or be clear from the site.
Figure 6.

Tumor targeting by PLGA-b-PEG NP and NP-Apt after systemic administration, (mean ± SD; n = 4; * P < 0.002)
Biodistribution patterns to the heart, lungs, and kidneys did not show substantial accumulation in either group and were not significantly different (Figure 7). The uptake by the RES, including the spleen and liver, was observed to be higher for the NP-Apt bioconjugates as compared to the control NPs. The outer PEG layer, while providing an excellent stealth shield for the NP group, was modified in the NP-Apt group by surface bioconjugation. Aptamers are not considered immunogenic, and thus the likely cause of the observed RES uptake was this disruption of the PEG shield. While not a focus of this study, the aptamer surface coating and length of PEG chains employed in the NP can be varied perhaps to improve on this outcome. Further, the bioconjugation resulted in a moderate increase in mean particle size compared to the NP group. The increased size can partially explain the increased uptake the spleen [9].
Figure 7.

Systemic biodistribution of (A) PLGA-b-PEG NP and (B) NP-Apt (mean ± SD; n = 4)
4. Conclusions
We studied the effect of solvent type, polymer concentration, and water / solvent ratio on the size of pegylated PLGA NP, during the nanoprecipitation method, and for the first time report a linear correlation between polymer concentrations and volumetric sizes of the resulting NPs. This observation was confirmed after evaluating four distinct solvents and may extend to other polymer systems, broadly enabling the reproducible tuning of polymeric NP sizes during their formulation.
We next conjugated the resulting pegylated PLGA NPs to the A10 aptamer and demonstrated enhanced NP delivery to prostate tumors as compared to equivalent non-targeted NPs. This is the first report of a tumor specific systemic targeting of a nanoparticle-aptamer bioconjugate system in vivo and may result in the development of therapeutically effective vehicles for disseminated prostate cancer for which the current therapies are largely ineffective.
Acknowledgments
This research was partially funded by NIH/NCI CA 119349, NIH/NIBIB EB 003647, the David H. Koch Foundation, and the Department of Defense Prostate Cancer Research Program PC 051156. Ines Sherifi was financially supported by the University of Toronto Summer Scholarship Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Allen TM, Cullis PR. Drug Delivery Systems: Entering the Mainstream. Science. 2004;303(5665):1818–22. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 2.LaVan DA, McGuire T, Langer R. Small-scale systems for in vivo drug delivery. Nat Biotechnol. 2003;21(10):1184–91. doi: 10.1038/nbt876. [DOI] [PubMed] [Google Scholar]
- 3.Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54(5):631–51. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 4.Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2004;56(11):1649–59. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 5.Fahmy TM, Fong PM, Goyal A, Saltzman WM. Targeted for drug delivery. Materials Today. 2005;8(8) 1:18–26. [Google Scholar]
- 6.Avgoustakis K. Pegylated Poly(Lactide) and Poly(Lactide-co-Glycolide) Nanoparticles: Preparation, Properties and Possible Application in drug Delivery. Curr Drug Deliv. 2004;1:321–33. doi: 10.2174/1567201043334605. [DOI] [PubMed] [Google Scholar]
- 7.Moghimi SM, Davis SS. Innovations in Avoiding Particle Clearance from Blood by Kupffer Cells - Cause for Reflection. Crit Rev Ther Drug. 1994;11(1):31–59. [PubMed] [Google Scholar]
- 8.Moghimi SM, Porter CJH, Muir IS, Illum L, Davis SS. Non-Phagocytic Uptake of Intravenously Injected Microspheres in Rat Spleen - Influence of Particle-Size and Hydrophilic Coating. Biochemical and Biophysical Research Communications. 1991;177(2):861–6. doi: 10.1016/0006-291x(91)91869-e. [DOI] [PubMed] [Google Scholar]
- 9.Storm G, Belliot SO, Daemen T, Lasic DD. Surface Modification of Nanoparticles to Oppose Uptake by the Mononuclear Phagocyte System. Adv Drug Deliv Rev. 1995;17(1):31–48. [Google Scholar]
- 10.Litzinger DC, Buiting AMJ, Vanrooijen N, Huang L. Effect of Liposome Size on the Circulation Time and Intraorgan Distribution of Amphipathic Poly(Ethylene Glycol)-Containing Liposomes. Bba-Biomembranes. 1994;1190(1):99–107. doi: 10.1016/0005-2736(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 11.Stolnik S, Heald CR, Neal J, Garnett MC, Davis SS, Illum L, Purkis SC, Barlow RJ, Gellert PR. Polylactide-poly(ethylene glycol) micellar-like particles as potential drug carriers: Production, colloidal properties and biological performance. J Drug Target. 2001;9(5):361–78. doi: 10.3109/10611860108998772. [DOI] [PubMed] [Google Scholar]
- 12.Porter CJH, Moghimi SM, Illum L, Davis SS. The Polyoxyethylene Polyoxypropylene Block Copolymer Poloxamer-407 Selectively Redirects Intravenously Injected Microspheres to Sinusoidal Endothelial-Cells of Rabbit Bone-Marrow. Febs Letters. 1992;305(1):62–6. doi: 10.1016/0014-5793(92)80655-z. [DOI] [PubMed] [Google Scholar]
- 13.Lupold SE, Hicke BJ, Lin Y, Coffey DS. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002;62(14):4029–33. [PubMed] [Google Scholar]
- 14.Farokhzad OC, Jon SY, Khadelmhosseini A, Tran TNT, LaVan DA, Langer R. Nanopartide-aptamer bioconjugates: A new approach for targeting prostate cancer cells. Cancer Res. 2004;64(21):7668–72. doi: 10.1158/0008-5472.CAN-04-2550. [DOI] [PubMed] [Google Scholar]
- 15.Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103(16):6315–20. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooperberg MR, Moul JW, Carroll PR. The changing face of prostate cancer. J Clin Oncol. 2005;23(32):8146–51. doi: 10.1200/JCO.2005.02.9751. [DOI] [PubMed] [Google Scholar]
- 17.Fonseca C, Simoes S, Gaspar RE. Paclitaxel-loaded PLGA nanoparticles: preparation, physicochemical characterization and in vitro anti-tumoral activity. J Controlled Release. 2002;83(2):273–86. doi: 10.1016/s0168-3659(02)00212-2. [DOI] [PubMed] [Google Scholar]
- 18.Galindo-Rodriguez S, Allemann E, Fessi H, Doelker E. Physicochemical parameters associated with nanoparticle formation in the salting-out, emulsification-diffusion, and nanoprecipitation methods. Pharm Res. 2004;21(8):1428–39. doi: 10.1023/b:pham.0000036917.75634.be. [DOI] [PubMed] [Google Scholar]
- 19.Bilati U, Allemann E, Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur J Pharm Sci. 2005;24(1):67–75. doi: 10.1016/j.ejps.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 20.Yu V, SV T. General principles governing dissolution of materials in solvents. ChemTec Publishing; 2001. [Google Scholar]
- 21.Suh HR, Jeong BM, Rathi R, Kim SW. Regulation of smooth muscle cell proliferation using paclitaxel-loaded poly(ethylene oxide)-poly(lactide/glycolide) nanospheres. J Biomed Mater Res. 1998;42(2):331–8. doi: 10.1002/(sici)1097-4636(199811)42:2<331::aid-jbm19>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 22.Dong YC, Feng SS. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25(14):2843–9. doi: 10.1016/j.biomaterials.2003.09.055. [DOI] [PubMed] [Google Scholar]
- 23.Dong Y, Feng SS. Poly(d,l-lactide-co-glycolide)/montmorillonite nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(30):6068–76. doi: 10.1016/j.biomaterials.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 24.Park TG. Degradation of Poly(D,L-Lactic Acid) Microspheres - Effect of Molecular-Weight. J Controlled Release. 1994;30(2):161–73. [Google Scholar]
- 25.De Jaeghere F, Allemann E, Feijen J, Kissel T, Doelker E, Gurny R. Freeze-drying and lyopreservation of diblock and triblock poly(lactic acid)poly(ethylene oxide) (PLA-PEO) copolymer nanoparticles. Pharmaceutical Development and Technology. 2000;5(4):473–83. doi: 10.1081/pdt-100102031. [DOI] [PubMed] [Google Scholar]
- 26.Konan YN, Cerny R, Favet J, Berton M, Gurny R, Allemann E. Preparation and characterization of sterile sub-200 nm meso-tetra(4-hydroxylphenyl)porphyrin-loaded nanoparticles for photodynamic therapy. European Journal of Pharmaceutics and Biopharm. 2003;55(1):115–24. doi: 10.1016/s0939-6411(02)00128-5. [DOI] [PubMed] [Google Scholar]