

Abstract
Selective inhibitors of neuronal nitric oxide synthase (nNOS) have the potential to develop into new neurodegenerative therapeutics. Recently, we described the discovery of novel nNOS inhibitors (1a and 1b) based on a cis-pyrrolidine pharmacophore. These compounds and related ones were found to have poor blood-brain barrier permeability, presumably because of the basic nitrogens in the molecule. Here, a series of monocationic compounds was designed on the basis of docking experiments using the crystal structures of 1a,b bound to nNOS. These compounds were synthesized and evaluated for their ability to inhibit neuronal nitric oxide synthase. Despite the excellent overlap of these compounds with 1a,b bound to nNOS, they exhibited low potency. This is because they bound in the nNOS active site in the normal orientation rather than the expected flipped orientation used in the computer modeling. The biphenyl or phenoxyphenyl tail is disordered and does not form good protein-ligand interactions. These studies demonstrate the importance of the size and rigidity of the side chain tail and the second basic amino group for nNOS binding efficiency and the importance of the hydrophobic tail for conformational orientation in the active site of nNOS.
Keywords: Neuronal nitric oxide synthase, cis-pyrrolidine pharmacophore, neurodegenerative therapeutics, enzyme inhibitors
Introduction
Many lines of biological evidence have shown that overproduction of nitric oxide (NO) in the central nervous system (CNS) is implicated in various types of neurodegenerative diseases, including Parkinson’s,1–3 Alzheimer’s,4 and Huntington’s disease.5 Neuronal nitric oxide synthase (nNOS), the enzyme responsible for the generation of neuronal NO, is, therefore, a promising target for the development of new neurodegenerative therapeutics.6–9 Over the past two decades, significant research has been devoted to developing nNOS selective inhibitors.10,11 Recently, we described the discovery of the novel nNOS inhibitors 1a,b (Figure 1)12 based on a cis-pyrrolidine pharmacophore.13,14 Inhibitor 1b has excellent potency (Ki = 15 nM), as well as high isozyme selectivity for nNOS (2100-fold over endothelial NOS; 630-fold over inducible NOS), which makes this compound a promising lead for neurodegenerative therapeutics.12
Figure 1.

Design of monocationic inhibitors 2 based on 1b.
Inhibition of an enzyme that is implicated in the CNS, such as nNOS, however, requires the designed molecules to cross the blood brain barrier (BBB).15 Generally, there are three criteria for a small molecule to cross the BBB: 1) molecular weight <450 Dalton;16 2) reasonably good lipid solubility;16 and 3) no affinity for one of BBB active efflux transporters (e.g., p-glycoprotein).17–21 Recent animal tests demonstrated that 1a,b have low BBB permeability, which strongly impedes further application of this compound as a neurodegenerative therapeutic.12 Among the mechanisms that could limit 1b from crossing the BBB, the dicationic character of this molecule under physiological conditions, as a result of the two high pKa secondary amino groups, is a crucial factor prohibiting passive diffusion.16 To improve the BBB permeability, prodrug approaches have been carried out to partially decrease the cationic character of 1b.22 Herein, we describe the structure-based design and synthesis of a new series of nNOS inhibitors (2, Fig.1) with the aminoalkyl tail of 1b replaced by a lipophilic biphenyl fragment or a phenoxyphenyl fragment. We reasoned that these compounds would be effective, given that they were designed based on a recent unexpected finding on how the (3R,4R) enantiomer of 1a binds to nNOS.23 It was anticipated that the (3R,4R)-1a would bind such that the aminopyridine ring stacks over the heme and H-bonds with the active site Glu592. Instead, we found that it binds in a flipped orientation with the aminopyridine extending out of the active site, where it H-bonds with a heme propionate. We also found that the (3R,4R)-1b binds the same as its counterpart of 1a (to be published).23,24 To make room for the aminopyridine, Tyr706 must swing out of the way, where it forms a π-stacking interaction with the aminopyridine (Fig.2).23,24 Because of this new binding orientation, compounds with fewer basic amino groups could overlay nicely on the flipped binding mode of 1b. There are two potential advantages to this new design. First, one of the two high pKa amino groups is removed from the lead compound (1b), resulting in a monocationic compound, which should have a better chance to cross the BBB by passive diffusion.25 Second, using a phenoxyphenyl or biphenyl fragment, new inhibitors 2 possess a more rigid structure compared to 1b, which can potentially improve the potency of inhibitors and be less susceptible to metabolic degradation. The 4-methyl group on the aminopyridine ring of 1b was removed in compounds 2a–f because the methyl group of the eutomer of 1b is stabilized at the hydrophobic pocket of rat nNOS lined with Met336, Leu337, and Trp306 (chain B).23,24 In human NOS, the residue that corresponds to rat nNOS Leu337 is His342, and it is the only residue in the active site that differs between rat nNOS and human nNOS.26 It was hypothesized that demethylated compound 2a–f might fit better in the active site of human nNOS, which does not have this hydrophobic pocket.
Figure 2.
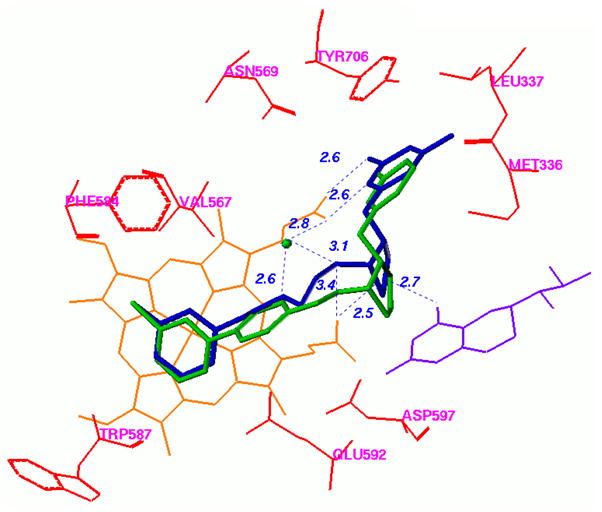
Docking conformation of 2h (green) in the active site of nNOS. The potent and selective inhibitor N1-[(3R,4R)-4′-((6″-amino-4″-methylpyridin-2″-yl)methyl)pyrrolidin-3′-yl]-N2-(3′-fluorophenethyl)ethane-1,2-diamine (blue, the (3R,4R) enantiomer of 1a in Figure 1 in the crystal structure of rat nNOS (PDB code: 3JWT)23 is overlaid for comparison.
2. Chemistry
The synthesis of key intermediate 9 began with 2-amino-6-methylpyridine (3) (Scheme 1). Protection of the amino group in 3 with (Boc)2O in the presence of triethylamine (TEA) gave 4 in an excellent yield. Boc-protected aminopyridine 4 was treated with 2 equiv of n-BuLi, and the resulting dianion was allowed to react with epoxide 513 to give secondary alcohol 6 in good yields. Next, 6 was converted to a bis-Boc-protected alcohol using a three-step procedure.27 First, the hydroxyl group was protected as the t-butyldimethylsilyl (TBS)-ether (7) using TBSCl in the presence of imidazole. Then, 7 was treated with (Boc)2O in the presence of N,N-dimethylpyridine (DMAP) to provide bis-Boc-protected silyl ether 8. Finally, the TBS-protecting group was removed using tetrabutylammonium fluoride (TBAF) to generate 9 in high yields.
Scheme 1.

Synthesis of 9.a
a Reagents and conditions: (i) (Boc)2O, TEA, rt, 12 h, 97%; (ii) (1) n-BuLi, −78 ºC – 0 ºC, (2) 5, −78 ºC – rt, 16 h, 70%; (iii) TBSCl, imidazole, rt, 16 h, 78%; (iv) (Boc)2O, DMAP, rt, 30 h, 92%; (v) TBAF, rt, 30 min, 97%.
The substituted phenoxyphenols (10b–f) were synthesized using a two-step procedure (Scheme 2). First, Ullmann couplings of m-methoxyphenol (11) with iodobenzene derivatives using Cs2CO3 in the presence of a catalytic amount of CuBr provided 12b–f.28 Then, the methyl ethers were cleaved using BBr3 to give 10b–f in high yields.
Scheme 2.

Synthesis of 10b–f.a
a Reagents and conditions: (i) iodobenzene, Cs2CO3, CuBr, 60 °C, 48 h, 87–95%; (ii) BBr3, −78 °C – rt, 85–92%.
With both 9 and 10a–f in hand, the syntheses of inhibitors 2a–f were completed (Scheme 3). After screening a series of reaction conditions, the Mitsunobu reaction using 9 and 10a–f as starting materials went smoothly at room temperature to generate 12a–f in modest to good yields. Then, the three Boc-protecting groups of 12a–f were removed in TFA to generate inhibitors 2a–f.
Scheme 3.

Syntheses of inhibitors 2a–f.a
a Reagents and conditions: (i) phenol, PPh3, DEAD, 0 °C – rt, 72 h, 35–51%; (ii) TFA/CH2Cl2 (1:2), rt, 4 h, 91–95%.
The synthesis of single enantiomer 14a and 14b is shown in Scheme 4. The free NH group on the pyridine ring of 15 was protected with a SEM-protecting group using SEM-Cl using NaH as a base to yield 16 in good yields. The two enantiomers were resolved through camphanic ester derivatives using a Mitsunobu reaction to generate two separable diastereomers 17a and 17b in reasonable yields. Finally, the ester linkage was hydrolyzed using Na2CO3 to provide chiral precursor 14a and 14b in excellent yields.
Scheme 4.

Synthesis of 14a and 14b.
a Reagents and conditions: (a) NaH, SEM-Cl, rt, 16 h, 68%; (b) (S)-(-) camphanic acid, PPh3, DEAD, rt, 16 h, 88%; (d) Na2CO3, rt, 4 h, 95%.
The bi-aryl fragment 19a–e was synthesized using a two-step procedure (Scheme 5). Suzuki coupling of 4-(bromomethyl)phenol with a series of boronic acid generated to 18a–e. Then, the hydroxyl group of 18a–e was converted to bromide (19a–e) by refluxing in HBr in high yields.
Scheme 5.

Synthesis of 19a–e.
aReagents and conditions: (a) ArB(OH)2, Pd(Ph3P)4, 2 N Na2CO3, DME, 80 °C, 48 h; (b) HBr, reflux, 3 h.
The syntheses of inhibitors 2g–k were finished as shown in Scheme 6. Compound 14b was treated with NaH, and the resulting anion was reacted with 19a–e to provide 13g–k in excellent yields. Then, global deprotection of SEM-protecting group and Boc-protecting groups generated inhibitors 2g–k in high yields.
Scheme 6.

Synthesis of 2g–k.
a Reagents and conditions: (a) NaH, 4-arylbenzyl bromide, rt, 6 h; (b) 4 N HCl in MeOH, rt, 16 h, 90%.
3. Results and Discussion
Inhibitors 2a–k were evaluated for in vitro inhibition activity against three isozymes of NOS: rat nNOS, bovine eNOS, and murine iNOS using known methods.22 The results are summarized in Table 1. Inhibitor 2a, with a biphenyl group in place of the aminoalkyl tail of 1b, had a Ki value of 10 μM for nNOS, while the phenoxyphenyl analog 2b had slightly better inhibitory activity (Ki = 3.0 μM for nNOS). Inhibitors 2c and 2d, with halogen-substituents at the para position of the terminal phenyl ring, are weaker inhibitors than the non-substituted analog (2b). However, the additional substituent at the meta position on the ring, inhibitors 2e and 2f, showed slightly enhanced potency relative to 2a against nNOS. We believe that the additional fluorine or methyl substituent fits into a small hydrophobic pocket at Met336, providing additional binding energy. Inhibitors 2g–k, with the installation of a 4-methyl group on the aminopyridine ring, indicated some improved inhibitory activity. All new inhibitors are significantly less potent (300–600 fold) than lead compound 1b, with Ki values in the low micromolar range, although the shape and size of the active site of nNOS can fit this series of compounds according to the docking calculation. These results were initially disappointing, given that this series overlaid very well with the crystal structure of the (3R,4R)-1b bound to nNOS in the flipped mode (Fig. 2). However, the crystal structure of a representative phenoxyphenyl analogue (2b) bound to nNOS showed that although the racemic mixture of 2b was used in crystal preparation, only the (3R,4R) enantiomer was observed in the structure, and it did not bind in the flipped orientation (Fig. 3). Instead, the aminopyridine was observed to bind in the normal orientation in which the aminopyridine interacts with Glu592, and Tyr706 remains in its native position. This is the first case in the pyrrolidine series of compounds in which the (3R,4R) stereochemistry does not bind in the flipped orientation. It appears that the configuration around the (3,4) positions of the pyrrolidine, therefore, is not the sole determinant controlling the binding orientation of these inhibitors; the size and rigidity of the tail also are very important. When the tail is bulky and rigid, as that in 2b, the inhibitor cannot adopt the flipped mode because its tail no longer fits into the active site over the heme distal surface. Instead, the aminopyridine ring interacts with Glu592, and the tail end of the inhibitor extends out of the active site. The absence of electron density for the phenoxyphenyl tail indicates poor interactions with protein groups, which may account, in part, for the relatively weak affinity seen for these compounds. It is interesting that the (3S,4S)-1b24 was bound in the same orientation as was observed for (3R,4R)-2b in this structure. The pyrrolidine nitrogen of (3S,4S)-2b would have H-bonded directly to Glu592, which is not observed here (Fig. 3). It is likely that the bulky phenoxyphenyl tail in (3S,4S)-2b would have an even poorer fit than what was seen for the tail in (3R,4R)-2b. The bulkiness of this hydrophobic tail, apparently, does not permit the molecule from assuming the flipped binding mode.
Table 1.
Kia values of inhibitors for rat nNOS, bovine eNOS, and murine iNOS.
| Compound | nNOS (μM) | eNOS (μM) | iNOS (μM) | selectivityb |
|
|---|---|---|---|---|---|
| n/e | n/i | ||||
| 2a | 10.0 | >200 | >200 | >20 | >20 |
| 2b | 3.0 | >200 | >200 | >67 | >67 |
| 2c | 7.8 | 18 | 221 | 2.3 | 28 |
| 2d | 8.4 | 16 | 79.5 | 1.9 | 10 |
| 2e | 2.2 | 8.3 | 81.8 | 1.8 | 18 |
| 2f | 4.3 | 15 | 79.1 | 1.7 | 9 |
| 2g | 0.75 | 13.3 | 39.5 | 18 | 53 |
| 2h | 1.2 | 26.0 | 55.6 | 22 | 46 |
| 2i | 0.82 | 13.6 | 37.3 | 17 | 45 |
| 2j | 0.62 | 8.0 | 22.1 | 13 | 36 |
| 2k | 0.76 | 13.3 | 50.0 | 18 | 66 |
The Ki values were calculated based on the directly measured IC50 values, which represent at least duplicate measurements with standard deviations of ±10%.
The ratio of Ki (eNOS or iNOS) to Ki (nNOS).
Figure 3.

The active site crystal structure of nNOS with 2b bound (PDB code: 3N2R) showing some relevant hydrogen bonds as dashed lines. The 2Fo-Fc (gray) and Fo-Fc (red) electron density map of 2b are contoured at 1.0 σ and −3.0 σ, respectively. Although crystals were soaked in a racemic mix of 2b, the electron density is consistent with only the (3R,4R) enantiomer. The pyrrolidine ring nitrogen of the (3S,4S) enantiomer would have directly hydrogen bonded to Glu592, replacing the position of Wat1. The electron density for the aminopyridine group is very clear and partially so for the pyrrolidine. However, there is no electron density for the phenoxyphenyl tail, indicating disordering and, therefore, the absence of any tight protein-phenoxyphenyl interactions.
4. Conclusion
A new series of nNOS inhibitors (2a–k) has been designed and synthesized based on docking experiments with the crystal structure of the lead compound, (3R,4R)-1b, which bound to nNOS in a flipped binding mode. Because of the bulky tail of 2a–k, they actually prefer the normal binding mode with a disordered tail, and, therefore, these compounds are only weak inhibitors of nNOS. The secondary amino group in the lipophilic tail also might be critical for tight binding of 1 to nNOS in the flipped mode, as compounds without this amino group, to date, bind poorly to nNOS.
5. Experimental procedures
5.1. General methods
All syntheses were conducted under anhydrous conditions in an atmosphere of argon, using flame-dried apparatus and employing standard techniques in handling air-sensitive materials. All solvents were distilled and stored under an argon or nitrogen atmosphere before use. All reagents were used as received. Aqueous solutions of sodium bicarbonate, sodium chloride (brine), and ammonium chloride were saturated. Analytical thin layer chromatography was visualized by ultraviolet light. Flash column chromatography was carried out under a positive pressure of nitrogen. 1H NMR spectra were recorded on 500 MHz spectrometers. Data are presented as follows: chemical shift (in ppm on the δ scale relative to δ = 0.00 ppm for the protons in TMS), integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constant (J/Hz). Coupling constants were taken directly from the spectra and are uncorrected. 13C NMR spectra were recorded at 125 MHz, and all chemical shift values are reported in ppm on the δ scale, with an internal reference of δ 77.0 or 49.0 for CDCl3 or CD3OD, respectively. High-resolution mass spectra were measured on liquid chromatography/time-of-flight mass spectrometry (LC-TOF).
5.2. General procedure A (Ullmann reaction)
To a mixture of Cs2CO3 (1.37 g, 4.2 mmol), CuBr (29 mg, 0.2 mmol), and ethyl 2-oxocyclohexanecarboxylate28 (64 μL, 0.4 mmol) was added DMSO (1.0 mL). The mixture was allowed to stir at room temperature for 30 min, followed by addition of a mixture of 3-methoxyphenol (2.0 mmol) and iodobenzene (2.0 mmol) as a solution in DMSO (1.0 mL) through a cannula. The flask was sealed and heated at 60 °C for 24 h and then cooled to room temperature. The reaction was filtered through Celite, and the resulting cake was washed with EtOAc (4 × 25 mL). The combined organic layers were washed with brine (50 mL), dried over Na2SO4, and concentrated. The crude product was purified by flash chromatography (EtOAc/hexanes, 1:9) to yield 12b–f (87–95%) as pale yellow oils.
5.3. General procedure B (demethylation)
To a solution of a methyl ether (12b–f, 1.0 mmol) in dichloromethane at −78 °C was added BBr3 (1 M solution, 1.2 mL, 1.2 mmol) dropwise. The mixture was warmed to room temperature over a period of 2 h and kept stirring overnight. The solvent was removed by rotary evaporation, and the resulting material was purified by flash chromatography (EtOAc/hexanes, 1:9) to yield 10b–f (85–92%) as colorless oils.
5.4. General procedure C (Mitsunobu reaction)
A mixture of 9 (100 mg, 0.2 mmol) and PPh3 (100 mg, 0.3 mmol) was dissolved in THF (5 mL). To the resulting solution was added a phenol (10a–f, 0.3 mmol) as a solution in THF (1.0 mL), followed by DEAD (60 μL, 0.3 mmol) dropwise. The mixture was allowed to stir at room temperature for 40 h and then concentrated. The crude product was purified by flash chromatography to yield 13a–f (35–51%) as white solids.
5.5. General procedure D (Boc-deprotection)
To a solution of 13a–f (50 μmol) in dichloromethane (1.0 mL) was added TFA (1.0 mL). The reaction mixture was allowed to sit at room temperature for 4 h and then concentrated. The crude product was purified by flash chromatography (5–10% methanol in dichloromethane) to yield 2a–f (91–95%) as white solids.
5.6. General procedure E (Suzuki coupling)
To a solution of 3-fluorophenylboronic acid (462 mg, 3.3 mmol) and 4-bromophenylmethanol (561 mg, 3 mmol) in DME (10 mL) was added Pd(Ph3P)4 (173 mg, 0.15 mmol) and 2 N Na2CO3 (1.5 mL). The mixture was stirred at 80 °C for 48h. After cooling to room temperature, the reaction mixture was filtered through Celite. The filtrate was concentrate, and the resulting residue was purified by flash column chromatography (EtOAc/hexane, 1:4) to generate 18a–e as white solids (80–84%).
5.7. General procedure F
To 2.0 mmol of 18a–e was added HBr solution (48%, 5 mL). The reaction was heated under reflux for three hours, and then cooled to room temperature. The mixture was partitioned between EtOAc (100 mL) and H2O (30 mL). The organic layer was washed with H2O (2 × 30 mL), dried over Na2SO4, and concentrated. The crude product was purified by flash column chromatography on silica gel to provide the product 19a–e (84–90%).
5.8. General procedure G
To a solution of 14b (0.1 mmol) in DMF (2 mL) was added NaH (60% in mineral oil, 10 mg, 0.25 mmol) at 0 °C. After 10 min, 19a–e (0.105 mmol) was added as a solution DMF (1 mL). The reaction was stirred at room temperature for 16 h, and concentrated in vacuo. The resulting residue was partitioned between EtOAc (100 mL) and H2O (10 mL). The organic layer was washed with H2O (2 × 10 mL), dried over Na2SO4, and concentrated. The crude product was purified by flash column chromatography on silica gel (EtOAc/hexanes, 5:2) to generate 13g–k as colorless oils (60–66%).
5.9. General procedure H
A solution of 13g–k (0.1 mmol) in 3 N HCl in methanol (3 mL) was stirred at room temperature for 24h. The mixture was concentrated, and the resulting crude material was purified by Sephdex LH-20 to give final inhibitors 2g–k (95–99%) as hydrochloride salts.
5.10. tert-Butyl 6-methylpyridin-2-ylcarbamate (4)
To a solution of 2-amino-6-methylpyridine (4.32 g, 40 mmol) in t-butanol (40 mL) was added (Boc)2O (10.9 g, 50 mmol) and triethylamine (7.0 mL, 50 mmol). The solution was heated at 50 °C for 24 h. The solvent was removed by rotary evaporation, and the crude product was purified using flash column chromatography (EtOAc/hexanes, 1:18-1:9) to generate 4 (7.5 g, 36 mmol, 97%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 1.42 (s, 9H), 2.40 (s, 3H), 7.46–7.50 (dd, J = 7.5, 8.5 Hz, 1H), 6.72–6.74 (d, J = 8.0 Hz, 1H), 7.71–7.73 (d, J = 8.5 Hz, 1H), 8.79 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 24.0, 28.4, 80.7, 109.6, 118.0, 138.6, 152.0, 153.0, 156.9; LCQ-MS (M + H+) calcd for C11H17N2O2 209, found 209.
5.11. tert-Butyl 3-((6-(tert-butoxycarbonylamino)pyridin-2-yl)-methyl)-4-hydroxypyrrolidine-1-carboxylate (6)
To a solution of 4 (1.0 g, 4.81 mmol) in THF (30 mL) at −78 °C was added n-BuLi (1.6 M in hexanes, 6.3 mL, 10.0 mmol) dropwise. The resulting dark red solution was stirred at the same temperature for an additional 1 h, and then the reaction flask was transferred to an ice-bath for another 30 min. The reaction was cooled to −78 °C, to which a solution of epoxide 5 (890 mg, 4.81 mmol) in THF (10 mL) was added dropwise over a period of 30 min along the side of the reaction flask. After addition, the Dry Ice-acetone bath was removed, and the reaction was kept stirring for an additional 2.5 h. The reaction was quenched with H2O (1.0 mL) and concentrated by rotary evaporation. The crude product was purified using flash column chromatography (EtOAc/hexanes, 2:3-1:1) to provide 6 (1.32 g, 3.37 mmol, 70%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 1.45 (s, 9H), 1.49 (s, 9H), 2.30–2.50 (m, 1H), 2.60–2.80 (m, 2H), 3.00–3.30 (m, 2H), 3.50–3.80 (m, 2H), 4.00–4.20 (m, 1H), 5.04 (br s, 1H), 6.80–6.90 (m, 1H), 7.50–7.80 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 28.3, 28.6, 39.2, 44.8, 45.2, 49.2, 49.7, 52.3, 52.7, 60.5, 64.5, 74.5, 75.2, 79.5, 81.1, 110.3, 118.2, 139.2, 151.6, 152.4, 154.6, 157.9; LCQ-MS (M + H+) calcd for C20H32N3O5 394, found 394.
5.12. tert-Butyl 3-((6-(tert-butoxycarbonylamino)pyridin-2-yl)-tert-butyldimethylsilyloxy)pyrrolidine-1-carboxylate (7)
To a solution of 6 (850 mg, 2.16 mmol) in DMF (5.0 mL) were added TBSCl (408 mg, 2.7 mmol) and imidazole (367 mg, 5.4 mmol). The reaction mixture was stirred at 40 °C for 24 h and then partitioned between EtOAc (150 mL) and aqueous NH4Cl (100 mL). The organic layer was washed with brine (100 mL), dried over Na2SO4, and concentrated. The crude product was purified using flash column chromatography (EtOAc/hexanes, 1:9-1:6) to provide 7 (855 mg, 1.68 mmol, 78%) as a white solid: 1H NMR (500 MHz, CDCl3) δ −0.03 (s, 6H), 0.81 (s, 9H), 1.40 (s, 9H), 1.47 (s, 9H), 2.40–2.50 (m, 2H), 2.75–2.85 (m, 1H), 2.98–3.14 (m, 2H), 3.38–3.62 (m, 2H), 3.90–3.98 (m, 1H), 6.73 (d, J = 7.5 Hz, 1H), 7.38–7.54 (m, 2H), 7.71 (m, 1H); 13C NMR (125 MHz, CDCl3) δ −4.6, 18.2, 25.9, 28.4, 28.7, 39.3, 46.1, 46.7, 48.8, 49.1, 52.7, 53.0, 74.5, 75.3, 79.4, 81.0, 109.9, 117.9, 138.7, 151.6, 152.5, 154.9, 158.3; LCQ-MS (M + H+) calcd for C26H46N3O5Si 508, found 508.
5.13. tert-Butyl 3-((6-(bis(tert-butoxycarbonyl)amino)pyridin-2-yl)-methyl)-4-(tert-butyldimethylsilyloxy)pyrrolidine-1-carboxylate (8)
To a solution of 7 (507 mg, 1.0 mmol) in THF (10 mL) was added (Boc)2O (327 mg, 1.5 mmol) and DMAP (30 mg, 0.25 mmol). The solution was stirred at room temperature for 16 h. The solvent was removed by rotary evaporation, and the crude product was purified using flash column chromatography (EtOAc/hexanes, 1:9-1:6) to generate 8 (558 mg, 0.92 mmol, 92%) as a white solid: 1H NMR (500 MHz, CDCl3) δ −0.01 (s, 6H), 0.84 (s, 9H), 1.40–1.45 (m, 27H), 2.45–2.55 (m, 2H), 2.83–3.14 (m, 3H), 3.40–3.64 (m, 2H), 3.98–4.04 (m, 1H), 6.98–7.02 (m, 1H), 7.06–7.14 (m, 1H), 7.55–7.65 (m, 1H); 13C NMR (125 MHz, CDCl3) δ −4.5, 18.2, 25.9, 28.1, 28.7, 39.1, 46.1, 46.7, 48.5, 48.9, 52.6, 53.2, 74.5, 75.1, 79.4, 83.0, 118.9, 121.5, 138.3, 151.5, 152.0, 154.9, 159.2; LCQ-MS (M + H+) calcd for C31H54N3O7Si 608, found 608.
5.14. tert-Butyl 3-((6-(bis(tert-butoxycarbonyl)amino)pyridin-2-yl)-methyl)-4-hydroxypyrrolidine-1-carboxylate (9)
To a solution of 8 (607 mg, 1.0 mmol) in THF (10 mL) was added TBAF (1 M in THF, 1.25 mL, 1.25 mmol). The solution was stirred at room temperature for 16 h. The solvent was removed by rotary evaporation, and the resulting crude product was purified using flash column chromatography (EtOAc/hexanes, 1:1) to generate 9 (476 mg, 0.97 mmol, 97%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 1.42–1.47 (m, 27H), 2.43–2.50 (m, 1H), 2.85–2.95 (m, 2H), 3.03–3.12 (m, 1H), 3.15–3.24 (m, 1H), ), 3.61–3.80 (m, 2H), 4.14–4.18 (m, 1H), 7.06–7.14 (m, 2H), 7.70 (dd, J = 7.5, 8.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 28.1, 28.7, 39.5, 39.7, 44.9, 45.7, 49.7, 50.0, 52.5, 53.0, 74.6, 75.3, 79.5, 83.7, 119.8, 120.0, 122.2, 122.4, 139.1, 151.5, 151.8, 154.7, 159.4; LCQ-MS (M + H+) calcd for C25H40N3O7 494, found 494.
5.15. 1-(4-Fluorophenoxy)-3-methoxybenzene (12c)
Compound 12c was synthesized using general procedure A (95%): 1H NMR (500 MHz, CDCl3) δ 3.76 (s, 3H), 6.52–6.54 (d, J = 8.5 Hz, 2H), 6.62–6.64 (d, J = 10.0 Hz, 1H), 6.90–7.10 (m, 4H), 7.18–7.22 (m, 1H);.13C NMR (125 MHz, CDCl3) δ 55.5, 104.5, 108.9, 110.5, 116.4, 116.6, 120.9, 121.0, 130.4; LC-MS (M + H+) calcd for C13H12FO2 219, found 219.
5.16. 1-(4-Chlorophenoxy)-3-methoxybenzene (12d)
Compound 12d was synthesized using general procedure A (87%): 1H NMR (500 MHz, CDCl3) δ 3.76 (s, 3H), 6.56–6.67 (m, 2H), 6.66–6.68 (d, J = 10.0 Hz, 1H), 6.94–6.96 (d, J = 11.0 Hz, 2H), 7.15–7.35 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 55.6, 105.2, 109.5, 111.2, 120.5, 129.9, 130.5; LC-MS (M + H+) calcd for C13H12ClO2 235, found 235.
5.17. 1-Chloro-2-fluoro-4-(3-methoxyphenoxy)benzene (12e)
Compound 12e was synthesized using general procedure A (88%): 1H NMR (500 MHz, CDCl3) δ 3.76 (s, 3H), 6.56–6.67 (m, 2H), 6.66–6.68 (d, J = 10.0 Hz, 1H), 6.94–6.96 (d, J = 11.0 Hz, 2H), 7.15–7.35 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 55.6, 105.2, 109.5, 111.2, 120.5, 129.9, 130.5; LC-MS (M + H+) calcd for C13H11ClFO2 253, found 253.
5.18. 1-Fluoro-4-(3-methoxyphenoxy)-2-methylbenzene (12f)
Compound 12f was synthesized using general procedure A (87%): 1H NMR (500 MHz, CDCl3) δ 3.76 (s, 3H), 6.56–6.67 (m, 2H), 6.66–6.68 (d, J = 10.0 Hz, 1H), 6.94–6.96 (d, J = 11.0 Hz, 2H), 7.15–7.35 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 55.6, 105.2, 109.5, 111.2, 120.5, 129.9, 130.5; LC-MS (M + H+) calcd for C14H14FO2 233, found 233.
5.19. 3-(4-Fluorophenoxy)phenol (10c)
Compound 10c was synthesized using general procedure B (85%): 1H NMR (500 MHz, CDCl3) δ 5.51 (br s, 1H), 6.44 (s, 1H), 6.49–6.55 (m, 2H), 6.90–7.10 (m, 4H), 7.12–7.16 (dd, J = 10.0, 10.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 105.6, 110.3, 110.6, 116.4, 116.7, 121.15, 121.24, 130.7; LC-MS (M + H+) calcd for C12H10FO2 205, found 205.
5.20. 3-(4-Chlorophenoxy)phenol (10d)
Compound 10d was synthesized using general procedure B (85%): 1H NMR (500 MHz, CDCl3) δ 5.79 (br s, 1H), 6.47 (s, 1H), 6.52–6.58 (dd, J = 11.0, 14.5, 2H), 6.91–6.94 (d, J = 11.0, 2H), 7.13–7.17 (dd, J = 10.0, 11.0 Hz, 1H), 7.23–7.25 (d, J = 11.0, 2H); 13C NMR (125 MHz, CDCl3) δ 106.4, 110.9, 111.2, 120.7, 128.8, 130.0, 130.8, 155.6, 157.1, 158.5; LC-MS (M + H+) calcd for C12H10ClO2 221, found 221.
5.21. 3-(4-Chloro-3-fluorophenoxy)phenol (10e)
Compound 10e was synthesized using general procedure B (92%): 1H NMR (500 MHz, CDCl3) δ 5.79 (br s, 1H), 6.47 (s, 1H), 6.52–6.58 (dd, J = 11.0, 14.5, 2H), 6.91–6.94 (d, J = 11.0, 2H), 7.13–7.17 (dd, J = 10.0, 11.0 Hz, 1H), 7.23–7.25 (d, J = 11.0, 2H); 13C NMR (125 MHz, CDCl3) δ 107.1, 107.6, 107.8, 110.7, 110.8, 115.27, 115.29, 131.0, 131.1, 157.2, 157.4; LC-MS (M + H+) calcd for C12H9ClFO2 239, found 239.
5.22. 3-(4-Fluoro-3-methylphenoxy)phenol (10f)
Compound 10f was synthesized using general procedure B (86%): 1H NMR (500 MHz, CDCl3) δ 5.79 (br s, 1H), 6.47 (s, 1H), 6.52–6.58 (dd, J = 11.0, 14.5, 2H), 6.91–6.94 (d, J = 11.0, 2H), 7.13–7.17 (dd, J = 10.0, 11.0 Hz, 1H), 7.23–7.25 (d, J = 11.0, 2H); 13C NMR (125 MHz, CDCl3) δ 14.9, 150.0, 105.6, 110.2, 110.5, 116.0, 116.2, 118.46, 118.52, 122.67, 122.71, 126.5, 126.7, 130.7, 152.09, 152.11, 157.0, 158.9, 159.5; LC-MS (M + H+) calcd for C13H12FO2 219, found 219.
5.23. (3R,4R/3S,4S)-tert-Butyl 3-(biphenyl-4-yloxy)-4-((6-(bis(tert-butoxycarbonyl)amino)pyridin-2-yl)methyl)pyrrolidine-1-carboxylate (13a)
Compound 13a was synthesized using general procedure C (40%): 1H NMR (500 MHz, CDCl3) δ 1.40–1.60 (m, 27H), 2.90–3.10 (m, 2H), 3.15–3.25 (m, 1H), 3.35–3.45 (m, 1H), 3.50–3.65 (m, 2H), 3.66–3.80 (m, 1H), 4.68–4.71 (m, 1H), 6.90–6.92 (d, J = 8.0 Hz, 2H), 7.02–7.06 (m, 1H), 7.07–7.12 (m, 1H), 7.30–7.35 (m, 1H), 7.40–7.65 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 24.9, 28.2, 28.7, 28.8, 30.0, 34.9, 42.8, 76.7, 79.7, 79.8, 83.2, 100.0, 116.1, 116.2, 119.3, 120.0, 126.9, 127.0, 128.5, 128.9, 129.0, 134.6, 138.6, 140.9, 151.5, 152.1, 154.6, 154.8, 157.0, 159.3; LCQ-MS (M + H+) calcd for C37H48N3O7 646, found 646.
5.24. (3R,4R/3S,4S)-tert-Butyl 3-((6-(bis(tert-butoxycarbonyl)amino)pyridin-2-yl)methyl)-4-(3-phenoxyphenoxy)pyrrolidine-1-carboxylate (13b)
Compound 13b was synthesized using general procedure C (35%): 1H NMR (500 MHz, CDCl3) δ 1.40–1.60 (m, 27H), 2.80–3.00 (m, 2H), 3.10–3.20 (dd, J = 7.5, 16.5 Hz, 1H), 3.25–3.35 (m, 1H), 3.45–3.50 (m, 1H), 3.55–3.60 (m, 1H), 3.61–3.70 (m, 1H), 4.57–4.60 (d, J = 10.5 Hz, 1H), 7.00–7.60 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 16.6, 25.1, 28.6, 32.9, 113.0, 113.2, 113.4, 113.6, 122.4, 129.9, 130.0, 130.1, 143.77, 143.83, 162.2, 164.1; LC-MS (M + H+) calcd for C37H48N3O8 662, found 662.
5.25. (3R,4R/3S,4S)-tert-Butyl 3-((6-(bis(tert-butoxycarbonyl)amino)pyridin-2-yl)methyl)-4-(3-(4-fluorophenoxy)phenoxy)pyrrolidine-1-carboxylate (13c)
Compound 13c was synthesized using general procedure C (35%): 1H NMR (500 MHz, CDCl3) δ 1.40–1.60 (m, 27H), 2.80–3.00 (m, 2H), 3.10–3.20 (dd, J = 7.5, 16.5 Hz, 1H), 3.25–3.35 (m, 1H), 3.45–3.50 (m, 1H), 3.55–3.60 (m, 1H), 3.61–3.70 (m, 1H), 4.57–4.60 (d, J = 10.5 Hz, 1H), 7.00–7.60 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 16.6, 25.1, 28.6, 32.9, 113.0, 113.2, 113.4, 113.6, 122.4, 129.9, 130.0, 130.1, 143.77, 143.83, 162.2, 164.1; LC-MS (M + H+) calcd for C37H48N3O8 662, found 662.
5.26. (3R,4R/3S,4S)-tert-Butyl 3-((6-(bis(tert-butoxycarbonyl)amino)pyridin-2-yl)methyl)-4-(3-(4-chlorophenoxy)phenoxy)pyrrolidine-1-carboxylate (13d)
Compound 13d was synthesized using general procedure C (46%): 1H NMR (500 MHz, CDCl3) δ 1.40–1.60 (m, 27H), 2.80–3.00 (m, 2H), 3.10–3.20 (dd, J = 7.5, 16.5 Hz, 1H), 3.25–3.35 (m, 1H), 3.45–3.50 (m, 1H), 3.55–3.60 (m, 1H), 3.61–3.70 (m, 1H), 4.57–4.60 (d, J = 10.5 Hz, 1H), 7.00–7.60 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 16.6, 25.1, 28.6, 32.9, 113.0, 113.2, 113.4, 113.6, 122.4, 129.9, 130.0, 130.1, 143.77, 143.83, 162.2, 164.1; LC-MS (M + H+) calcd for C37H48N3O8 662, found 662.
5.27. (3R,4R/3S,4S)-tert-Butyl 3-((6-(bis(tert-butoxycarbonyl)amino)pyridin-2-yl)methyl)-4-(3-(4-chloro-3-fluorophenoxy)phenoxy)pyrrolidine-1-carboxylate (13e)
Compound 13e was synthesized using general procedure C (48%): 1H NMR (500 MHz, CDCl3) δ 1.40–1.60 (m, 27H), 2.80–3.00 (m, 2H), 3.10–3.20 (dd, J = 7.5, 16.5 Hz, 1H), 3.25–3.35 (m, 1H), 3.45–3.50 (m, 1H), 3.55–3.60 (m, 1H), 3.61–3.70 (m, 1H), 4.57–4.60 (d, J = 10.5 Hz, 1H), 7.00–7.60 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 16.6, 25.1, 28.6, 32.9, 113.0, 113.2, 113.4, 113.6, 122.4, 129.9, 130.0, 130.1, 143.77, 143.83, 162.2, 164.1; LC-MS (M + H+) calcd for C37H48N3O8 662, found 662.
5.28. (3R,4R/3S,4S)-tert-Butyl 3-((6-(bis(tert-butoxycarbonyl)amino)pyridin-2-yl)methyl)-4-(3-(4-fluoro-3-methylphenoxy)phenoxy)pyrrolidine-1-carboxylate (13f)
Compound 13f was synthesized using general procedure C (51%): 1H NMR (500 MHz, CDCl3) δ 1.40–1.60 (m, 27H), 2.80–3.00 (m, 2H), 3.10–3.20 (dd, J = 7.5, 16.5 Hz, 1H), 3.25–3.35 (m, 1H), 3.45–3.50 (m, 1H), 3.55–3.60 (m, 1H), 3.61–3.70 (m, 1H), 4.57–4.60 (d, J = 10.5 Hz, 1H), 7.00–7.60 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 16.6, 25.1, 28.6, 32.9, 113.0, 113.2, 113.4, 113.6, 122.4, 129.9, 130.0, 130.1, 143.77, 143.83, 162.2, 164.1; LC-MS (M + H+) calcd for C37H48N3O8 662, found 662.
5.29. 6-(((3R,4R/3S,4S)-4-(Biphenyl-4-yloxy)pyrrolidin-3-yl)methyl)pyridin-2-amine (2a)
Compound 2a was synthesized using general procedure D (95%): 1H NMR (500 MHz, CD3OD) δ 3.00–3.20 (m, 2H), 3.21–3.30 (dd, J = 9.0, 14.0 Hz, 1H), 3.61–3.72 (m, 3H), 5.01 (s, 1H), 6.69–6.71 (d, J = 7.5 Hz, 1H), 6.81–6.83 (d, J = 9.0 Hz, 1H), 7.05–7.07 (d, J = 7.5 Hz, 2H), 7.29–7.32 (dd, J = 7.0, 7.5 Hz, 1H), 7.40–7.43 (dd, J = 7.5, 8.0 Hz, 2H), 7.55–7.59 (m, 3H), 7.73–7.77 (dd, J = 7.5, 9.0 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 21.3, 41.5, 47.7, 50.4, 75.6, 99.9, 116.2, 119.3, 122.0, 127.0, 129.0, 134.6, 138.6, 140.9, 151.5, 152.1, 154.6, 154.8, 157.0, 158.3; LC-MS (M + H+) calcd for C22H24N3O 346.1919, found 346.1911.
5.30. 6-(((3R,4R/3S,4S)-4-(3-Phenoxyphenoxy)pyrrolidin-3-yl)methyl)pyridin-2-amine (2b)
Compound 2b was synthesized using general procedure D (91%): 1H NMR (500 MHz, D2O) δ 2.70–3.00 (m, 3H), 3.14–3.19 (dd, J = 11.0, 11.0 Hz, 1H), 3.25–3.40 (m, 2H), 3.45–3.55 (dd, J = 7.5, 11.5 Hz, 1H), 6.29 (s, 1H), 6.41–6.50 (m, 2H), 6.51–6.55 (dd, J = 1.5, 8.0 Hz, 1H), 6.56–6.60 (d, J = 8.5 Hz, 1H), 6.79–6.81 (d, J = 8.0 Hz, 2H), 6.98–7.01 (dd, J = 7.0, 7.0 Hz, 1H), 7.06–7.10 (dd, J = 8.0, 8.0 Hz, 1H), 7.17–7.21 (d, J = 8.0 Hz, 2H), 7.45–7.55 (dd, J = 7.5, 8.5 Hz, 1H); 13C NMR (125 MHz, D2O) δ 21.3, 41.5, 47.7, 50.4, 75.6, 106.1, 110.9, 111.5, 112.4, 119.4, 124.4, 130.3, 131.1, 144.7, 146.7, 154.2, 156.2, 157.5, 158.4; LC-MS (M + H+) calcd for C22H24N3O2 362.18685, found 362.18597.
5.31. 6-(((3R,4R/3S,4S)-4-(3-(4-Fluorophenoxy)phenoxy)pyrrolidin-3-yl)methyl)pyridin-2-amine (2c)
Compound 2c was synthesized using general procedure D (95%): 1H NMR (500 MHz, CD3OD) δ 2.99–3.08 (m, 2H), 3.09–3.16 (dd, J = 8.5, 14.5 Hz, 1H), 3.30–3.35 (m, 1H), 3.50–3.52 (m, 2H), 3.60–3.72 (dd, J = 8.0, 11.5 Hz, 1H), 4.94–4.95 (t, J = 2.0 Hz, 1H), 6.56 (s, 1H), 6.64–6.66 (d, J = 7.0 Hz, 1H), 6.67–6.70 (m, 1H), 6.80–6.82 (d, J = 8.5 Hz, 1H), 6.98–7.00 (m, 2H), 7.07–7.11 (m, 2H), 7.23–7.27 (dd, J = 8.0, 8.5 Hz, 1H), 7.72–7.74 (dd, J = 7.0, 8.0 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 28.5, 30.5, 43.4, 51.5, 77.2, 107.2, 111.0, 112.7, 112.8, 112.9, 117.4, 117.5, 122.15, 122.22, 132.0, 145.5, 148.7, 153.91, 153.93, 156.8, 159.0, 159.6, 160.8, 161.5, 162.4, 162.6; LC-MS (M + H+) calcd for C22H23FN3O2 380.1774, found 380.1777.
5.32. 6-(((3R,4R/3S,4S)-4-(3-(4-Chlorophenoxy)phenoxy)pyrrolidin-3-yl)methyl)pyridin-2-amine (2d)
Compound 2d was synthesized using general procedure D (95%): 1H NMR (500 MHz, CD3OD) δ 2.99–3.16 (m, 3H), 3.30–3.35 (m, 1H), 3.50–3.52 (m, 2H), 3.60–3.72 (m, 1H), 4.96 (s, 1H), 6.60–6.95 (m, 6H), 6.96–6.97 (d, J = 2.5 Hz, 2H), 6.97–6.98 (d, J = 2.5 Hz, 2H), 7.27–7.29 (m, 1H), 7.73–7.76 (dd, J = 7.5, 9.0 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 27.7, 30.5, 43.4, 51.5, 77.2, 107.8, 111.6, 112.7, 112.9, 113.0, 116.5, 118.8, 121.6, 130.9, 132.1, 145.5, 145.6, 147.9, 148.7, 156.8, 156.9, 157.0, 159.0, 159.9, 162.3, 162.6; LC-MS (M + H+) calcd for C22H23ClN3O2 396.1479, found 396.1465.
5.33. 6-(((3R,4R/3S,4S)-4-(3-(4-Chloro-3-fluorophenoxy)phenoxy)pyrrolidin-3-yl)methyl)pyridin-2-amine (2e)
Compound 2e was synthesized using general procedure D (92%): 1H NMR (500 MHz, CD3OD) δ 3.00–3.20 (m, 3H), 3.30–3.35 (m, 1H), 3.50–3.75 (m, 3H), 5.02 (s, 1H), 6.64–6.75 (m, 3H), 6.80–6.90 (m, 3H), 6.88–6.91 (m, 2H), 7.34–7.37 (dd, J = 8.0, 8.5 Hz, 1H), 7.75–7.79 (dd, J = 7.0, 8.0 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 28.5, 30.5, 43.4, 51.5, 77.2, 108.3, 108.4, 108.5, 112.4, 112.7, 112.8, 113.0, 114.2, 116.0, 116.1, 116.4, 116.7, 119.0, 132.3, 132.4, 145.5, 145.6, 147.9, 148.7, 156.8, 156.9, 158.5, 158.6, 158.7, 158.9, 159.1, 160.8, 162.6, 162.9; LC-MS (M + H+) calcd for C22H22ClFN3O2 414.1385, found 414.1397.
5.34. 6-(((3R,4R/3S,4S)-4-(3-(4-Fluoro-3-methylphenoxy)phenoxy)pyrrolidin-3-yl)methyl)pyridin-2-amine (2f)
Compound 2f was synthesized using general procedure D (95%): 1H NMR (500 MHz, CD3OD) δ 2.24 (s, 3H), 2.95–3.18 (m, 3H), 3.30–3.35 (m, 1H), 3.50–3.75 (m, 3H), 4.95–4.96 (t, J = 2.0 Hz, 1H), 6.56–6.58 (m, 1H), 6.67–6.70 (m, 1H), 6.80–6.85 (m, 3H), 6.86–6.90 (m, 3H), 7.01–7.07 (m, 1H), 7.72–7.74 (dd, J = 7.0, 8.0 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 14.6, 27.7, 30.5, 43.4, 51.5, 77.1, 107.1, 110.8, 111.8, 112.7, 112.8, 113.0, 116.6, 116.8, 117.0, 118.9, 119.4, 119.5, 123.5, 127.5, 127.6, 131.9, 145.5, 147.9, 148.7, 153.5, 156.8, 156.9, 158.1, 158.9, 161.0, 162.4, 162.6, 162.9; LC-MS (M + H+) calcd for C23H25FN3O2 394.1931, found 394.1920.
5.35. (±)-tert-Butyl -{{6-{tert-butoxycarbonyl{[2-(trimethylsilyl)ethoxy]methyl}amino}-4-methylpyridine-2-yl}methyl}-4-hydroxypyrrolidine-1-carboxylate (16)
To an ice-cooled solution of 1512 (1.328 g, 3.26 mmol) in DMF (10 mL) was added sodium hydride (0.157 g, 60% in mineral oil, 3.92 mmol). After 15 min, SEM-Cl (0.59 g, 0.64 mL, 3.59 mmol) was added dropwise. The mixture was stirred at room temperature for 16 h, and then quenched with. H2O (5 mL). The solvent was removed by rotary evaporation, and the resulting material was partitioned between EtOAc (30 mL) and H2O (20 mL). The organic layer was extracted with EtOAc (2 × 30 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, EtOAc/hexanes, 2:3) to yield a white foamy solid (1.25 g, 68%): 1H NMR (400 MHz, CDCl3) δ 0.0 (s, 9H), 0.93 (t, J = 8.0 Hz, 2H), 1.47 (s, 9H), 1.52 (s, 9H), 2.35 (s, 3H), 2.38–2.58 (m, 1H), 2.78–2.98 (m, 2H), 3.01–3.30 (m, 2H), 3.63 (t, J = 8.0 Hz, 2H), 3.64–3.90 (m, 2H), 4.10–4.25 (m, 1H), 4.85+5.12 (brs, OH), 5.29 (s, 2H), 6.82 and 6.84 (s, 1H), 7.17 (s, 1H); LC-TOF (M + H+) calcd for C27H47N3O6Si 537.3234, found 537.3244.
5.36. (3S,4S)-tert-Butyl 3-((6-(tert-butoxycarbonyl((2-(trimethylsilyl)ethoxy)methyl)amino) -4-methylpyridin-2-yl)methyl)-4-((4S)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carbonyloxy)pyrrolidine-1-carboxylate (17a) and (3R,4R)-tert-butyl 3-((6-(tert-butoxycarbonyl((2-(trimethylsilyl)ethoxy)methyl)amino)-4-methylpyridin-2-yl)methyl)-4-((4S)-4,7,7-trimethyl-3-oxo-2-oxabicyclo[2.2.1]heptane-1-carbonyloxy)pyrrolidine-1-carboxylate (17b)
To an ice-cooled solution of 16 (0.935 g, 1.74 mmol), (1S)-(-)-camphanic acid (0.4 g, 2.02 mmol) and Ph3P (0.75 g, 2.86 mmol) in THF (10 mL) was added DEAD (0.9 mL, 3.0 mmol) dropwise. The mixture was stirred at room temperature for 6 h, and then concentrated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc/hexanes, 1:5) to obtain the two diastereomers in 88% overall yield.
17a (Foamy solid, 44%; 0.55 g): 1H NMR (400 MHz, CDCl3) δ 0 (s, 9H), 0.92 (t, J = 8.0 Hz, 2H), 0.97+0.99 (s, 3H), 1.07+1.09 (s, 3H), 1.16 (s, 3H), 1.25–1.4 (m, 1H), 1.46 (s, 9H), 1.54 (s, 9H), 1.65–1.80 (m, 1H), 1.90–2.10 (m, 2H), 2.326+2.336 (s, 3H), 2.40–2.55 (m, 1H), 2.75–3.05 (m, 2H), 3.18–3.30 (m, 1H), 3.50–3.70 (m, 3H), 4.10–4.44 (m, 2H), 5.36 (s, 2H), 6.79 (s, 1H), 7.18 (s, 1H); LC-MS (M + H+) calcd for C37H59N3O9Si 718, found 718.
17b (Foamy solid, 0.55 g, 44%): 1H NMR (400 MHz, CDCl3) δ 0 (s, 9H), 0.92 (t, J = 8.8 Hz, 2H), 0.99+1.01 (s, 3H), 1.08 (s, 3H), 1.34+1.60 (s, 3H), 1.25–1.40 (m, 1H), 1.70–1.80 (m, 1H), 1.90–2.04 (m, 1H), 2.06–2.19 (m, 1H), 2.33 (s, 3H), 2.39–2.54 (m, 1H), 2.70–3.04 (m, 2H), 3.25 (t, J =10.4 Hz, 1H), 3.50–3.70 (m, 3H), 4.22–4.42 (m, 3H), 5.36 (s, 1H), 6.74 (s, 1H), 7.17 (s, 1H); LC-MS (M + H+) calcd for C37H59N3O9Si 718, found 718.
5.37. (3R,4R)-tert-Butyl-3-((6-(tert-butoxycarbonyl((2-(trimethylsilyl)ethoxy)methyl) amino)-4-methylpyridin-2-yl)methyl)-4-hydroxypyrrolidine-1-carboxylate (14b)
To a solution of 17b (397 mg, 0.55 mmol) in methanol (5 mL) and water (0.5 mL) was added Na2CO3 (0. 11 g, 1 mmol). The mixture was stirred at room temperature overnight, and concentrated. The resulting residue was partitioned between EtOAc (20 mL) and H2O (20 mL). The organic layer was dried over Na2SO4, and concentrated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc/hexanes, 2:3) to yield compound 17b as a foamy solid (0.291 g, 98%): 1H NMR (400 MHz, CDCl3) δ 0 (s, 9H), 0.95 (t, J = 8.0 Hz, 2H), 1.46 (s, 9H), 2.35 (s, 3H), 2.80 (t, J = 12.0 Hz, 1H), 2.96 (t, 1H), 3.22 (t, 1H), 3.40–3.70 (m, 4H), 4.05–4.25 (m, 2H), 4.53+4.63 (br s, OH), 5.25 (s, 2H), 6.85 (s, 1H), 7.21 (s, 1H). 13C NMR (100 MHz, CDCl3): 17.7, 20.8, 28.0, 28.3, 34.8, 44.3, 45.0, 48.8, 49.2, 53.3, 53.6, 61.6, 65.6, 69.8, 70.6, 76.6, 78.8, 81.4, 118.8, 120.8, 120.9, 149.5, 149.6, 153.1, 153.7, 154.2, 157.8; ESI-MS: calcd for C27H47N3O6Si 537.3234, found 537.32437.
5.38. (3′-Chlorobiphenyl-4-yl)methanol (18a)
Compound 18a was synthesized using general procedure E (87%): 1H NMR (400 MHz, CDCl3) δ 4.75 (s, 2H), 7.30–7.34 (m, 1H), 7.37 (t, J = 8.0 Hz, 1H), 7.42–7.48 (m, 3H), 7.54–7.58 (m, 3H).
5.39. (3′-Fluorobiphenyl-4-yl)methanol (18b)
Compound 18b was synthesized using general procedure E (87%): 1H NMR (400 MHz, CDCl3) δ 4.75 (s, 2H), 7.00–7.08 (m, 1H), 7.22–7.32 (m, overlapped by CDCl3, 1H), 7.34–7.42 (m, 2H), 7.45 (d, J = 8.0 Hz, 2H), 7.57 (d, J = 8.0 Hz, 2H).
5.40. (3′-(Trifluoromethyl)biphenyl-4-yl)methanol (18c)
Compound 18c was synthesized using general procedure E (87%): 1 H NMR (400 MHz, CDCl3) δ 4.75 (s, 2H), 7.46 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 7.2 Hz, 1H), 7.59 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 7.2 Hz, 1H), 7.83 (s,1H).
5.41. (3′-Chloro-4′-fluorobiphenyl-4-yl)methanol (18d)
Compound 18d was synthesized using general procedure E (87%): 1H NMR (400 MHz, CDCl3) δ 4.75 (s, 2H), 7.20 (t, J = 8.8 Hz, 1H), 7.40–7.47 (m, 3H), 7.49–7.55 (m, 2H), 7.61 (dd, J = 7.8, 2.4 Hz, 1H).
5.42. (4′-Fluoro-3′-methylbiphenyl-4-yl)methanol (18e)
Compound 18e was synthesized using general procedure E (87%): 1H NMR (400 MHz, CDCl3) δ 2.33 (s, 3H), 4.73 (s, 2H), 7.06 (t, J = 9.2 Hz, 1H), 7.25–7.54 (m, 6H).
5.43. 4′-(Bromomethyl)-3-chlorobiphenyl (19a)
Compound 19a was synthesized using general procedure F (87%): 1H NMR (400 MHz, CDCl3) δ 4.54 (s, 2H), 7.29–7.33 (m,1H), 7.35 (t, J = 8.0 Hz, 1H), 7.41–7.43 (m, 1H), 7.45 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 8.0 Hz, 2H), 7.53–7.55 (m, 1H).
5.44. 4′-(Bromomethyl)-3-fluorobiphenyl (19b)
Compound 19b was synthesized using general procedure F (87%): 1H NMR (400 MHz, CDCl3) δ 4.55 (s, 2H), 7.0–7.10 (m, 1H), 7.22–7.3 (m, 1H, overlapped by CDCl3), 7.35–7.44 (m, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.55 (d, J = 8.0 Hz, 2H).
5.45. 4′-(Bromomethyl)-3-(trifluoromethyl)biphenyl (19c)
Compound 19c was synthesized using general procedure F (87%): 1H NMR (400 MHz, CDCl3) δ 4.55 (s, 2H), 7.50 (d, J = 7.6 Hz, 2H), 7.57 (d, J = 7.6 Hz, 2H), 7.62 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.82 (s, 1H).
5.46. 4′-(Bromomethyl)-3-chloro-4-fluorobiphenyl (19d)
Compound 19d was synthesized using general procedure F (87%): 1H NMR (400 MHz, CDCl3) δ 4.52 (s, 2H), 7.18 (t, J = 8.8 Hz, 1H), 7.38–7.47 (m, 5H), 7.57 (dd, J = 8.8, 2.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): 33.0, 116.8 (d, JC–F = 84 Hz), 121.2, 126.6 (d, JC–F = 29.2 Hz), 127.3, 129.1, 129.6, 137.3, 137.5 (d, JC–F = 12 Hz), 138.9, 156.4, 158.9; 19F NMR (376 MHz, CDCl3): −119.5.
5.47. 4′-(Bromomethyl)-4-fluoro-3-methylbiphenyl (19e)
Compound 19e was synthesized using general procedure F (87%): 1H NMR (400 MHz, CDCl3) δ 2.32 (s, 3H), 4.53 (s, 2H), 7.05 (t, J = 8.8 Hz, 1H), 7.30–7.40 (m, 2H), 7.44 (d, J = 8.8 Hz, 2H), 7.49 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): 314.5, 32.2, 115.2 (d, JC–F =90.8 Hz), 125.0 (d, JC–F =69.2 Hz), 125.8 (d, JC–F =29.2 Hz), 127.2, 129.4, 130.0 (d, JC–F =18.4 Hz), 136.0 (d, JC–F =14.4 Hz), 136.5, 140.3, 159.8, 162.3; 19F NMR (376 MHz, CDCl3): −117.8.
5.48. (3S,4S)-tert-Butyl 3-((6-(tert-butoxycarbonyl((2-(trimethylsilyl)ethoxy)methyl)amino)-4-methylpyridin-2-yl)methyl)-4-((3′-chlorobiphenyl-4-yl)methoxy)pyrrolidine-1-carboxylate (13g)
Compound 13g was synthesized using general procedure G (87%): 1H NMR (400 MHz, D2O) δ 0 (s, 9H), 0.94 (t, J = 8.0 Hz, 2H), 1.49 (s, 9H), 1.53 (s, 9H), 2.31+2.32 (s, 3H), 2.66–2.82 (m, 1H), 2.83–2.94 (m, 1H), 3.08 (dd, J = 14.0, 6.8 Hz, 1H), 3.22–3.33 (m, 1H), 3.35–3.42 (m, 1H), 3.48–3.60 (m, 1H), 3.63 (t, J = 8.0 Hz, 2H), 3.78 (d, J = 12.0 Hz, 1H), 3.97–4.03 (m, 1H), 4.38–4.48 (m, 1H), 4.68 (t, J = 11.6 Hz, 1H), 5.37 (s, 2H), 6.79 (s, 1H), 7.16 (d, J = 9.6 Hz, 1H), 7.33–7.46 (m, 4H), 7.50 (d, J = 7.2 Hz, 1H), 7.55–7.63 (m, 3H); 13C NMR (100 MHz, CDCl3) δ −1.48, 18.0, 21.0, 28.2, 28.5, 28.9, 34.9, 42.6, 43.3, 49.0, 49.3, 50.2, 50.8, 65.7, 70.6+70.7, 76.5, 78.0, 79.0, 79.16+79.21, 81.3, 119.2, 121.3, 125.2, 127.0, 127.1, 127.2, 127.9, 128.2, 130.0, 134.6, 137.8, 138.0, 139.1, 142.7, 148.8, 153.4, 154.1, 154.5, 154.8, 158.4; ESI-MS: m/z = 738 [M+H]+; HR-ESI-MS: 737.3627 calcd for C40H56ClN3O6Si, found: 737.3634.
5.49. (3S,4S)-tert-Butyl 3-((6-(tert-butoxycarbonyl((2-(trimethylsilyl)ethoxy)methyl)amino)-4-methylpyridin-2-yl)methyl)-4-((3′-fluorobiphenyl-4-yl)methoxy)pyrrolidine-1-carboxylate (13h)
Compound 13h was synthesized using general procedure G (87%): 1H NMR (400 MHz, D2O) δ 0 (s, 9H), 0.92 (t, J = 8.0 Hz, 2H), 1.49 (s, 9H), 1.54 (s, 9H), 2.31+2.33 (s, 3H), 2.68–2.83 (m, 1H), 2.84–2.93 (m, 1H), 3.04–3.13 (m, 1H), 3.22–3.34 (m, 1H), 3.35–3.43 (m,1H), 3.48–3.60 (m,1H), 3.63 (t, J = 8.0 Hz, 2H), 3.78 (d, J = 12.0 Hz, 1H), 3.96–4.04 (m, 1H), 4.38–4.49 (m, 1H), 3.68 (t, J = 11.2 Hz, 1H), 5.37 (s, 2H), 6.80 (s, 1H), 7.04–7.12 (m, 1H), 7.16 (d, J = 8.8 Hz, 1H), 7.32–7.35 (m, 1H), 7.38–7.50 (m, 4H), 7.55–7.62 (m, 2H); 13C NMR (100 MHz, CDCl3) δ −1.46, 18.0, 21.0, 28.2, 28.5, 29.7, 34.9+35.0, 42.6, 43.3, 49.0, 49.3, 50.2, 50.8, 64.8, 65.7, 70.6+70.7, 76.5, 78.0, 78.9, 79.17+79.22, 81.23+81.28, 113.8, 113.9, 114.0, 114.1, 119.2, 121.3, 122.6+122.7, 127.0, 127.2, 127.4, 127.9, 128.2, 130.16+130.24, 137.9, 138.0, 139.2, 143.0, 148.8, 153.4, 154.2, 154.5, 154.8, 158.4, 161.9, 164.4; ESI-MS: m/z = 700 [M+H]+; HR-ESI-MS: 699.4119 calcd for C41H57F3N3O6Si, found: 699.4113.
5.50. (3S,4S)-tert-Butyl 3-((6-(tert-butoxycarbonyl((2-(trimethylsilyl)ethoxy)methyl)amino)-4-methylpyridin-2-yl)methyl)-4-((3′-(trifluoromethyl)biphenyl-4-yl)methoxy)pyrrolidine-1-carboxylate (13i)
Compound 13i was synthesized using general procedure G (87%): 1H NMR (400 MHz, D2O) δ 0 (s, 9H), 0.92 (t, J = 8.0 Hz, 2H), 1.49 (s, 9H), 1.54 (s, 9H), 2.31+2.33 (s, 3H), 2.65–2.83 (m, 1H), 2.84–2.93 (m, 1H), 3.02–3.13 (m, 1H), 3.22–3.34 (m, 1H), 3.35–3.43 (m, 1H), 3.47–3.60 (m, 1H), 3.63 (t, J = 8.0 Hz, 2H), 3.79 (d, J = 12.8 Hz, 1H), 4.00 (s, 1H), 4.40–4.50 (m, 1H), 4.69 (t, J = 10.8 Hz, 1H), 5.37 (s, 2H), 6.80 (s, 1H), 7.16 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 7.2 Hz, 2H), 7.55–7.67 (m, 4H), 7.80 (d, J = 7.2 Hz, 1H), 7.86 (s, 1H); 13C NMR (100 MHz, CDCl3) δ −1.47, 18.0, 21.0, 28.2, 28.5, 29.7, 34.85+34.95, 42.6, 43.3, 49.0, 49.3, 50.2, 50.8, 65.7, 70.6+70.7, 76.5, 78.1, 79.0, 79.16+79.23, 81.23+81.27, 119.2, 121.3, 123.8+123.9, 127.1, 128.0, 128.3, 129.2, 130.3, 138.1, 138.2, 139.0, 141.6, 148.8, 153.4, 154.2, 154.8, 158.4; 19F NMR (100 MHz, CDCl3): −63.0; ESI-MS: m/z = 772 [M+H]+; HR-ESI-MS: 771.3891 calcd for C41H57F3N3O6Si, found: 771.3898.
5.51. (3S,4S)-tert-Butyl 3-((6-(tert-butoxycarbonyl((2-(trimethylsilyl)ethoxy)methyl)amino)-4-methylpyridin-2-yl)methyl)-4-((4′-fluoro-3′-methylbiphenyl-4-yl)methoxy)pyrrolidine-1-carboxylate (13j)
Compound 13j was synthesized using general procedure G (87%): 1H NMR (400 MHz, D2O) δ 0 (s, 9H), 0.92 (t, J =8.0 Hz, 2H), 1.49 (s, 9H), 1.54 (s, 9H), (2.31+2.33) (s, 3H), 2.37 (s, 3H), 2.67–2.83 (m, 1H), 2.84–2.94 (m,1H), 3.08 (dd, J = 14.0, 6.8 Hz, 1H), 3.22–3.34 (m, 1H), 3.35–3.42 (m, 1H), 3.48–3.59 (m, 1H), 3.63 (t, J = 8.0 Hz, 2H), 3.78 (d, J = 12.0 Hz, 1H), 3.96–4.04 (m, 1H), 4.38–4.46 (m, 1H), 4.67 (t, J = 11.2, 10.8 Hz, 1H), 5.37 (s, 2H), 6.79 (s, 1H), 7.10 (t, J = 8.8 Hz, 1H), 7.16 (d, t, J = 9.2 Hz, 1H), 7.36–7.45 (m, 4H), 7.51–7.58 (m, 2H); 13C NMR (100 MHz, CDCl3) δ −1.46, 14.669+14.696, 18.0, 20.983+20.965, 28.2, 28.5, 29.7, 34.9+35.0, 42.6, 43.3, 49.0, 49.3, 50.2, 50.8, 65.7, 70.7+70.8, 76.5, 77.9, 78.9, 79.1, 79.2, 81.22+81.26, 115.1, 115.3, 119.2, 121.3, 125.80+125.88, 126.9, 127.9, 128.1, 130.09+130.14, 136.6, 137.0, 137.1, 139.8, 148.8, 153.4, 154.2, 154.5, 154.8, 158.4, 159.8, 162.2; ESI-MS: m/z = 736 [M+H]+; HR-ESI-MS: 735.4079 calcd for C24H26ClN3OS, found: 735.4082.
5.52. (3S,4S)-tert-Butyl 3-((6-(tert-butoxycarbonyl((2-(trimethylsilyl)ethoxy)methyl)amino)-4-methylpyridin-2-yl)methyl)-4-((3′-chloro-4′-fluorobiphenyl-4-yl)methoxy)pyrrolidine-1-carboxylate (13k)
Compound 13k was synthesized using general procedure G (87%):: 1H NMR (400 MHz, D2O) δ 0 (s, 9H), 0.92 (t, J =8.0 Hz, 2H), 1.49 (s, 9H), 1.54 (s, 9H), 2.31+2.33 (s, 3H), 2.65–2.82 (m,1H), 2.83–2.94 (m, 1H), 3.08 (dd, J =14.0, 7.2 Hz, 1H), 3.21–3.34 (m, 1H), 3.34–3.43 (m, 1H), 3.48–3.60 (m, 1H), 3.63 (t, J = 8.0 Hz, 2H), 3.78 (d, J = 12.0 Hz, 1H), 3.96–4.05 (m, 1H), 4.38–4.49 (m, 1H), 4.67 (t, J = 12.0, 11.6, 9.2 Hz, 1H), 5.38 (s, 2H), 6.79 (s, 1H), 7.16 (d, J = 9.2 Hz, 1H), 7.24 (t, J = 8.8 Hz, 1H), 7.40–7.49 (m, 3H), 7.50–7.56 (m, 2H), 7.64 (dd, J = 6.8, 2.1 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ −1.47, 17.9, 21.0, 28.2, 28.5, 29.6, 34.9, 42.6, 43.3, 48.9, 49.3, 50.2, 50.8, 65.7, 70.5+70.6, 76.5, 78.0, 79.0, 79.1+79.2, 81.3, 116.7, 116.9, 119.2, 121.2, 126.6, 126.7, 126.9, 128.0, 128.2, 129.1, 137.8, 138.0, 138.1, 138.3, 148.8, 153.4, 154.1, 154.8, 156.3, 158.4, 158.8; 19F NMR (100 MHz, CDCl3): −120.4; ESI-MS: m/z = 756 [M+H]+; HR-ESI-MS: 755.3533 calcd for C24H26ClN3OS, found: 755.3540.
5.53. 6-(((3S,4S)-4-((3′-Chlorobiphenyl-4-yl)methoxy)pyrrolidin-3-yl)methyl)-4-methylpyridin-2-amine hydrochloride salt. (2g)
Compound 2g was synthesized using general procedure H (100%): 1H NMR (400 MHz, D2O) δ 1.80 (s, 3H), 2.50–2.72 (m, 3H), 3.04–3.10 (m, 1H), 2.32–2.34 (m, 1H), 3.48–3.59 (m, 1H), 3.71 (d, J = 12.8 Hz, 1H), 4.01 (s, 1H), 4.15 (d, J = 12 Hz, 1H), 4.62 (d, J = 12 Hz, 1H), 5.86 (s, 1H), 6.24 (s, 1H), 7.09 (d, J = 7.2 Hz, 1H), 7.17–7.35 (m, 7H); 13C NMR (100 MHz, D2O) δ 21.1, 28.4, 40.7, 47.4, 49.8, 70.1, 75.5, 110.0, 113.3, 124.9, 126.2, 126.4, 127.4, 130.5, 134.2, 136.8, 138.2, 141.4, 145.4, 153.4, 157.1; ESI-MS: m/z = 408 [M+H]+; HR-ESI-MS: 407.1764 calcd for C24H26ClN3OS, found: 407.1773.
5.54. 6-(((3S,4S)-4-((3′-Fluorobiphenyl-4-yl)methoxy)pyrrolidin-3-yl)methyl)-4-methylpyridin-2-amine hydrochloride salt (2h)
Compound 2h was synthesized using general procedure H (100%): 1H NMR (400 MHz, D2O) δ 1.86 (s, 3H), 2.55–2.80 (m, 3H), 3.04–3.15 (m, 1H), 3.30–3.40 (m, 1H), 3.50–3.61 (m, 1H), 3.68–3.75 (m, 1H), 4.06 (s, 1H), 4.17 (d, J = 12 Hz, 1H), 4.62 (d, J = 12 Hz, 1H), 6.97 (s, 1H), 6.20 (s, 1H), 6.95 (s, 1H), 7.11–7.44 (m, 7H); 13C NMR (100 MHz, D2O) δ 21.0, 28.1, 40.0, 47.5, 49.9, 70.0, 75.0, 109.8, 113.0, 114.2, 122.3, 126.4, 129.6, 130.8, 136.6, 138.4, 141.8, 145.5, 153.4, 157.2, 161.8, 164.3; 19F NMR (100 MHz, D2O): −113.7; ESI-MS: m/z = 392 [M+H]+; HR-ESI-MS: 391.2071 calcd for C24H26FN3OS, found: 391.2072.
5.55. 4-Methyl-6-(((3S,4S)-4-((3′-(trifluoromethyl)biphenyl-4-yl)methoxy)pyrrolidin-3-yl)methyl)pyridin-2-amine hydrochloride salt (2i)
Compound 2i was synthesized using general procedure H (100%): 1H NMR (400 MHz, D2O) δ 1.67 (s, 3H), 2.42–2.68 (m, 3H), 2.94–3.08 (m, 1H), 3.24–3.32 (m, 1H), 3.42–3.54 (m, 1H), 3.69 (d, J = 12.4 Hz, 1H), 3.91 (s, 1H), 4.07 (d, J = 11.2 Hz, 1H), 4.57 (d, J = 11.2 Hz, 1H), 5.70 (s, 1H), 6.23 (s, 1H), 7.08–7.28 (m, 6H), 7.37–7.50 (m, 2H); 13C NMR (100 MHz, D2O) δ 20.8, 28.7, 41.4, 47.3, 49.5, 70.1, 76.2, 110.1, 113.5, 122.6, 122.8, 123.7, 126.4, 129.1, 129.5, 129.9, 130.3, 130.6, 137.3, 137.9, 140.4, 145.6, 153.6, 156.8; 19F NMR (100 MHz, D2O): −63.2; ESI-MS: m/z = 442 [M+H]+; HR-ESI-MS: 441.2028 calcd for C25H26F3N3O, found: 441.2032.
5.56. 6-(((3S,4S)-4-((4′-Fluoro-3′-methylbiphenyl-4-yl)methoxy)pyrrolidin-3-yl)methyl)-4-methylpyridin-2-amine hydrochloride salt (2j)
Compound 2j was synthesized using general procedure H (100%): 1H NMR (400 MHz, D2O) δ 1.84 (s, 3H), 2.06 (s, 3H), 2.50–2.78 (m, 3H), 3.05–3.12 (m, 1H), 3.32–3.34 (m, 1H), 3.51–3.60 (m, 1H), 3.72 (d, J = 12.8 Hz, 1H), 4.04 (s, 1H), 4.17 (d, J =12.0 Hz, 1H), 4.63 (d, J = 12.0 Hz, 1H), 5.92 (s, 1H), 6.25 (s, 1H), 6.86 (t, J = 8.8 Hz, 1H), 7.14–7.29 (m, 6H); 13C NMR (100 MHz, D2O) δ 13.9, 21.1, 28.4, 40.6, 47.4, 49.8, 70.1, 75.3, 110.0, 113.3, 115.1, 115.3, 125.0, 125.2, (125.34+125.42), 126.2, 129.4, 135.5, 135.9, 138.9, 145.5, 153.4, 157.2, 159.6, 162.1; ESI-MS: m/z = 406 [M+H]+; HR-ESI-MS: 405.2216 calcd for C25H28FN3O, found: 405.2285.
5.57. 6-(((3S,4S)-4-((3′-Chloro-4′-fluorobiphenyl-4-yl)methoxy)pyrrolidin-3-yl)methyl)-4-methylpyridin-2-amine hydrochloride salt (2k)
Compound 2k was synthesized using general procedure H (100%): 1H NMR (400 MHz, D2O) δ 1.86 (s, 3H), 2.55–2.74 (m, 3H), 3.06–3.12 (m, 1H), 3.32 (d, J = 12.8 Hz, 1H), 3.50–3.57 (m, 1H), 4.05 (s, 1H), 4.17 (d, J = 12.0 Hz, 1H), 4.62 (d, J = 12.0 Hz, 1H), 5.95 (s, 1H), 6.24 (s, 1H), 7.06 (t, J = 8.8 Hz, 1H), 7.20–7.40 (m, 6H); 13C NMR (100 MHz, D2O) δ 21.0, 28.6, 40.7, 47.4, 49.8, 70.1, 75.5, 109.8, 113.3, 116.8, 117.0, (120.5+120.7), 126.3, 126.5, 128.2, 129.4, 136.6, 137.0, 137.5, 146.1, 153.7, 156.1, 156.8, 158.5; ESI-MS: m/z = 426 [M+H]+; HR-ESI-MS: 425.1670 calcd for C24H25ClFN3O, found: 425.1674.
5.58. Enzyme assays
IC50 values for inhibitors 2a–d were measured for the three different isoforms of NOS, including rat nNOS, bovine eNOS, and murine macrophage iNOS using L-arginine as the substrate. The three isozymes were recombinant enzymes, which were overexpressed (in E. coli) and isolated as reported23. The formation of nitric oxide was measured using a hemoglobin capture assay described previously29. All NOS isozymes were assayed at room temperature in a 100 mM Hepes buffer (pH 7.4) containing 10 μM L-arginine, 1.6 mM CaCl2, 11.6 μg/mL calmodulin, 100 μM DTT, 100 μM NADPH, 6.5 μM H4B, 3.0 mM oxyhemoglobin (for iNOS assays, no Ca2+ and calmodulin was added). The assay was initiated by the addition of enzyme, and the initial rates of the enzymatic reactions were determined by monitoring the formation of NO-hemoglobin complex at 401 nm from 0 to 60 s after. The corresponding Ki values of inhibitors were calculated from the IC50 values using equation 1 with known Km values (rat nNOS, 1.3 μM; iNOS, 8.3 μM; eNOS, 1.7 μM).
| (1) |
5.59. Molecular docking
Docking simulations were carried out as described by Ji et al. with AutoDock 3.0.5.30 Polar hydrogen atoms were added to the nNOS crystal structure (PDB code: 3JWT),24 and Kollman united atom charges31 were assigned. Hydrogens were also added to the heme and H4B, and charges were calculated by the Gasteiger–Marsili method.32 The charge of the Fe atom bound to heme was assigned +3. The nonpolar hydrogen atoms of heme and H4B were removed manually, and their charges were united with the bonded carbon atoms. Atomic solvation parameters and fragmental volumes were assigned using the AddSol utility. The 3D structures of the ligands were built and partial atomic charges were also calculated using the Gasteiger–Marsili method. The rotatable bonds in the ligands were defined using AutoTors, which also unites the nonpolar hydrogens and partial atomic charges to the bonded carbon atoms. The grid maps were calculated using AutoGrid. The dimensions of the grid box was 31 × 28 × 31 Å, and the grid spacing was set to 0.375 Å. Docking was performed using the Lamarckian genetic algorithm (LGA), and the pseudo-Solis and Wets method were applied for the local search.
5.60. Crystal preparation, X-ray diffraction data collection, and structure refinement
The nNOS heme domain protein used for crystallographic studies was produced by limited trypsin digest from the corresponding full length enzyme and further purified through a Superdex 200 gel filtration column (GE Healthcare) as described previously.33, 34 The nNOS heme domain at 7–9 mg/mL containing 20mM histidine was used for the sitting drop vapor diffusion crystallization setup under the conditions reported before.33 Fresh crystals (1–2 day old) were first passed stepwise through cryo-protectant solutions described 33 and then soaked with 10 mM inhibitor for 4–6 h at 4 °C before being flash cooled by plunging into liquid nitrogen.
The cryogenic (100K) X-ray diffraction data were collected remotely at the Stanford Synchrotron Radiation Lightsource through the data collection control software Blu-Ice35 and the crystal mounting robot. Raw data frames were indexed, integrated, and scaled using HKL2000.36 The binding of inhibitors was detected by the initial difference Fourier maps calculated with REFMAC.37 The inhibitor molecules were then modeled in O(38 or COOT39 and refined using REFMAC. Water molecules were added in REFMAC and checked by COOT. The TLS40 protocol was implemented in the final stage of refinements with each subunit as one TLS group. The refined structures were validated in COOT before deposition to RCSB protein data bank with PDB accession code 1OM4. The crystallographic data collection and structure refinement statistics are summarized in Table 2.
Table 2.
Crystallographic data collection and refinement statistics
| nNOS-2b | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 52.28, 111.56, 164.85 |
| Resolution (Å) | 1.90 (1.93–1.90) |
| Rsym | 0.075 (0.572) |
| I/σI | 24.2 (2.0) |
| No. unique reflections | 75,896 |
| Completeness (%) | 99.3 (99.7) |
| Redundancy | 4.0 (4.0) |
| Refinement | |
| Resolution (Å) | 1.90 |
| No. reflections used | 71,875 |
| Rwork/Rfree* | 0.182/0.215 |
| Mean B value | 44.53 |
| No. atoms | |
| Protein | 6,671 |
| Ligand/ion | 186 |
| Water | 380 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.014 |
| Bond angles (°) | 1.350 |
The 5% of reflections were set aside for the free R calculation throughout the refinement according to the free R flags used in the refinement of the starting model (1OM4).
Acknowledgments
The authors are grateful to the National Institutes of Health for financial support to R.B.S. (GM49725), T.L.P (GM57353), and Dr. Bettie Sue Masters (GM52419, with whose laboratory P.M. and L.J.R. are affiliated). B.S.S.M. also is grateful to the Welch Foundation for a Robert A. Welch Distinguished Professorship in Chemistry (AQ0012). P.M. is supported by grants 0021620806 and 1M0520 from MSMT of the Czech Republic.
Abbreviations
- nNOS
neuronal nitric oxide synthase
- CNS
central nervous system
- BBB
blood brain barrier
- TEA
triethylamine
- TBAF
tetrabutylammonium fluoride
- TBS
t-butyldimethylsilyl
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM. Proc Natl Acad Sci USA. 1996;93:4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hantraye P, Brouillet E, Ferrante R, Palfi S, Dolan R, Matthews RT, Beal MF. Nat Med. 1996;2:1017–1021. doi: 10.1038/nm0996-1017. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Dawson VL, Dawson TM. Role of nitric oxide in Parkinson’s disease. Pharmacol Ther. 2006;109:33–41. doi: 10.1016/j.pharmthera.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Dorheim MA, Tracey WR, Pollock JS, Grammas P. Biochem Biophys Res Commun. 1994;205:659–665. doi: 10.1006/bbrc.1994.2716. [DOI] [PubMed] [Google Scholar]
- 5.Norris PJ, Waldvogel HJ, Faull RLM, Love DR, Emson PC. Neuroscience. 1996;72:1037–1047. doi: 10.1016/0306-4522(95)00596-x. [DOI] [PubMed] [Google Scholar]
- 6.Sims NR, Anderson MF. Neurochem Int. 2002;40:511–526. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 7.Southan GJ, Szabo C. Biochem Pharmacol. 1996;51:383–394. doi: 10.1016/0006-2952(95)02099-3. [DOI] [PubMed] [Google Scholar]
- 8.Babu BR, Griffith OW. Curr Opin Chem Biol. 1998;2:491–500. doi: 10.1016/s1367-5931(98)80125-7. [DOI] [PubMed] [Google Scholar]
- 9.Hobbs AJ, Higgs A, Moncada S. Annu Rev Pharmacol. 1999;39:191–220. doi: 10.1146/annurev.pharmtox.39.1.191. [DOI] [PubMed] [Google Scholar]
- 10.Silverman RB. Acc Chem Res. 2009;42:439–451. doi: 10.1021/ar800201v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tafi A, Angeli L, Venturini G, Travagli M, Corelli F, Botta M. Curr Med Chem. 2006;13:1929–1946. doi: 10.2174/092986706777585031. [DOI] [PubMed] [Google Scholar]
- 12.Lawton GR, Ranaivo HR, Chico LK, Ji H, Xue F, Martesek P, Roman LJ, Watterson DM, Silverman RB. Bioorg Med Chem. 2009;17:2371–2380. doi: 10.1016/j.bmc.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ji H, Stanton BZ, Igarashi J, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. J Am Chem Soc. 2008;130:900–3914. doi: 10.1021/ja0772041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji H, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. J Med Chem. 2009;52:779–797. doi: 10.1021/jm801220a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Begley DJ. Pharmacol Ther. 2004;104:29–45. doi: 10.1016/j.pharmthera.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 16.Scherrmann JM. Vasc Pharmacol. 2002;38:349–354. doi: 10.1016/s1537-1891(02)00202-1. [DOI] [PubMed] [Google Scholar]
- 17.Löscher W, Potschka H. NeuroRx. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linnet K, Ejsing TB. Eur Neuropsychopharmacol. 2008;18:157–169. doi: 10.1016/j.euroneuro.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Juliano RL, Ling VA. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 20.Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, van der Valk MA, Voordouw AC, Spits H, van Tellingen O, Zijlmans JM, Fibbe WE, Borst P. Proc Natl Acad Sci USA. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schinkel AH. Adv Drug Deliv Rev. 1999;36:179–194. doi: 10.1016/s0169-409x(98)00085-4. [DOI] [PubMed] [Google Scholar]
- 22.(a) Silverman RB, Lawton GR, Ralay Ranaivo H, Seo J, Watterson DM. Bioorg Med Chem. 2009;17:7593–7605. doi: 10.1016/j.bmc.2009.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xue F, Fang J, Lewis WW, Martásek P, Roman LJ, Silverman RB. Bioorg Med Chem Lett. 2010;20:554–557. doi: 10.1016/j.bmcl.2009.11.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delker SL, Ji H, Li H, Jamal J, Fang J, Xue X, Silverman RB, Poulos TL. J Am Chem Soc. 2010;132:5437–5442. doi: 10.1021/ja910228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji H, Delker SL, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. manuscript in preparation. [Google Scholar]
- 25.Oldendorf WH. Ann Rev Pharmaco. 1974;14:239–248. [Google Scholar]
- 26.Fang J, Ji H, Lawton GR, Xue F, Roman LJ, Silverman RB. J Med Chem. 2009;52:4533–4537. doi: 10.1021/jm900380j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawton GR, Ji H, Silverman RB. Tetrahedron Lett. 2006;47:6113–6115. [Google Scholar]
- 28.Lu X, Bao WA. J Org Chem. 2007;72:3863–3867. doi: 10.1021/jo070443m. [DOI] [PubMed] [Google Scholar]
- 29.Hevel JM, Marletta MA. Method Enzymol. 1994;233:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 30.Ji H, Li H, Flinspach M, Poulos TL, Silverman RB. J Med Chem. 2003;46:5700–5711. doi: 10.1021/jm030301u. [DOI] [PubMed] [Google Scholar]
- 31.Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta S, Weiner P. J Am Chem Soc. 1984;106:765–84. [Google Scholar]
- 32.Gasteiger J, Marsili M. Tetrahedron. 1980;36:3219–3222. [Google Scholar]
- 33.Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. Biochemistry. 2002;41:13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 34.Flinspach ML, Li H, Jamal J, Yang W, Huang H, Hah JM, Gomez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Nat Struct Mol Biol. 2004;11:54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- 35.McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Murshudov GN, Vagin AA, Dodson EJ. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 38.Jones TA, Zou JY, Cowan SW, Kjeldgaarrd M. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Winn MD, Isupov MN, Murshudov GN. Acta Cryst. 2001;D57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]