

Abstract
 The use of low polarity aromatic solvents (benzene or toluene) and/or the removal of inorganic salts results in dramatically improved yields of fluorinated arenes from diaryliodonium salts. This methodology is shown to “scale down” to the conditions used typically for radiotracer synthesis.
The use of low polarity aromatic solvents (benzene or toluene) and/or the removal of inorganic salts results in dramatically improved yields of fluorinated arenes from diaryliodonium salts. This methodology is shown to “scale down” to the conditions used typically for radiotracer synthesis.
The inability to functionalize electron-rich arenes with fluoride ion sources narrows the scope and increases the expense of practical 18F-labeled imaging agents for positron emission tomography.1 Two fluoride-based approaches to fluoroarenes have been heavily investigated of late: 1) transition-metal-catalyzed fluorination,2-6 and 2) elimination of aryl fluorides from diaryliodonium salts.7,8 Stang and coworkers have stressed the similarities of iodonium ion and late transition metal ion chemistries,9 thus we thought that the use of low polarity, non-coordinating media, which often suppresses ionic (disproportionation, ligand exchange, electron transfer) side reactions of transition metal complexes,10 might be a strategy to improve diaryliodonium fluoride chemistry. Here we show that the use of benzene solvent and/or removal of “inert” electrolyte is associated with substantially improved yields of fluorinated aromatic compounds from the decomposition of diaryliodonium fluorides. These results have significant ramifications for the preparation of fluorinated radiotracers.
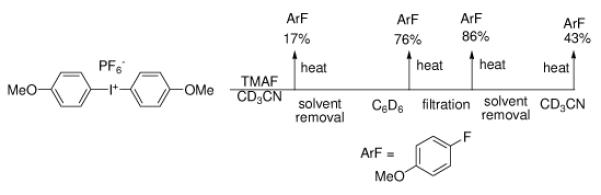
Previously we reported the preparation of phenyliodonium difluoride from anhydrous tetrabutylammonium fluoride11 (TBAF) and phenyliodonium diacetate.12 In the course of this work we noted that excess fluoride ion led to the eventual reduction of PhIF2 to PhI and bifluoride ion by an unknown mechanism. This reaction was largely suppressed in nonpolar solvents such as benzene. Similarly, if bis(4-methoxyphenyl)iodonium hexafluorophosphate 1 is treated with excess anhydrous TBAF or anhydrous tetramethylammonion fluoride13 (TMAF) in acetonitrile at ambient temperature, 4-iodoanisole is formed without the generation of any fluorinated arenes. If substoichiometric amounts of fluoride are present the decomposition reaction is largely suppressed at room temperature, but upon heating of the acetonitrile solution 1 undergoes thermal decomposition to form 4-fluoroanisole (4FA) in poor yield (17%) along with 4-iodoanisole and anisole. Given that the redox chemistry of PhIF2 was suppressed in benzene, we investigated the thermal decomposition of diaryliodonium fluoride salts in nonpolar media as well. Initial experiments (Figure 1) indicated that this approach might be fruitful.
Figure 1.

Solvent effect upon the decomposition of bis(4-methoxyphenyl)iodonium fluoride.
To test the generality of this approach, diaryliodonium hexafluorophosphates 2-7 (Figure 2) were prepared by two established methods: 3 and 5-7 were prepared by TsOH catalyzed electrophilic aromatic substitution of anisole by ArI(OAc)2, 14,15 and compounds 2 and 4 were prepared from the corresponding tributylstannanes and p-OMePhI(OH)(OTs) (Figure 3).16,17 The iodonium salts were isolated by precipitation from aqueous solutions using NaPF6. (Full experimental procedures are provided in the Supporting Information.)
Figure 2.

Direct decomposition of diaryliodonium fluorides in C6D6 and CD3CN at 140 °C.
Figure 3.

Preparation of diaryliodonium salts 2-7. a) NaBO3, AcOH, 40 °C, 8 h; b) TsOH·H2O, anisole, CH3CN; c) H2O, NaPF6; d) Pd2(dba)3, P(t-Bu)3, benzene, Sn2Bu6, 100 °C; e) p-(OMe)PhI(OTs)(OH), CH3CN.
Several procedures for diaryliodonium fluoride decomposition were investigated. In the first, compounds 1-7 were treated with 1 equivalent of anhydrous TMAF in CD3CN and heated (140 °C, 15 min) in sealed NMR tubes. Yields of fluorinated arenes were determined by 1H NMR spectroscopy and confirmed by GC-MS analysis. In the second method, ion exchange with TMAF was performed in acetonitrile, the solvent was evaporated, and the remainder was suspended in d6-benzene and heated (140 °C, 15 min) in sealed NMR tubes. A comparison of the results from these two procedures is given in Table 1. In all cases, the yields of fluorinated arenes were superior for the reactions conducted in benzene. In acetonitrile, the formation of HF2− and BF4− (from the borosilicate NMR tube) was observed, whereas no inorganic fluoride byproducts were seen when benzene was the reaction solvent. Inspection of the results shows that the 4-methoxyphenyl group strongly directs the fluoride nucleophile to the electron-poor aromatic ring, as is generally observed when diaryliodonium salts are fluorinated with cyclotron-derived 18F-fluoride ion.1
Table 1.
Yields of fluorinated arenes obtained from direct decomposition of salts 1-7.
| Cmpd. | C6D6 (ArF + 4FA)a |
CD3CN (ArF + 4FA) |
|---|---|---|
| 1 | 76 | 17 |
| 2 | (59 + 19) | (2 + 1) |
| 3 | (59 + 25) | (2 + 1) |
| 4 | (76 + 14) | (2 + 1) |
| 5 | (41 + 10) | (3 + 1) |
| 6 | (86 + 0) | (33 + 0) |
| 7 | (93 + 5) | 0 |
The numbers inside the parentheses indicate the percentage yields of the desired fluorinated arenes followed by the amount of 4-fluoroanisole (4FA) produced during the reaction. All solutions were heated at 140 °C for 15 minutes in sealed NMR tubes.
In addition to reducing the solvent polarity, the use of benzene as the reaction solvent effectively removes the inorganic spectator ions (tetramethylammonium and hexafluorophosphate) from solution. While these salts are not expected to participate directly in the thermal decomposition reaction, their presence might facilitate ligand exchange reactions and increase the I-F dissociation rate and dissociation constants. To probe whether the observed improvement in fluorination yield was a function of solvent polarity or extraneous salt, the thermal decomposition of the diaryliodonium fluorides was also conducted under “salt free” conditions in both solvents.
Salt removal was performed by a simple solvent exchange process; compounds 2-7 were dissolved in CH3CN, treated with TMAF and the solvent was evaporated. The remaining salts were dissolved in benzene and passed through a 0.20 μm PTFE syringe filter (Scheme 1). Upon evaporation of the benzene, clean samples of the diaryliodonium salts were obtained. 1H and 19F NMR spectra of the isolated diaryliodonium fluorides showed no residual TMAPF6 in the samples of diaryliodonium fluorides prepared using this solvent exchange method.
Scheme 1.

Salt removal by solvent exchange process.
The results of thermal decomposition reactions (140 °C, 15 min) of the “salt free” diaryliodonium fluorides are summarized in Table 2. For reactions conducted in benzene, salt-free conditions were associated with a modest enhancement in the yields of fluorinated arenes. In contrast, the yields of reactions conducted in CD3CN improved dramatically after the salt was removed; in some instances the yields obtained for the thermal decomposition of salt-free diaryliodonium fluorides in acetonitrile approached those seen for reactions conducted in benzene. These results suggest that fluoride ion dissociation may be responsible for some degradation of the arene fluorination efficiency observed in polar aprotic solvents.
Table 2.
Yields of fluorinated arenes obtained from decomposition of salts 1-7 after removal of TMAPF6.
| “Salt Free” Conditions | ||
|---|---|---|
| Cmpd. | C6D6 (ArF + 4FA)a |
CD3CN (ArF + 4FA) |
| 1 | 86 | 43 |
| 2 | (77 + 14) | (30 + 8) |
| 3 | (49 + 23) | (40 + 20) |
| 4 | (78 + 12) | (49 + 32) |
| 5 | (57 + 20) | (40 + 15) |
| 6 | (85 + 10) | (68 + 0) |
| 7 | (89 + 0) | (78 + 0) |
The numbers inside the parentheses indicate the percentage yields of the desired fluorinated arene followed by the amount of 4-fluoroanisole (4FA) produced during the reaction. All solutions were heated at 140 °C for 15 minutes in sealed NMR tubes.
Deuterated benzene is a particularly convenient and relatively inexpensive solvent for conducting these iodonium salt decomposition experiments, but its carcinogenicity makes benzene unattractive for practical preparations. Thus, we also investigated the thermal decomposition reactions of compounds 1-7 in d8-toluene under salt free conditions. The yields and selectivities for formation of fluorinated arenes from 1-7 (Table S1, Supporting Information) were essentially identical to those obtained for reactions conducted in C6D6, indicating that a wide range of nonpolar aromatic solvents might be readily used for this process.
Compounds 8 and 9, suitably protected precursors of previously investigated radiotracers 18F-6-fluorodopamine18 and 18F-2-fluoroestradiol,19 respectively, were decomposed to the corresponding fluorinated arenes in excellent (80% for 8) and fair (42% for 9) yields in benzene. The reduced fluorination yield for the latter compound is accompanied by a concomitant increase in the amount of 4-fluoroanisole produced, consistent with the similar “directing group” abilities of the 2-methoxy and 4-methoxy substituted arenes. (Recently, our laboratory reported a potential solution to this directing group problem.20) Successful fluorination of these arenes indicates that the methodology is sufficiently broad to tolerate suitably protected alcohol and amine functionality.
To more closely mimic the conditions of radiotracer synthesis, we examined the thermal decomposition reactions of 1 at three concentrations (1 mM, 5 μM, and 5 nM); radiotracer synthesis is typically conducted in the nM-μM 18F-fluoride concentration range. Since our studies were conducted exclusively with 19F-fluoride, we needed an appropriate analytical tool to test the impact of dilution upon the yield of fluorinated arenes. For product analysis we used a GC-TOF-MS; this technique was sufficiently sensitive to detect and quantify reproducibly 500 femtograms of injected 4-fluoroanisole. To minimize fluoride losses due to adsorption on glass surfaces during the dilution process, 1 was first treated with TBAF and the bis(4-methoxyphenyl)iodonium fluoride 1(F) was isolated.
Under standard thermal decomposition conditions (140 °C, 15 min, 0.5 mL of benzene) the yield of 4-fluoroanisole declined sharply with the concentration of 1(F): 1 mM – 90%, 5 μM – 30%, 5 nM – 0%. In contrast, thermal decomposition of a mixture of 5 ng 1(F) and excess (1 mg) bis(4-methoxyphenyl)iodonium trifluoroacetate under standard conditions gave excellent and reproducible yields of 4-fluoroanisole (80 ± 10% by GC-TOF-MS). (Control experiments in which bis(4-methoxyphenyl)iodonium trifluoroacetate was heated in benzene without added 1(F) produced no detectable fluoroanisole.)
Taken together, the results reported here show that the perhaps counterintuitive choice of nonpolar media for the pyrolysis of highly polar diaryliodonium fluorides has an important and practical impact on the success of arene fluorination, and that these new conditions give excellent yields of fluorinated arenes. Even when the total concentration of fluoride ion is vanishingly small, as it is when 18F-fluorinated radiotracers are synthesized, the presence of excess diaryliodonium substrate leads to excellent conversion to the fluorinated arene. In addition to suppressing deleterious side reactions, an important role of the nonpolar solvent is to precipitate inorganic salts that may catalyze ligand exchange, electron transfer, and disproportionation reactions. These new conditions have their genesis in the hypothesis that I(III) behaves like a transition metal ion, and long-standing observations of fluoride's propensity to promote the disproportionation of low-valent transition metal ions. Studies to demonstrate the direct application of these new reaction conditions in 18F-radiotracer synthesis are currently underway.
Supplementary Material
Figure 4.

Syntheses of compounds 8 and 9. a) Br2, AcOH, rt, KOH; b) diisopropylethylamine, phthaloyl dichloride, CH3CN, rt; c) Pd2(dba)3, t-Bu3P, benzene, Sn2Bu6, 100 °C; d) p-OMePhI(OTs)(OH), CH3CN; e. H2O, NaPF6; f) NBS, CH3CN/CCl4 (3:7) ; g) n-BuLi, Bu3SnCl, THF, −78 °C to rt.
Figure 5.

a) Total ion chromatogram of the decomposition reaction mixture of 1(F) in acetonitrile; b) Selected ion chromatogram of the same reaction mixture (mass range = 126.03-126.04).
Acknowledgment
We thank the National Science Foundation (CHE 0717562) for support and the National Institutes of Health (RR016544-01) for infrastructure to conduct this research.
Footnotes
Supporting Information Available: Experimental procedures and analytical data for all new compounds and synthetic intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cai L, Lu S, Pike VW. Eur. J. Org. Chem. 2008:2853–2873. [Google Scholar]
- 2.Watson DA, Su M, Teverovskiy G, Zhang Y, Garcia-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:1661–1664. doi: 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ball ND, Sanford MS. J. Am. Chem. Soc. 2009;131:3796–3797. doi: 10.1021/ja8054595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hull KL, Anani WQ, Sanford MS. J. Am. Chem. Soc. 2006;128:7134–7135. doi: 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]
- 5.Furuya T, Ritter T. Org. Lett. 2009;11:2860–2863. doi: 10.1021/ol901113t. [DOI] [PubMed] [Google Scholar]
- 6.Furuya T, Strom AE, Ritter T. J. Am. Chem. Soc. 2009;131:1662–1663. doi: 10.1021/ja8086664. [DOI] [PubMed] [Google Scholar]
- 7.Pike VW, Aigbirhio FI. J. Chem. Soc., Chem. Commun. 1995:2215–6. [Google Scholar]
- 8.For functionalization of arenes using diaryliodonium salts and other nucleophiles see Thiele J, Umnoff A. J. Liebigs Ann. Chem. 1910;369:147–9. Fletcher CJM, Hinshelwood CN. J. Chem. Soc. 1935:596–9. Lucas HJ, Kennedy ER, Wilmot CA. J. Am. Chem. Soc. 1936;58:157–60. Sandin RB, Kulka M, McCready R. J. Am. Chem. Soc. 1937;59:2014–15. Beringer FM, Brierley A, Drexler M, Gindler EM, Lumpkin CC. J. Am. Chem. Soc. 1953;75:2708–2712. Olah GA, Sakakibara T, Asensio G. J. Org. Chem. 1978;43:463–8.
- 9.Stang PJ. J. Org. Chem. 2003;68:2997–3008. doi: 10.1021/jo030022e. [DOI] [PubMed] [Google Scholar]
- 10.Sun H, Xue F, Nelson AP, Redepenning J, DiMagno SG. Inorg. Chem. 2003;42:4507–4509. doi: 10.1021/ic0345830. [DOI] [PubMed] [Google Scholar]
- 11.Sun H, DiMagno SG. J. Am. Chem. Soc. 2005;127:2050–2051. doi: 10.1021/ja0440497. [DOI] [PubMed] [Google Scholar]
- 12.Sun H, Wang B, DiMagno SG. Org. Lett. 2008;10:4413–4416. doi: 10.1021/ol8015429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christe KO, Wilson WW, Wilson RD, Bau R, Feng JA. J. Am. Chem. Soc. 1990;112:7619–25. [Google Scholar]
- 14.Kitamura T, Matsuyuki J, Taniguchi H. Synthesis. 1994:147–8. [Google Scholar]
- 15.Shah A, Pike VW, Widdowson DA. J. Chem. Soc., Perkin Trans. 1. 1997:2463–2465. [Google Scholar]
- 16.Koser GF, Wettach RH, Smith CS. J. Org. Chem. 1980;45:1543–4. [Google Scholar]
- 17.Pike VW, Butt F, Shah A, Widdowson DA. J. Chem. Soc., Perkin Trans. 1. 1999:245–248. [Google Scholar]
- 18.Ding YS, Fowler JS, Gatley SJ, Dewey SL, Wolf AP, Schlyer DJ. J. Med. Chem. 1991;34:861–3. doi: 10.1021/jm00106a055. [DOI] [PubMed] [Google Scholar]
- 19.Hostetler ED, Jonson SD, Welch MJ, Katzenellenbogen JA. J. Org. Chem. 1999;64:178–185. doi: 10.1021/jo981619a. [DOI] [PubMed] [Google Scholar]
- 20.Wang B, Graskemper JW, Qin L, DiMagno SG. Angew. Chem., Int. Ed. 2010;49:4079–4083. doi: 10.1002/anie.201000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.