

Abstract
Linear tetrapyrroles (bilins) perform important antioxidant and light harvesting functions in cells from bacteria to humans. To explore the role of the propionate moieties in bilin metabolism, we report the semisynthesis of mono- and di-amides of biliverdin IXα and those of its non-natural XIIIα isomer. Initially, these were examined as substrates of two types of NADPH-dependent biliverdin reductase, BVR and BvdR, and of the representative ferredoxin-dependent bilin reductase, phycocyanobilin:ferredoxin oxidoreductase (PcyA). Our studies indicate that the NADPH-dependent biliverdin reductases are less accommodating to amidation of the propionic acid sidechains of biliverdin IXα than PcyA, which does not require free carboxylic acid sidechains to yield its phytobilin product, phycocyanobilin. Bilin amides were also assembled with BV-type and phytobilin-type apophytochromes, demonstrating a role for the 8-propionate in formation of the spectroscopically native Pr dark states of these biliprotein photosensors. Neither ionizable propionate sidechain proved essential to primary photoisomerization for both classes of phytochromes, but an unsubstituted 12-propionate was required for full photointerconversion of phytobilin-type phytochrome Cph1. Taken together, these studies provide insight into the roles of the ionizable propionate sidechains in substrate discrimination by two bilin reductase families while further underscoring the mechanistic differences between the photoconversions of BV-type and phytobilin-type phytochromes.
Heme-derived linear tetrapyrroles (bilins) perform important roles as antioxidants and as light harvesting and photosensory pigments in many organisms (1, 2). Owing to the ability of free heme to generate strong intracellular oxidants in the presence of oxygen, excess heme accumulation is averted by feedback regulation of its biosynthesis and through its enzymatic conversion to biliverdin IXα (BV)1 by heme oxygenases (3). Heme oxygenase is strongly product inhibited, so turnover is sustained by coupled bilin reductase activity or by transfer to bilin-binding proteins. The NAD(P)H-dependent biliverdin reductase A (BVR) of animals is well tailored to this role, as formation of its lipophilic bilirubin IXα (BR) product is essentially irreversible (4, 5). Oxygenic photosynthetic organisms can instead use ferredoxin-dependent bilin reductases (FDBRs) to convert BV to the more reduced phytobilins, phytochromobilin (PФB), phycocyanobilin (PCB) and phycoerythrobilin (PEB) (6, 7). BV transfer from heme oxygenase to a bacteriophytochrome in Pseudomonas aeruginosa (8) suggests that these widespread two component kinases contribute to heme turnover in eubacteria in addition to their role as bilin and light sensors (9, 10).
A greater understanding of the substrate specificity of bilin reductases is expected to presage development of new reagents of clinical and agricultural significance. In this regard, neonatal jaundice arising from delayed induction of the BR-specific UDP-glucuronyl transferase expression is responsible for profound neurological disorders in newborns. While treatment of neonatal jaundice entails blue light exposure that triggers BR photoisomerization from the thermally favored 4Z, 15Z form to produce more readily excreted isomers such as 4Z, 15E (11-13), treatments for other hepatopathological and congenital forms of bilirubinemia could benefit from new drugs that specifically inhibit BVR activity (14). Inhibitors of FDBRs also are desirable for use as potential algicides and as novel regulators of plant growth and development. In cyanobacteria and some eukaryotic algae, the phytobilin products of FDBRs are direct precursors of the covalently bound chromophores of the abundant light-harvesting phycobiliproteins (15, 16). The red (R) and far-red (FR) light sensing phytochromes use BV, PCB or PФB, whose covalent association with a phytochrome apoprotein permit their hosts to adapt to unfavorable light conditions (10, 17-20). As potential antimicrobial agents and plant growth regulators, bilin-based inhibitors of phytochrome biogenesis thus hold considerable promise for both agriculture and medicine.
Phytochromes are attracting increasing attention as targets for protein engineering, due to their potential uses as gene photoswitches, as fluorescent reporters and in agricultural biotechnology (21-27). In this regard, the cyanobacterial phytochrome Cph1 was converted from a photochemically active red/far-red photosensor into a photochemically inert, intensely fluorescent protein by a single point mutation (23, 28). Similar results have subsequently been obtained for several related proteins using either directed evolution or site-directed mutagenesis (26, 29-31). The photoswitching properties of such proteins arise via a combination of the intrinsic photochemical properties of the chromophore and the effects of the surrounding protein matrix, so the use of site-directed mutagenesis to modify that protein matrix has also provided invaluable information about the effect of chromophore-protein interactions on the photocycle and on subsequent transduction of the light signal (24, 30, 32-34). The broad photosensing range of the phytochrome-related cyanobacteriochrome family reflects their molecular evolution and natural selection for new regulatory functions using a common bilin precursor (35).
The converse experiment in which the structure and function of a holophytochrome is perturbed by modification of the chromophore has also been undertaken. Indeed, nature has already performed this experiment, yielding phytochromes with BV-, PФB- and PCB-derived chromophores. While more extensive chromophore modifications require synthetic chemistry expertise and hence are more labor-intensive than site-directed mutagenesis (36-39), the ability to incorporate semi-synthetic BV isomers and unnatural phytobilins into plant phytochromes has proven particularly insightful to probe bilin-protein interactions (21, 40-44). Chromophore analogs can provide information about the roles played by specific moieties that cannot be obtained in other ways, and hence such studies provide a valuable counterpoint to approaches based on mutagenesis. We previously used such an approach to explore the importance of the bilin 12-propionate sidechain in two model phytochromes, Cph1 from Synechocystis sp. PCC6803 and DrBphP from Deinococcus radiodurans (45).
Over the years, many BV analogs have been synthesized, but surprisingly, syntheses of simple BV amides have not been described. Here, we report the semisynthesis of mono- and di-amides of BV and those of the unnatural XIIIα isomer employing BR as the starting material. These biliverdin amides were used for investigations that initially focus on their ability to serve as substrates for the representative bilin reductases, BVR and the FDBR phycocyanobilin:ferredoxin oxidoreductase (PcyA). Our studies indicate that BVR is considerably more sensitive to amidation of the propionic acid sidechains than PcyA, which does not require free carboxylic acid sidechains for catalysis. We have also exploited the ability of PcyA to reduce the 8- and 12-monoamides of BV to their corresponding 3E/3Z-PCB phytobilin products for comparative studies examining distinct roles played by the propionate sidechains in phytochrome assembly and photointerconversion between their red-absorbing (Pr) and far red-absorbing (Pfr) forms. Our results demonstrate the utility of such compounds in studying bilin-protein interactions and provide further support for the proposal that Cph1 and DrBphP have different requirements for formation of Pfr.
MATERIALS AND METHODS
Reagents and General Procedures
All reagents/solvents were ACS reagent grade unless otherwise specified, except that HPLC-grade solvents were used for reversed phase (RP-HPLC) chromatography. Bilirubin IXα (BR) was purchased from Frontier Scientific (Supp. Fig. 1A for rubin structures). Bilirubin XIIIα (BR13) was synthesized according to the method of Lightner and colleagues (46). Biliverdins were obtained by oxidizing the corresponding bilirubins with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ, Aldrich) as described by McDonagh and co-workers (41, 47) using DMF as solvent in place of DMSO. Preparative column chromatography was performed using Merck silica gel 60 (70-230 mesh). Analytical thin layer chromatography (TLC) was performed on Merck 60 F254 plastic silica gel sheets (0.1 mm layer). Analytical RP-HPLC (Fig. 2 and Supp. Fig. 2) used a Phenomenex Ultracarb ODS(20) column (250 × 4.6 mm, 5 μm particle size) on an Agilent 1100 series HPLC system equipped with an Agilent G1315B diode-array detector. A Phenomenex Ultracarb ODS(20) column (250 × 10 mm, 5 μm particle size) was used on the same HPLC system for preparative purification of bilins. HPLC and TLC solvent systems used for the various compounds under study are described in the sections pertaining to each compound. Protein concentrations were determined with bicinchoninic acid (48) or Bradford (49) protocols using bovine serum albumin as a protein standard. Pigment concentrations were estimated using previously reported extinction coefficients for the diacid.
Figure 2.
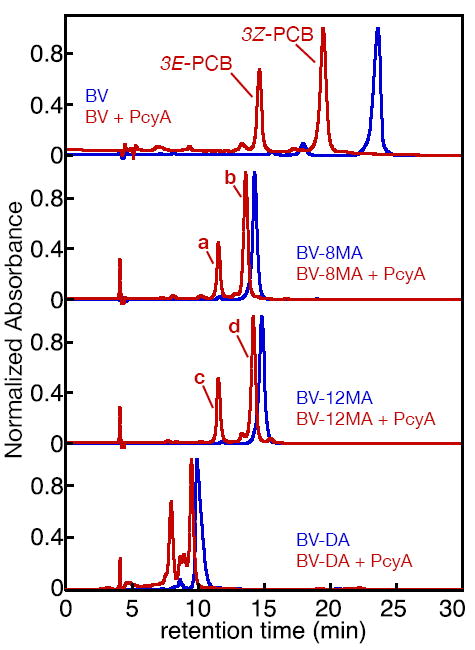
Reaction of BV with PcyA. BV derivatives were analyzed by RP-HPLC in 50% aqueous acetone/20 mM formic acid either with (red) or without (blue) prior treatment with PcyA. Absorbance at 650 nm was monitored using a diode-array detector. Top, reduction of BV gave rise to a mix of 3E-PCB and 3Z-PCB, as expected (52). Second from top, reduction of BV-8MA gave rise to two product peaks (a and b) identified as 3E-PCB-8MA and 3Z-PCB-8MA, respectively. Third from top, reduction of BV-12MA produced products c and d, similarly identified as 3E-PCB-12MA and 3Z-PCB-12MA. Absorbance spectra for 3E- and 3Z-PCB and products a-d are presented in Supp. Fig. 6, and peak wavelengths are reported in Supp. Table 5. Bottom, reduction of BV-DA by PcyA resulted in formation of several apparent products that were not well resolved in this mobile phase. Further characterization of these products (Supp. Fig. 7) suggested the presence of PCB diamide, but it was not possible to obtain pure products.
Characterization of bilin amides
Compounds synthesized in the course of this study were characterized by 1H-NMR spectroscopy, UV-Vis absorption spectroscopy, and mass spectrometry. Supp. Table 1 reports peak absorbance wavelengths and m/z values. 1H-NMR spectra were acquired on a 500 MHz Bruker-500 spectrometer in d5-pyridine (99.96%, Aldrich), and assignments are reported in Supp. Tables 2 and 3. UV-visible spectra (Supp. Fig. 3) were recorded in methanol/5% conc. HCl (v/v), 50% aqueous acetone/20 mM formic acid, or 40% aqueous acetone/20 mM formic acid. Spectra in acidic methanol were recorded on Hewlett-Packard 8453 or Cary 50 spectrophotometers, while spectra in aqueous acetone/formic acid were obtained in real time during RP-HPLC analysis. Mass spectra were recorded on an Applied Biosystems Electron/Spray Q-trap mass spectrometer with methanol as a sample solvent. For estimation of bilin pKa values, bilins (3.4 μM in 50 mM tetramethylammonium chloride) were characterized by absorbance spectroscopy at various pHs (buffers at 200 mM). Buffers used were potassium phosphate (pH 3 - 7), tris(hydroxymethyl)methylamine (Tris-HCl, pH 8 - 9.5), N-cyclohexyl-3-aminopropanesulfonic acid (CAPS-KOH, pH 10 - 11.5), and methylamine-HCl (pH 12).
Representative procedures for semisynthesis of biliverdin IXα diamide (BV-DA), biliverdin XIIIα diamide (BV13-DA) and bilirubin dimethyl esters
Bilirubin IXα (BR: 50 mg, 0.09 mmol) was suspended in 10 ml DMF under argon with stirring. Di(isopropyl)ethylamine (DIEA, Aldrich; 120 μl, 8 eq.) was added, and the solution was stirred for 10 min before addition of a mixture of solid O-benzotriazole-N,N,N’,N’-tetramethyl-uronium hexafluorophosphate (HBTU, Novabiochem: 98 mg, 0.26 mmol, 3.0 eq.) and hydroxybenzotriazole (HOBt, Novabiochem: 40 mg, 0.26 mmol, 3.0 eq.) under argon. The reaction mixture was stirred in the dark for 1 h or until the solution clarified. Solid crystalline NH4Cl (Aldrich, 37 mg, 0.69 mmol, 8 eq.) was then added, and the solution was stirred in the dark under argon for 10-12 h. The reaction mixture (containing a light yellow precipitate) was then poured into a solution of 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ: 41 mg, 0.18 mmol, 2.1 eq.) in DMF (70 ml) with stirring and argon bubbling. After 5 min, argon-saturated water (400 ml) was added to stop the reaction, and the green product was extracted several times with chloroform (100 ml) until the aqueous layer was colorless. The combined chloroform layer was washed with water (5 × 100 ml) and once with brine (100 ml). The resulting organic phase was evaporated to dryness to yield crude biliverdin IXα diamide (BV-DA) as a dark green solid. Silica gel TLC (CHCl3: MeOH, 9: 1 vol/vol) showed one main spot (Rf = 0.3). BV-DA was further purified by gravity flow silica gel chromatography to afford 20 mg final product (40% yield). A similar procedure was used with BR13 as starting material for synthesis of biliverdin XIIIα diamide (BV13-DA). The bilirubin dimethylesters, BR-DME and BR13-DME, were also prepared by similarly activating the corresponding bilirubin isomer with HBTU/HOBt followed by introducing methanol in excess.
In addition to proceeding with oxidation in a one-pot reaction, we were able to isolate bilirubin diamide by adding excess water to the reaction mixture and then collecting BR diamide as a yellow solid via filtration. However, BR diamide exhibited extremely poor solubility in a number of solvents, including chloroform, DMF, acidic DMSO, and water. The poor solubility of BR diamide in DMSO is likely to explain its inert character in acid co-scrambling reactions.
Representative procedure for semisynthesis of biliverdin IXα 8-monoamide (BV-8MA) and biliverdin XIIIα monoamide (BV13-MA)
A mixture of BR-DME (40 mg, 0.07 mmol) and BR XIIIα (BR13: 40 mg, 0.07 mmol) was dissolved in dimethylsulfoxide (DMSO: 30 ml). Concentrated hydrochloric acid (3 ml) was rapidly added to the solution with stirring. After stirring for 5 min at room temperature under argon, the reaction mixture was poured into ice-cold water (300 ml) and stirred for 15 min. The resulting brown precipitate was collected by vacuum filtration and washed with water (3 × 100 ml). The product was dried over phosphorous pentoxide in a vacuum desiccator overnight to afford mixed rubins (Fig. 1) as crude product (70 mg, ~0.12 mmol). Crude mixed rubins (70 mg) were suspended in DMF (14 ml), and DIEA (100 μl, 5 eq.) was added with stirring. After 10 min, HBTU (79 mg, 0.21 mmol) and HOBt (32 mg, 0.21 mmol) were directly added to the reaction mixture. The solution was stirred for 1 h before solid ammonium hydrochloride (30 mg, 0.56 mmol) was added. After overnight incubation with stirring in darkness under argon, the reaction solution was poured into a solution of DDQ (58 mg, 0.25 mmol) in DMF (100 ml). Argon-saturated water (500 ml) was added to the mixture after 5 min, and products were isolated by repeated extraction with chloroform (100 ml each) until the aqueous layer was colorless. The combined organic phases were washed with water (3 × 100 ml) and evaporated to dryness to yield 40 mg mixed bilin products as dark green solid. This mixture showed three major blue-green spots on silica gel TLC (CHCl3:MeOH, 9:1). The fastest eluting species (Rf = 0.45) corresponded to mixed biliverdin dimethylesters (Group I), the middle species (Rf = 0.25) corresponded to monomethylesters of both BV-8MA and BV13-MA (Group II), and the slowest migrating species (Rf = 0.11) contained biliverdin diamides (Group III). The three components were resolved by silica gel column chromatography (CHCl3: MeOH, 20:1 or 10:1) to yield group I (4 mg), group II (10 mg) and group III (4 mg). The Group II biliverdin-MA monoester mixture was subjected to a modified ester cleavage method of Lindner et al (50). This entailed dissolving the monoamide/monoester mixture (4 mg, ~ 7 μmol) in 50% aqueous trifluoroacetic acid (TFA, 6.4 ml) to which pre-washed DOWEX 50WX8-200 ion-exchange resin (1.4g) was added with stirring in darkness. Ester cleavage was monitored by TLC (CHCl3:MeOH, 9:1) and by HPLC (mobile phase: 50% aqueous acetone containing 20 mM formic acid). After hydrolysis was complete (~ 2 days), the resin was removed by filtration and the filtrate was diluted with distilled water (60 ml). The resulting solution was applied to a C18 Sep-Pak solid phase cartridge (Waters) pre-equilibrated with 0.1% aqueous TFA. The loaded Sep-Pak column was washed with water (10 ml) and then with 0.1% aqueous TFA (10 ml). Biliverdin products were eluted with acetonitrile followed by vacuum drying to yield biliverdin monoamides as a green solid. Preparative HPLC was used to separate biliverdin XIIIα monoamide (BV13-MA: 0.3 mg, 0.5 μmole, 15% theoretical yield assuming 1:1 mix of monoamides) and biliverdin IXα 8-monoamide (BV-8MA; 0.2 mg, 0.3 μmol, 10% theoretical yield).
Figure 1.
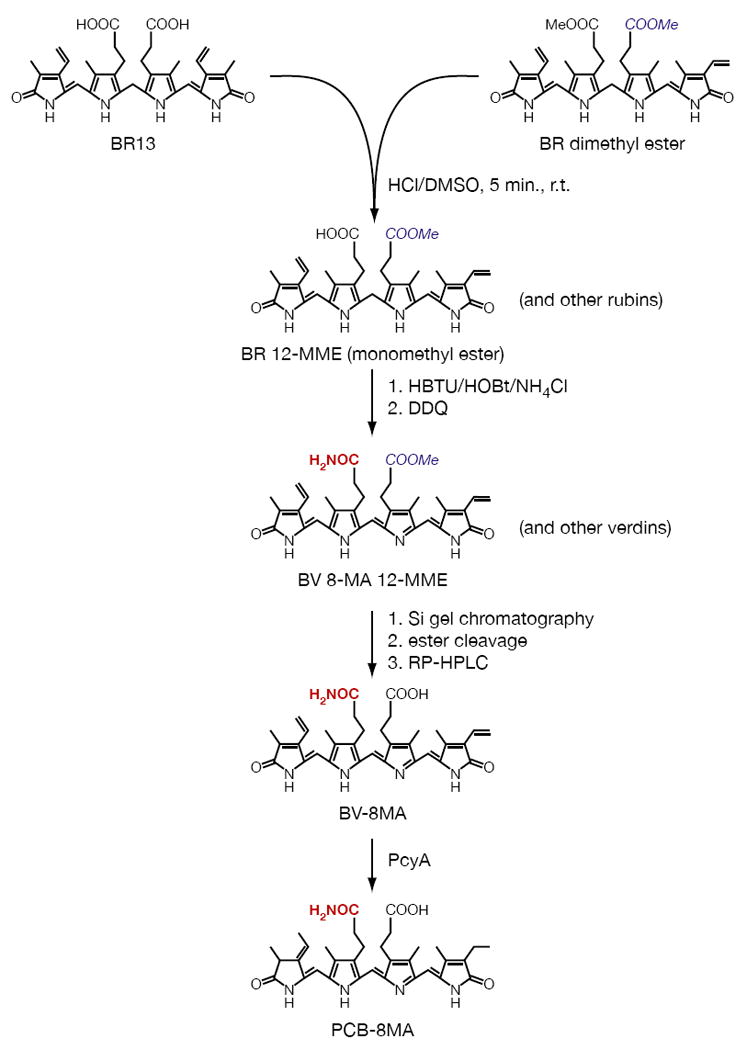
Synthesis of bilin 8-monoamides. BR13 and BR13 dimethyl ester were co-acid-scrambled to yield mixed rubins including BR-12MME. Amidation of free propionate sidechains and subsequent oxidation yielded mixed verdins including BV-8MA12MME. Ester cleavage and purification yielded BV-8MA, which could be enzymatically converted to PCB-8MA by the ferredoxin-dependent bilin reductase PcyA.
Semisynthesis of biliverdin IXα 12-monoamide (BV-12MA)
A similar procedure was used as described above using bilirubin XIIIα dimethylester (BR13-DME) and bilirubin IXα (BR) as starting materials to generate biliverdin IXα 12-monoamide (BV-12MA).
Preparation of recombinant proteins
Recombinant PcyA from Anabaena sp. PCC 7120 and recombinant ferredoxin from Synechococcus sp. PCC7002 were expressed in E. coli as described previously (51, 52). PcyA was purified as a glutathione-S-transferase fusion protein on glutathione-agarose (Sigma) (51) while ferredoxin was purified by ion-exchange chromatography on DEAE-Sephadex (53). Plasmids pBAD-Cph1N514-CBD and pBAD-DrBphPFL-CBD (45) were used for expression of the N-terminal 514 amino acids of Cph1 and of full-length DrBphP as apoproteins, respectively. Purified PcyA, ferredoxin, Cph1, and DrBphP were each dialyzed against TKKG buffer (25 mM TES-KOH pH 8.5, 100 mM KCl, 10% (v/v) glycerol). For expression of mammalian (rat) BVR, E. coli strain BL21 was transformed with pGEX6P1-ratBVR, a GE Healthcare GST gene fusion plasmid, and grown in LB. Plasmid pGEX6P1-ratBVR (generous gift of Prof. Mahin D. Maines) encodes GST-tagged BVR. This plasmid was found to contain two mutations relative to the rat BVR sequence established by X-ray crystallography (54) D263G, which is a known polymorphism, and G141W. Neither mutation is predicted to lie close to the active site. IPTG (1 mM) was used to induce BVR expression. Clarified lysate was loaded onto a glutathione affinity column equilibrated in phosphate buffered saline (PBS). After extensive washing with PBS, BVR protein was eluted from the column in 50 mM Tris-HCl, pH 8, supplemented with 10 mM reduced glutathione. Eluted BVR was concentrated by centrifugal ultrafiltration (10K MWCO) and dialyzed in phosphate buffer (100 mM potassium phosphate, pH 7.4, 10% v/v glycerol). To obtain Synechocystis biliverdin reductase (BvdR, (59)), E. coli strain BL21 was transformed with plasmid pASK75B-BvdR (obtained by inserting the PCR-amplified Synechocystis BvdR locus into the Biometra Strep-tag vector pASK75B (55)) and grown in LB media. BvdR protein was expressed and purified using strep-tag resin (IBA) in accordance with the manufacturer’s instructions. Purified protein was dialyzed against phosphate buffer (100 mM potassium phosphate, pH 7.4, 10% v/v glycerol). After dialysis, all recombinant proteins were flash-frozen in liquid nitrogen, and stored at -80°C prior to use.
Biliverdin reductase assays
Mammalian BVR assays were performed in absorbance cuvettes containing 1 ml of 0.1 M Tris-HCl buffer, pH 8.7, 0.1 mM NADP+, 1 mM glucose 6-phosphate, 0.1 units/ml glucose-6-phosphate dehydrogenase, 1 mg/ml BSA, 1.25-5 μM bilin substrate, and 34 nM BVR. BvdR assays were performed in absorbance cuvettes containing 1 ml BvdR buffer (0.1M citrate, pH 5.8; 10% glycerol (v/v); 0.2 mg/ml BSA; 100 μM NADPH; 1-5 μM bilin substrate; 0.5-2 μM BvdR). Enzyme concentrations for BV IXα substrate controls were adjusted to 17 nM. Reactions were initiated by addition of enzyme, and absorbance was monitored at 450 nm at 20-second intervals. All assays were performed at room temperature (25°C) in duplicate. The amount of rubin product formed was calculated based on an absorption coefficient at 450 nm of 53,000 M-1 cm-1. Initial rates of reaction were determined at substrate concentrations of 1.25 μM, 2.5 μM and 5 μM. Reaction velocity was linear with substrate concentration in this regime, indicating that the reaction was proceeding under kcat/KM conditions for all substrates (i.e., that total enzyme concentration is approximately equal to free enzyme concentration). The linear slope is therefore approximately equal to kcat/KM times total enzyme concentration, permitting determination of kcat/KM under the assumptions that the enzyme is purified to homogeneity and is entirely active. These assumptions do not affect the measurement of specificity, which is determined by the ratio of kcat/KM values.
Reaction of biliverdin amides with PcyA
PcyA was assayed under steady-state conditions (52), with all solutions degassed under vacuum prior to use in a closed vessel. Standard assays used TKK buffer (25 mM TES-KOH pH 8.5/100 mM KCl), while Mes-NaOH (pH 6.0) was used for the reaction of BV diamide with PcyA at pH 6. Assays contained 0.1 μM PcyA, 5 μM biliverdin, 0.025 U/ml ferredoxin-NADP+ reductase (FNR), 5 μM ferredoxin, 10 μM bovine serum albumin, and an oxygen scavenging system (25 U/ml glucose oxidase, 50 mM glucose and 25 U/ml catalase). Reactions were initiated by addition of NADPH from a 30 mM stock with immediate incubation of the reaction tube at 30°C. After 30 min, reactions were iced. Crude bilins were extracted with a C18 Sep-Pak C18 cartridge and eluted as described above, followed by evaporation to dryness using a SpeedVac concentrator (Savant). Analytical RP-HPLC analyses were performed as described above with monitoring at 650 nm, 480 nm, and 380 nm. Complete UV-visible spectra were obtained for peaks of interest. This procedure was performed on a larger scale for preparative production of PCB amides for assembly with apophytochromes.
Assembly of bilin amides with apophytochromes and characterization of monoamide adducts
In vitro assembly reactions were performed in TKKG buffer as described (45), with tris(2-carboxyethyl)phosphine hydrochloride (TCEP) added to a final concentration of 1 mM immediately before assembly reactions were set up. Apophytochromes (Cph1 or DrBphP) were diluted in freshly prepared buffer to a final volume of 1 ml, and bilin was added such that the apophytochrome was present in 2-4X excess. After incubation in darkness for 2 h at room temperature, reactions were dialyzed in TKKG buffer overnight at 4°C. The presence of covalently attached chromophore following dialysis was determined by SDS-PAGE and zinc blotting (Supp. Fig. 4). Phytochrome photochemistry was assayed using a Cary 50 spectrophotometer equipped with a cuvette holder permitting irradiation of the sample from above with light from a 75W xenon source used with appropriate filters (45). Circular dichroism (CD) spectra were acquired on a Chirascan CD spectrometer (Applied Photophysics). For denaturation experiments, holophytochrome (150 μl) was combined with concentrated guanidinium chloride (900 μl) and concentrated hydrochloric acid (10 μl). Final guanidinium concentration always exceeded 6 M. Denatured samples were typically characterized by absorbance spectroscopy on the Cary 50 spectrophotometer with or without illumination to examine photochemistry over a period of 10 min or less to avoid unwanted side reactions associated with the low pH.
RESULTS
Semisynthesis and characterization of biliverdin amides
Bilirubin IXα (BR) is commercially available, inexpensive, and can readily be converted to BV via mild oxidation. We therefore chose BR as the starting point for the synthesis of biliverdin amides. BR could readily be converted to its diamide (BR-DA) by activation of the carboxylate moieties with the coupling reagent HBTU. It was possible to similarly generate the diamide of BR13 (BR13-DA). Unfortunately, both rubin diamides proved difficult to characterize due to extremely low solubility in several solvent systems. It was also not possible to generate BR monoamides via co-acid scrambling of BR and BR-DA (56), because BR-DA proved inert in this reaction, presumably due to its low solubility. We were able to generate BV diamide (BV-DA) from BR via amidation and subsequent oxidation in a one-pot reaction (representative procedure in the Methods), and BV-DA proved more soluble and hence more amenable to study. A similar approach could be used to prepare BV13-DA. Using lower amounts of ammonium chloride in this procedure resulted in formation of a mixture of BV monoamides, but it was not possible to purify either monoamide satisfactorily after such reactions.
We therefore devised an alternative approach for preparing BV monoamides in which BR and BR13 were used as starting materials (Figure 1; detailed procedures are presented in the Methods). BR was converted to its dimethyl ester (BR-DME) and then co-acid-scrambled with BR13. This reaction regenerated starting materials along with BR 12-monomethylester (BR-12MME) and BR13 monomethylester (BR13-MME). This mixture was then reacted with HBTU, HOBt, and ammonium chloride as for synthesis of the diamide, with subsequent oxidation in a one-pot reaction to obtain a mix of biliverdin esters and amides that included BV-8MA/12-MME and BV13-MA/MME. After ester cleavage and purification by preparative HPLC, this route yielded BV-8MA and BV13-MA. By changing the starting materials in this scheme to BR and BR13 dimethylester, we were similarly able to synthesize BV-12MA.
The purity of the resulting bilin amides was assessed by analytical RP-HPLC (Fig. 2 and Supp. Fig. 2). All compounds were found to be of good quality. Compared with the parent BV diacid molecules, the corresponding amides elute with shorter retention times: BV diamides eluted first, followed by monoamides and then diacids. BV diamides were more soluble in chloroform or in mixed aqueous/organic media (10% DMSO or 10% DMF) than their parent diacids. This is in marked contrast to the equivalent chloroform–soluble rubins, as BR diamides were largely insoluble in all solvents examined.
The structures of BV amides were confirmed by mass spectrometry, absorbance spectroscopy, and 1H-NMR spectroscopy (Supp. Fig. 2; Supp. Tables 1-3). All compounds had correct m/z values. Peak absorbance wavelengths and absorbance lineshapes were essentially identical in BV diacids and amides (Supp. Fig. 3; Supp. Table 1), providing evidence that the bilin π system active in absorbance spectroscopy is not intrinsically affected by loss of carboxylate moieties. Moreover, the 1H-NMR spectra of monoamides and diamides showed almost no changes relative to the parent diacids (Supp. Tables 2 & 3). The exceptions are the appearance of amide protons in the BV amide spectra and the appearance of additional peaks in the 1H-NMR spectrum of BV13-MA relative to the spectra of BV13 or BV13-DA (Supp. Table 3). Such additional peaks are expected for the asymmetrical BV13-MA relative to the symmetrical BV13 and BV13-DA isomers (Supp. Fig. 2C). The NMR and absorbance spectra thus provide evidence that the aromatic π system is not substantially affected by the amide substitutions.
Further support for this conclusion comes from estimation of the pKa value of the bilin π system. We chose to evaluate this by absorbance spectroscopy for the parent BV diacid and for BV diamide. This approach can produce complicated results due to conformational changes (57), so we carried out these measurements in the presence of 50 mM tetramethylammonium chloride to minimize effects of the propionate or amide sidechains on chromophore geometry. We found that the peak wavelength for the bilin blue (Soret) absorbance band exhibited a clear transition consistent with an apparent single spectroscopically sensitive titratable group for both compounds (Supp. Fig. 5). We measured identical pKa values of 5.7 for biliverdin diacid and diamide. This pKa is close to that of pyridine (5.2), consistent with titration of the proton responsible for the positively charged bilin π system (formally the C-ring nitrogen: Supp. Fig. 1) and not with the propionates, which are absent in the diamide. We therefore conclude that the substitution of one or two amides for propionates has very little effect on the bilin π system per se and hence could be expected to have little effect on its intrinsic behavior as a chromophore, while permitting assessment of the importance of the propionate sidechains for bilin-protein interactions.
Biliverdin amides as substrates for NAD(P)H-dependent biliverdin reductases
In some organisms, biliverdins are metabolized by biliverdin reductases that target the C10 position to generate bilirubins. Mammalian bilirubin production mainly relies on the activity of NAD(P)H-dependent biliverdin reductase A (BVR) (58), an enzyme with an apparent ortholog designated as BvdR in many cyanobacteria (59). While BVR and BvdR both target biliverdin alpha isomers, they exhibit very different pH dependence profiles (59-61). To assess the importance of the bilin propionates in substrate binding and catalysis by these NAD(P)H-dependent reductases, we examined the reaction of biliverdin amides with recombinant rat BVR and BvdR from the cyanobacterium Synechocystis sp. PCC 6803 (59, 62). The limited water solubility of biliverdin and its amides precluded satisfactory saturation kinetics. However, we were able to determine Vmax/KM values, which were used to calculate kcat/KM values by assuming that total protein concentration is an accurate measurement of active enzyme concentration. The results demonstrate the importance of the 8- and 12- propionates for BVR and BvdR specificity (Suppl. Table 4). Both enzymes displayed similar substrate preference for the various bilin amides. For BVR, all three monoamide substrates exhibited slight defects of 5- to 10-fold relative to the physiological substrate, BV (Supp. Table 4). For BvdR, the three monoamides exhibited slightly larger catalytic defects of 50- to 70-fold. Rubin formation by either enzyme could not be detected for either BV diamide or BV13 diamide within the sensitivity of our spectrophotometric assay. We therefore conclude that one propionate is sufficient to permit turnover by BvdR, but that loss of both propionates profoundly inhibits catalysis by both types of NAD(P)H-dependent biliverdin reductase.
Biliverdin amides as substrates for the ferredoxin-dependent biliverdin reductase PcyA
While BV is reduced at the C10 position to yield BR in mammalian tissue, its reduction by FDBRs in plants and cyanobacteria yields the phytobilins PФB, PCB, and PEB (6). To assess the importance of the BV propionates in initial substrate binding (i.e., formation of the Michaelis complex) and subsequent catalysis by this class of enzymes, we examined the utilization of biliverdin amides by the representative FDBR PcyA, for which there are available crystal structures with and without bound bilin (63-66). PcyA readily converted BV to PCB, which accumulated as a characteristic mixture of the 3Z and 3E isomers (Fig. 2). PcyA also proved competent to convert the monoamides BV-8MA and BV-12MA to the corresponding PCB monoamides, again with a mix of the 3E and 3Z forms (Fig. 2, Supp. Fig. 6, and Supp. Table 5). By contrast, PcyA was unable to utilize BV-DA as substrate under standard reaction conditions of pH 8.5 (data not shown). However, at a lower reaction pH, PcyA appeared to sustain reduction of BV-DA to the corresponding PCB-DA product mixture (Fig. 2). Using a different HPLC mobile phase (40% acetone / 20 mM formic acid) to improve resolution (Supp. Fig. 7), three main peaks and at least four additional minor species could be resolved. The majority species exhibited an absorbance spectrum similar to that of the starting BV-DA (Supp. Fig. 7 Supp. Table 5), despite its apparent change in elution time. The species whose spectra best matched that of PCB in this solvent system were only present at low levels (<50% of total) and were only poorly resolved from minor contaminants, so it was not possible to obtain pure products for detailed characterization. Based on these results, we conclude that PcyA can recognize BV-DA as substrate only at low pH, but that its metabolism is inefficient.
We also examined the ability of PcyA to utilize BV13-DA and BV13-MA as substrates (Supp. Fig. 2A). While BV13 diacid was converted into iso-PФB product with moderate yield, the diamide proved inert at both low and high pH (Supp. Fig. 2A and data not shown). By contrast, BV13-MA gave rise to a mixture of products upon reaction with PcyA. With biliverdins containing a single endo-vinyl group, such as BV IXα, PcyA can generate a mixture of 3E and 3Z products (Fig. 2; Supp. Fig. 2B). BV13 contains two endo-vinyl groups (Supp. Fig. 2C), so PcyA could convert either (or both) to an ethylidene. Since BV13 and BV13-DA are both symmetrical (Supp. Fig. 2C), due to their equivalent endo-vinyl groups, only two products (3E and 3Z) are expected upon 2-electron reduction. BV13-MA is no longer symmetrical, so equivalent reduction of one of the two endo-vinyl groups can give rise to 4 possible products. In practice, it was not possible to resolve these products from each other for detailed characterization. We conclude that the propionate sidechains are not critical for BV reduction by PcyA, in keeping with the paucity of conserved interactions with these moieties in FDBR crystal structures (63-66), but that at least one propionate is important for efficient binding and/or catalysis.
Assembly of bilin monoamides with apophytochromes
The efficient reduction of BV-8MA and BV-12MA by PcyA allowed us to prepare PCB-8MA and PCB-12MA in sufficient quantity for incorporation into phytochromes. Along with the corresponding 8- and 12-monoamide isomers of BV, these compounds enabled comparative studies of the roles of the propionates in assembly and photochemistry of both BV-type (DrBphP) and PCB-type (Cph1) phytochromes. In a previous study, we reported that bilin 12-monoamides could assemble with both Cph1 and DrBphP apoproteins to yield novel “dual-Pr” ground states, apparently arising from formation of two spectroscopically distinct species in equilibrium with each other (45). We also reported that the dual-Pr states of DrBphP:BV-12MA could be simultaneously converted to dual-Pfr states by irradiation of either dual-Pr peak, indicating that equilibration of the two states was rapid; photoconversion of the dual-Pfr states of this adduct demonstrated that they were also in rapid equilibrium with each other (45). To extend these results, both classes of apophytochrome were incubated with the corresponding 8-monoamides (Fig. 3), followed by overnight dialysis to remove any residual chromophore that had not formed a covalent holophytochrome adduct. BV-8MA and PCB-8MA were both able to form covalent adducts with the appropriate apophytochrome (Supp. Fig. 4), although formation of DrBphP:BV-8MA adduct was clearly less efficient than formation of DrBphP:BV and DrBphP:BV-12MA adducts.
Figure 3.
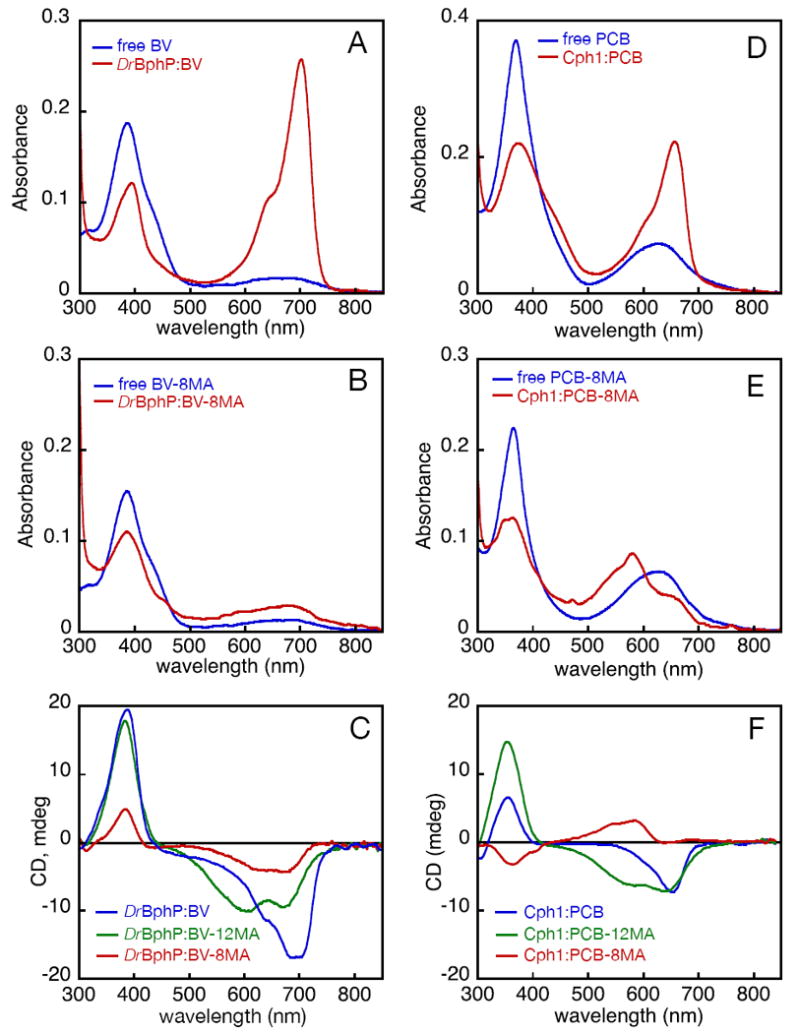
Assembly of apophytochromes with bilin 8-monoamides. (A) Absorbance spectra are shown for free BV (blue) and BV assembled with DrBphP and then dialyzed to remove unincorporated chromophore (DrBphP:BV, red) Starting amounts of BV were equal. (B) Absorbance spectra are shown for assembly of BV-8MA with DrBphP. Colors and sample handling are as in (A). (C) Adducts formed by assembly of DrBphP with BV (blue), BV-8MA (red), and BV-12MA (green) were analyzed by CD spectroscopy. (D) Absorbance spectra are shown for assembly of Cph1 with PCB. Colors, relative concentrations, and sample handling are as in (A). (E) Absorbance spectra are shown for assembly of Cph1 with PCB-8MA. Colors and sample handling are as in (A). (F) Adducts formed by assembly of Cph1 with PCB (blue), PCB-8MA (red), and PCB-12MA (green) were analyzed by CD spectroscopy.
As controls, assembly of DrBphP and Cph1 with diacid bilins was shown to give rise to the expected enhancement of the red absorbance band (Fig. 3), consistent with adoption of the extended, protonated Pr structure (28, 67-70). This enhancement was much weaker for DrBphP:BV-8MA (Fig. 3B). Indeed, the ratio of the red absorbance band intensity to the blue absorbance band intensity (R:B ratio) was 2.6 for in vitro assembled DrBphP with BV free acid, but only 0.5 with BV-8MA. The peak absorbance wavelengths of the 8-monoamide adduct were also blue-shifted relative to the diacid adduct (Supp. Table 6). The blue-shifted peak wavelengths and very low R:B ratio of DrBphP:BV-8MA are consistent with a cyclic and/or deprotonated chromophore (28). Incorporation of PCB-8MA into apoCph1 resulted in formation of a covalent adduct with an even more unusual spectrum (Fig. 3E). The Cph1:PCB-8MA adduct exhibited a much shorter peak absorbance wavelength than the diacid adduct (Supp. Table 6), with a reduced R:B ratio and a pronounced long-wavelength shoulder. This stands in marked contrast to native holophytochromes, which possess short-wavelength vibronic shoulders (71). The long-wavelength shoulder thus raises the possibility that Cph1:PCB-8MA adduct possesses a mix of multiple chromophore species, as appears to be the case for the 12-monoamide adducts (45).
To further characterize monoamide adducts of phytochromes, we characterized them by CD spectroscopy. The bilin π system is CD active, and it is known that phytobilin phytochromes such as Cph1 exhibit inversion of CD upon photoconversion from Pr to Pfr, while bacteriophytochromes such as DrBphP do not (45). Both DrBphP and Cph1 exhibit negative CD for the red band and positive CD for the blue band in the Pr state, in keeping with the similar Pr chromophore geometries observed in their crystal structures (68, 70). DrBphP:BV-8MA and DrBphP:BV-12MA also followed this pattern (Fig. 3C), with clear assignment of both dual-Pr peaks of DrBphP:BV-12MA as negative.
Cph1:PCB-12MA exhibited a similar CD spectrum for the dual-Pr ground state with both peaks exhibiting negative rotations (Fig. 3F). Surprisingly, Cph1:PCB-8MA possessed a different CD spectrum with its red absorbance maximum exhibiting a positive rotation, and the blue band exhibiting a negative rotation. This CD spectrum is similar to those of PCB and 15Z-PVB adducts of phycobiliproteins (72-74). We have proposed that PCB and BV CD spectra report disposition of the D-ring on one face of the bilin ring system or the other (29, 45), although other interpretations have also been proposed (75). Interestingly, the region of the CD spectrum corresponding to the longest-wavelength absorbance shoulder gave very weak signals (Fig. 3F), indicating the presence of either two species cancelling each other out or of a chromophore geometry associated with very weak CD. The distinct behavior of this region of the spectrum is consistent with a mix of at least two Cph1:PCB-8MA populations, one of which exhibits reversed CD relative to the diacid Pr state. These results demonstrate that a free 8-propionate is required for normal formation of the Pr state in both DrBphP and Cph1. In DrBphP, substitution of the appropriate 8-monoamide results in less efficient incorporation and in formation of a cyclic adduct, while in Cph1, a mix of species arises, including at least one species with opposite CD.
The propionates are nonessential for Pfr formation by DrBphP
We have previously demonstrated that the dual-Pr state of DrBphP-BV12MA can readily photoconvert to an apparent dual-Pfr state, while the dual-Pr state of Cph1 cannot (45). We were interested in further characterization of this process, so we examined the dual-Pfr state by CD spectroscopy (Fig. 4). In wild-type DrBphP, both Pr and Pfr exhibit negative CD for the red (Q) band and positive CD for the blue (Soret) band (45), with the Pfr state exhibiting weaker CD signals than the Pr state. We found that DrBphP:BV-12MA exhibits a similar pattern (Fig. 4C): both dual-Pfr peaks in the red/far-red region are associated with negative CD, and the CD signals for dual-Pfr are weaker than those of dual-Pr. The peak wavelengths measured in absorbance and CD spectroscopy are in reasonable agreement with each other (Supp. Table 6). In this study, most peak CD wavelengths for bands in the red/far-red region were red-shifted slightly relative to the corresponding absorbance peak wavelengths. This difference was also observed when CD spectra were compared to the absorbance spectra acquired simultaneously on the CD spectrometer itself (data not shown), indicating that it does not arise due to differences in machine calibration.
Figure 4.
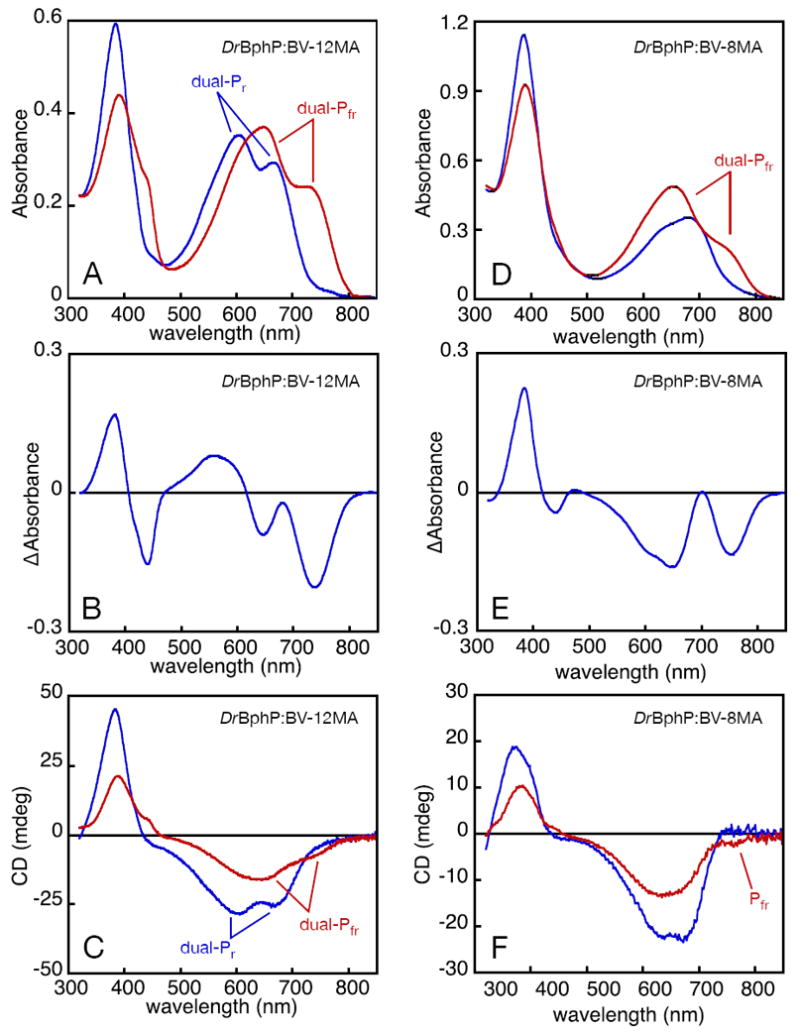
Photochemistry of DrBphP assembled with bilin monoamides. DrBphP:BV-12MA (A-C) and DrBphP:BV8-MA (D-F) were characterized by absorbance and CD spectroscopy. (A) DrBphP:BV-12MA was converted from dual-Pr (blue) to dual-Pfr (red) with 600 nm light (±5 nm), giving the difference spectrum shown in (B). (C) Both dual-Pr (blue) and dual-Pfr (red) were characterized by CD spectroscopy. Inversion of CD upon photoconversion was not observed, with both dual-Pfr red-band peaks exhibiting negative CD. (D) DrBphP:BV-8MA (blue) was illuminated with 670 nm light (± 20 nm), giving enhanced red absorbance and reduced blue absorbance (red). (E) The difference spectrum is shown for the spectra presented in (D). (F) Both the DrBphP:BV-8MA dark state (blue) and photoproduct (red) were characterized by CD spectroscopy. The photoproduct region corresponding to the Pfr state of DrBphP:BV was associated with negative CD, indicating no inversion of CD occurred. The photoproduct CD peak at shorter wavelength does not match the lineshape of the corresponding absorbance peak well (compare D), and the two peak wavelengths are not in very good agreement (Supp. Table 6). Assignment of this peak is therefore uncertain, as discussed in the text.
To assess the importance of the 8-propionate in DrBphP photoconversion, we examined the response of the thermally stable DrBphP:BV-8MA state to illumination of the red absorbance peak (Fig. 4). Interestingly, this illumination gave rise to enhanced red absorbance on either side of the illumination region, including far-red absorbance. This enhancement in red absorbance was balanced by a loss of absorbance in the Soret region, consistent with the idea that illumination produced either a more extended conformation or an increase in the ratio of extended to cyclic species in a mixed population. The photochemical difference spectrum shows the appearance of product(s) at 650 nm and 754 nm, with the latter being identical to the Dr-BV Pfr wavelength (Supp. Table 6). CD spectroscopy showed that this long-wavelength peak was associated with very weak but detectable negative CD, consistent with formation of a small amount of an authentic Pfr species. Interestingly, the region of the CD spectrum associated with the photochemical product at 650 nm exhibited a different peak wavelength (634 nm) and markedly different lineshape relative to the absorbance spectrum, raising the possibility that the spectra reflect multiple components in this region.
In both DrBphP:BV-8MA and DrBphP:BV-12MA, the amount of product formed at actual Pfr wavelengths was apparently less than that formed at other wavelengths. Such substoichiometric formation of far-red absorbance has also been reported in PcyA, in which it is not associated with photoisomerase activity but instead reflects the formation of lactim tautomers of the bilin chromophore (52, 76). We therefore sought to demonstrate the existence of authentic 15E chromophore in the Pfr states of the monoamide adducts. Denaturation of the holoprotein adduct under acidic conditions provides a useful assay for this purpose, both because of changes in peak wavelengths upon denaturation and because the 15E photoproduct can be photoconverted to the thermally stable 15Z form under acidic conditions (73, 74). We therefore examined the response of DrBphP:BV, DrBphP:BV-8MA, and DrBphP:BV-12MA to acidic guanidinium chloride (Fig. 5).
Figure 5.
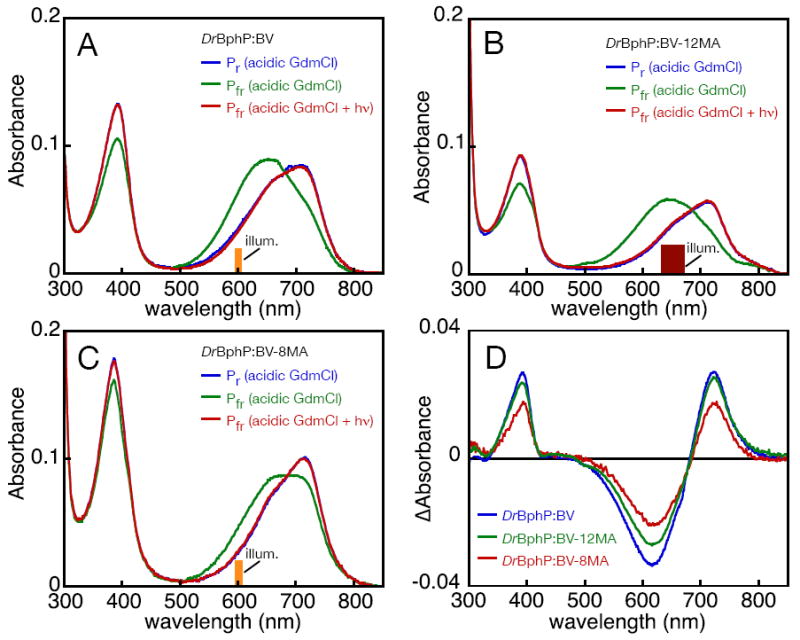
Denaturation analysis of DrBphP adducts. (A) DrBphP:BV was denatured in acidic guanidinium chloride in the native Pr (blue) or Pfr (green) states. Illumination of the denatured Pfr state with 600±5 nm light resulted in formation of a product (red) whose absorbance spectrum was equivalent to that of the denatured Pr sample. Similar results could be obtained by illumination of a denatured Pfr sample with 650 ±20 nm light (not shown). (B) Similar results were obtained with DrBphP:BV-12MA. Here, illumination with 650±20 nm light is shown; illumination with 600±5 nm light (not shown) gave similar results. (C) Similar experiments were performed with DrBphP:BV-8MA. The denatured Pfr DrBphP:BV-8MA seemed to be a mix of the denatured Pr and Pfr spectra seen with other adducts, suggesting that primary photochemistry was less efficient with DrBphP:BV-8MA than with other adducts. (D) Difference spectra are shown for photoconversion of the denatured Pfr adducts of DrBphP:BV (blue), DrBphP:BV-12MA (green), and DrBphP:BV-8MA (red).
Denaturation of the three DrBphP adducts in the thermally stable state gave rise to very similar spectra (Fig. 5), indicating that the distinct Pr spectra of DrBphP:BV-8MA and DrBphP:BV-12MA reflect structural differences that do not persist upon denaturation. Denaturation of the Pfr states of DrBphP:BV and DrBphP:BV-12MA also produced nearly identical blue-shifted spectra, consistent with the formation of the 15E isomer (73, 74). These results also demonstrate that the dual Pfr species of DrBphP:BV-12MA collapsed into a single species upon denaturation, as was the case for the two Pr species of this adduct. Photoconversion of the denatured Pfr DrBphP:BV and DrBphP:BV-12MA resulted in conversion of these species to species which were equivalent to those obtained by denaturation of the Pr states. The difference spectra for photoconversion of these adducts were strikingly similar (Fig. 5D), with very close peak/trough wavelengths (Supp. Table 7). Denaturation of DrBphP:BV-8MA in the Pfr state resulted in a somewhat different spectrum (Fig. 5C), although some absorbance did appear at shorter wavelengths. However, illumination of this sample resulted in formation of a spectrum identical to that of denatured Pr DrBphP:BV-8MA, with a comparable difference spectrum to those of DrBphP:BV and DrBphP:BV-12MA (Fig. 5D and Supp. Table 7). We therefore conclude that DrBphP:BV-8MA is capable of forming the 15E photoproduct, albeit less efficiently. The equivalent photochemical difference spectra observed in denatured DrBphP:BV, DrBphP:BV-8MA, and DrBphP:BV-12MA are consistent with a single primary photochemical event in all three adducts. Taken together with the spectroscopic characterization of the native adducts, we conclude that both DrBphP:BV-8MA and DrBphP:BV-12MA are able to form some Pfr, implying that neither propionate sidechain is essential for Pfr formation by DrBphP.
The propionates are dispensable for photoisomerization in Cph1
We have previously shown that Cph1:PCB-12MA is incapable of forming stable, detectable Pfr (45). To examine photoconversion in Cph1:PCB-8MA, we illuminated this adduct with several light sources matching regions corresponding to different populations detected by CD spectroscopy (see above). Irradiation of the long-wavelength shoulder of this adduct with 650±20 nm light resulted in detectable photochemistry (Fig. 5A), with a main product band appearing at 628 nm and a smaller band appearing at 705 nm in the photochemical difference spectrum (Fig. 5B). Interestingly, irradiation of this adduct at shorter wavelengths produced a much smaller amount of the 705 nm product (despite greater light absorption at this wavelength) along with little to no product at 628 nm (Fig. 5B and data not shown). This differential product formation as a function of illumination wavelength is consistent with our hypothesis that the Cph1:PCB-8MA adduct contains multiple populations of chromophore, although we cannot rule out the possibility that the shorter wavelength irradiation yields a reduced Pfr/Pr photoequilibrium ratio due to greater overlap of Pr and Pfr states.
A similar analysis of Cph1:PCB-12MA was undertaken to see whether this adduct might also contain such a mixture. As previously reported, illumination of Cph1:PCB-12MA with 650±20 nm light resulted in depletion of both dual-Pr peaks, with no loss of absorbance in a narrow region at approximately 610 nm (Fig. 6). Irradiation of this adduct with 550±35 nm light produced a smaller difference spectrum (Fig. 6D), but with similar features: depletion of both dual-Pr absorbance peaks and a similar constant absorbance at 610 nm. Thus, by contrast to the different populations of Cph1:PCB-8MA, the two populations of Cph1:PCB-12MA produce similar photochemical products.
Figure 6.
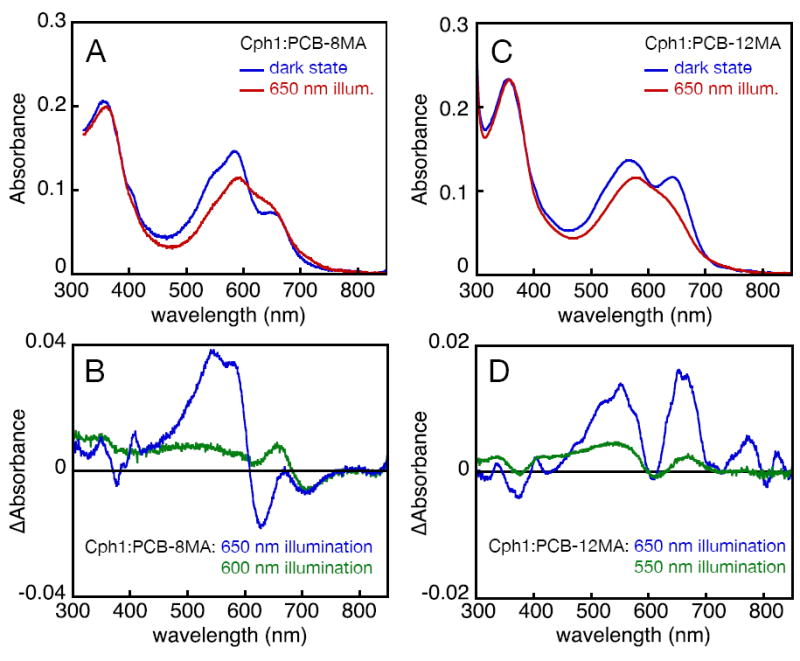
Photochemistry of Cph1 assembled with PCB monoamides. (A) Cph1:PCB-8MA (blue) was illuminated with 650±20 nm light to form photoproducts (red). (B) The difference spectrum for 650±20 nm illumination (blue) revealed appearance of products at 628 and 705 nm (Supp. Table 6). The difference spectra obtained upon illumination with 600±5 nm light (green) displayed different product ratios. (C) Cph1:PCB-12MA (blue) was illuminated with 650±20 nm light to form photoproducts (red). (D) The difference spectra obtained upon illumination of Cph1:PCB-12MA with 650±20 nm light (blue) or 550±35 nm light (green) indicated the presence of comparable changes.
The unchanging absorbance at 610 nm upon illumination of Cph1:PCB-12MA raised the possibility that this adduct produced a bona fide 15E bilin with peak absorbance in this region. To test this possibility and to further characterize Cph1:PCB-8MA, we subjected both adducts and wild-type Cph1-PCB to acidic denaturation (Fig. 7 and Supp. Table 7). The Pr and Pfr states of all three adducts gave rise to comparable absorbance spectra, and photoconversion of all three denatured Pfr samples resulted in formation of products with spectra equivalent to those of the denatured Pr samples. The resulting photochemical difference spectra were equivalent (Fig. 7D), indicating that all three chromophores were able to undergo primary photoisomerization in native Cph1 to give rise to 15E bilins. However, Cph1:PCB-12MA is unable to form detectable Pfr after primary photochemistry occurs, indicating a requirement for the 12-propionate sidechain in subsequent thermal steps leading to formation of stable Pfr. Cph1:PCB-8MA may be able to form very small amounts of Pfr, although the difficulty in producing PCB monoamides in large amounts preclude further characterization at this time.
Figure 7.
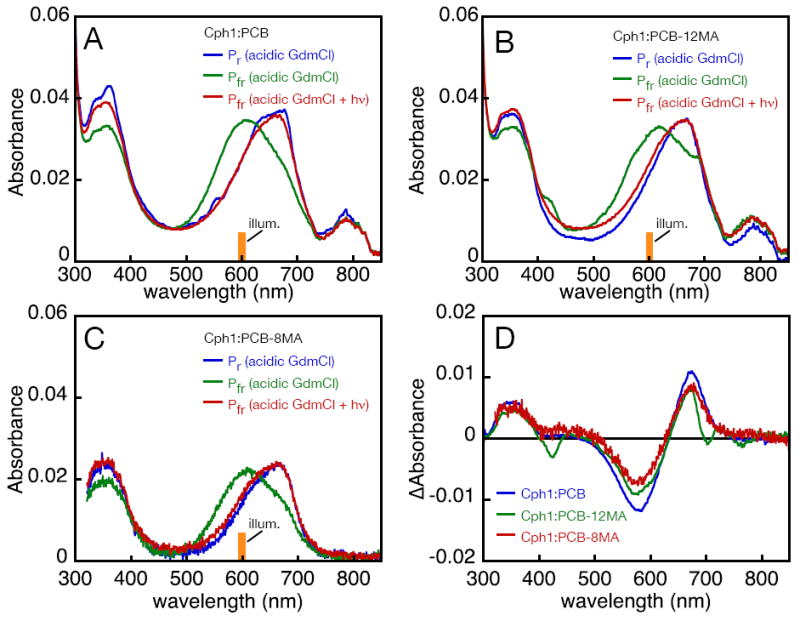
Denaturation analysis of Cph1 adducts. (A) Cph1:PCB was denatured in acidic guanidinium chloride in the native Pr (blue) or Pfr (green) states. Illumination of the denatured Pfr state with 600±5 nm light resulted in formation of a product (red) whose absorbance spectrum was equivalent to that of the denatured Pr sample. (B) Similar results were obtained with Cph1:PCB-12MA. The apparent absorbance peak at approximately 800 nm in (A) and (B) is associated with a contaminant in some lots of guanidine hydrochloride and is photochemically inert (not shown). (C) Similar results were obtained with Cph1:PCB-8MA. (D) Difference spectra are shown for photoconversion of the denatured Pfr adducts of Cph1:PCB (blue), Cph1:PCB-12MA (green), and Cph1:PCB-8MA (red).
DISCUSSION
Using co-acid-scrambling of appropriate rubin precursors with subsequent oxidation, we have successfully synthesized BV monoamides for examining the roles of tetrapyrrole propionic acid sidechains in ligand-protein interactions. BV monoamides are well-suited to such experiments, because they do not exhibit radically different solubility than the parent bilins in aqueous buffers and because amidation of one or both sidechains seems to have minimal effects on the electronic structure of the tetrapyrrole π system as judged by NMR spectroscopy, absorbance spectroscopy, and pKa measurements. Since the FDBR PcyA can readily convert BV monoamides into PCB monoamides, comparative studies using bilin monoamides in phytochromes thought to differ in their Pr-Pfr interconversion are also enabled (45). Interestingly, we were unable to generate monoamides via co-acid scrambling of BR diamide, because it exhibited very low solubility, which may explain why it also proved to be inert in co-acid-scrambling reactions. Bilirubin itself is known to form intramolecular hydrogen bonds between the propionate sidechains and the lactam moieties of the A- and D-rings in solution, giving rise to the ridge-tile conformation in organic solvents (77). BR diamide can still form such hydrogen bonds; indeed, the sidechain amides and the lactams can both supply donors and acceptors in BR diamide. Were such H bonds to form intermolecularly in BR diamide, the result could be poor solubility, as we have observed.
NADPH-dependent biliverdin reductases are less accommodating to amidation of the propionic acid sidechains of their BV substrate than the FDBR PcyA
We have assessed BV monoamides as substrates for representative NAD(P)H-dependent and ferredoxin-dependent biliverdin reductases. With rat BVR, substitution of either propionate resulted in a modest catalytic defect of approximately 10-fold, while biliverdin diamides were not metabolized. Cyanobacterial BvdR was also unable to recognize diamides as substrate; additionally, this enzyme exhibited more substantial catalytic defects (>50-fold) for reduction of the monoamides. The lack of reaction with either enzyme with biliverdin diamides indicates that one of the propionic acid sidechains must be ionizable for efficient binding to, or catalysis by, this family of enzymes. The lower pH optimum of BvdR (59) implies possible protonation of titratable groups in the substrate and/or active site, perhaps explaining the lower activity of this enzyme compared with BVR (Supp. Table 4). The more severe defect observed in reduction of monoamide substrates by BvdR than by BVR could arise for a similar reason: protonation of a single propionate in the monoamide at the lower pH optimum effectively mimics the inactive diamide substrate. Thus, with BVR, the high pH means that the propionate in the monoamide is more charged on average than at the low pH of BvdR. The lower activity of BvdR with monoamides thus reflects a lower concentration of the usable substrate (with deprotonated propionate). The requirement for at least one free propionate is consistent with the extensive literature on the substrate specificity of mammalian BVRs (78-84).
In contrast with the NAD(P)H-dependent enzymes, the FDBR PcyA is less specific for substrates with free propionate sidechains. BV monoamides are quite good substrates, giving effectively quantitative production of PCB monoamides. Moreover, PcyA can also catalyze reactions with the diamide analog of BV; however, reduction of BV-DA yields a mixture of unnatural products, and the reaction requires a lower pH. PcyA was unable to metabolize the unnatural BV13-DA isomer, which likely reflects its reduced binding affinity compared with the natural IXα isomer. The complex product mixture observed with BV13-MA as substrate appears to reflect distinct modes of binding to the enzyme. PcyA binds its substrate in a cyclic C5-Z,syn C10-Z,syn C15-Z,syn configuration, with the propionates facing out of the active site into solvent (64). In this configuration, BV13 could bind in either of two orientations, with the 3-vinyl group or the 17-vinyl group occupying the position of the 3-vinyl group of BV. For the symmetrical diacid, the two orientations would give rise to only two products, i.e. 3Z and 3E isomers of isoPФB. By contrast, BV13-MA is asymmetrical (Supp. Fig. 2C), so the two PcyA-binding orientations are expected to produce at least four distinct products if only one vinyl group is reduced. It is thus not surprising that BV13-MA gave rise to a number of products upon treatment with PcyA.
Phytochrome assembly experiments reveal distinct roles for 8- and 12-propionates in assembly and photoconversion of DrBphP and Cph1
The efficient production of PCB monoamides by PcyA has facilitated comparative examination of the roles played by the 8- and 12-propionates in two representative phytochromes, Cph1 and DrBphP. Both exhibit characteristic red/far-red reversible Pr-Pfr interconversions when associated with their natural chromophores, PCB for Cph1 and BV for DrBphP. Like plant phytochrome, Cph1 exhibits inversion of the red band CD upon formation of the Pfr state, while DrBphP and other BphPs do not (45, 67, 75, 85). Additionally, we have previously shown that the 12-propionate is required for formation of stable Pfr in Cph1, but not DrBphP. Thus, these two phytochromes serve as representatives for the two known classes of red/far-red photochemistry utilizing the knotted PAS-GAF-PHY photosensory core module (45).
In DrBphP, both monoamides are able to support formation of a small amount of the bona fide Pfr state as judged by absorbance and CD spectroscopy and by denaturation analysis. However, neither exhibits normal photochemistry. In this regard, DrBphP:BV-12MA exhibits dual-Pr and dual-Pfr, so the actual ability to sense the ratio of red to far-red light is altered. By comparison, DrBphP:BV-8MA adopts a more cyclic Pr state and also generates multiple products in the Pfr state. Denaturation analysis demonstrates that the primary photochemistry is the same as that of the parent diacid adduct for both monoamide adducts, but this photoisomerization seems less efficient for the 8-monoamide adduct, possibly due to the altered ground state conformation. This analysis also demonstrates that the dual peaks seen in the 12MA adduct collapse to a single peak upon denaturation, indicating that they differ in a property that is sensitive to denaturation, such as the degree of protonation or a thermally accessible isomerization, rather than due to some property that is unchanged by denaturation in aqueous acid, such as a shorter conjugated system eg. phycoviolobilin or phycoerythrobilin. In phytochromes utilizing BV as precursor, such as DrBphP, crystal structures provide good evidence that primary photochemistry occurs at the 15/16 bond (70, 86, 87). Therefore, neither propionate is required for photoisomerization of the 15/16 double bond in DrBphP, but both play roles in tuning the interconversion between Pr and Pfr. The 8-propionate is thus important for formation of the extended, red-enhanced Pr state and hence is very important for sensing red light physiologically, while the 12-propionate narrows the region that is sensed and reduces spectral overlap between the Pr and Pfr states.
For Cph1, we previous showed that the 12-monoamide adduct does not sustain formation of stable Pfr (45), even though its primary photochemistry proceeds efficiently (Figs 6 & 7). NMR and vibrational spectroscopy provide good evidence that the primary photochemistry entails Z-to-E isomerization of the 15/16 double bond (39, 88-91), although a controversial recent report has argued for photoisomerization of the 4/5 double bond in a “knotless phytochrome” of the Cph2 type (19, 92, 93) The 8-monoamide adduct gives rise to a complicated mixture in the ground state (Fig. 3). The components of this mixture apparently have different photochemical responses (Fig. 7), with at least one of these components giving rise to a very small amount of a species with a peak wavelength comparable to that of Pfr for the diacid chromophore. We have not been able to characterize the Cph1 monoamide photoproducts by CD spectroscopy due to the spectral overlap of the Pr and Pfr states. However, denaturation analysis confirms that both monoamide adducts are able to undergo apparently normal primary photochemistry, thus indicating that the multiple species observed in the ground state collapse to a single, common conformation upon denaturation. Therefore, like DrBphP, neither propionate is required for double bond photoisomerization for Cph1. Nevertheless, both propionates influence the interconversion between Pr and Pfr. The 12-propionate specifically tunes the spectral region sensed in the dark state, and also is required for formation of a bona fide Pfr. This implicates the 12-propionate in one or more of the light-independent steps that occur after initial formation of the 15E photoproduct to form Pfr in Cph1, which contrasts with no critical involvement for the 12-propionate in DrBphP.
The 8-propionate appears critical to the spectral range sensed by Cph1 in the Pr state. Additionally, the 8-monoamide Pr adduct possesses at least one population with inverted CD relative to the diacid and 12-monoamide adducts. Such an inversion suggests that the 8-monoamide D-ring is located on the opposite face of the bilin ring system, filling space normally occupied only in the Cph1 Pfr state (45, 68). Alternately, this signal could arise due to a drastic twist about C5, moving the A-ring much further to the α-face such that the positive CD signal from the α-facial A-ring is able to override the negative signal from the α-facial D-ring, or due to a “flipped” population that places the D-ring in the pocket normally occupied by the A-ring. This would invert the facial disposition of the D-ring and hence the CD of the chromophore. While such a population is not expected to be covalently bound, it remains formally possible that such a conformation could bind with sufficient affinity that it would remain associated with the Cph1 protein during dialysis.
Future perspectives
Our results indicate that bilin monoamides are useful research tools for characterizing protein-ligand interactions in biliproteins. They also show that the roles played by the propionates can vary within phytochromes, emphasizing the fact that red/far-red sensing by this important family of photosensory proteins can take place in more than one way. Fluorescent phytochrome mutants also hold promise as tools for in vivo imaging (21, 24, 26). Engineering such phytochromes to incorporate bilin diamides efficiently and preferentially might prove a valuable addition to such approaches, because the diamide chromophores are resistant to turnover by mammalian biliverdin reductases and hence may offer improved stability and sensitivity in imaging of mammalian tissue.
Supplementary Material
Acknowledgments
We thank Prof. Richard Vierstra (U. of Wisconsin-Madison) for the original DrBphP expression construct, Abigail Jang for construction of plasmid pBAD-Cph1FL-CBD, and Alex King for technical assistance in this project.
Footnotes
This work was supported by a grant from the National Institutes of Health GM068552 to J.C.L., by a subcontract from the National Science Foundation Center for Biophotonics Science and Technology PHY-0120999 to J. C. L., and by a research award from the UC Davis NMR facility.
Abbreviations: ΔAbsorbance, change in absorbance (in difference spectra, which are reported as 15Z - 15E); BR, bilirubin IXα; BR13, bilirubin XIIIα; BV, biliverdin IXα; BV-DA, biliverdin IXα diamide; BV-8MA, biliverdin IXα 8-monoamide; BV-12MA, biliverdin IXα 12-monoamide; BV13-DA, biliverdin XIIIα diamide; BV13-DA, biliverdin XIIIα monoamide; CD, circular dichroism; DDQ, 2,3-dichloro-5,6-dicyanobenzoquinone; DIEA, diisopropylethylamine; DME, dimethylester; DMF, dimethylformamide; FDBR, ferredoxin dependent bilin reductase; FNR, ferredoxin-NADP+ reductase; HBTU, O-benzotriazole-N,N,N’,N’-tetramethyluronium hexafluorophosphate; HOBt, hydroxybenzotriazole; HPLC, high performance liquid chromatography; MME, monomethylester; PCB, phycocyanobilin; PCB-8MA, phycocyanobilin 8-monoamide; PCB-12MA, phycocyanobilin 12-monoamide; PФB, phytochromobilin; Pr, redabsorbing state of phytochrome; Pfr, far-red-absorbing state of phytochrome, defined as a 15E chromophore red-shifted relative to the 15Z state; TLC, thin layer chromatography. Holoproteins are described throughout as the name of the protein followed by a colon and then by the bilin, as in DrBphP:BV (the BV adduct of DrBphP).
SUPPORTING INFORMATION AVAILABLE
Supporting information available online includes 7 figures and 7 tables. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Frankenberg NF, Lagarias JC. Biosynthesis and biological function of bilins. In: Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook Chlorophylls and Bilins: Biosynthesis Structure and Degradation. Academic Press; New York: 2003. pp. 211–235. [Google Scholar]
- 2.Foresti R, Green CJ, Motterlini R. Generation of bile pigments by haem oxygenase: a refined cellular strategy in response to stressful insults. Biochem Soc Symp. 2004:177–192. doi: 10.1042/bss0710177. [DOI] [PubMed] [Google Scholar]
- 3.Wilks A. Heme oxygenase: evolution, structure, and mechanism. Antioxid Redox Signal. 2002;4:603–614. doi: 10.1089/15230860260220102. [DOI] [PubMed] [Google Scholar]
- 4.McDonagh AF. Turning green to gold. Nature Struct Biol. 2001;8:198–200. doi: 10.1038/84915. [DOI] [PubMed] [Google Scholar]
- 5.Kapitulnik J, Maines MD. Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Tr Pharm Sci. 2009;30:129–137. doi: 10.1016/j.tips.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Frankenberg N, Mukougawa K, Kohchi T, Lagarias JC. Functional genomic analysis of the HY2 family of ferredoxin-dependent bilin reductases from oxygenic photosynthetic organisms. Plant Cell. 2001;13:965–978. doi: 10.1105/tpc.13.4.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dammeyer T, Frankenberg-Dinkel N. Function and distribution of bilin biosynthesis enzymes in photosynthetic organisms. Photochem Photobiol Sci. 2008;7:1121–1130. doi: 10.1039/b807209b. [DOI] [PubMed] [Google Scholar]
- 8.Wegele R, Tasler R, Zeng Y, Rivera M, Frankenberg-Dinkel N. The heme oxygenase(s)-phytochrome system of Pseudomonas aeruginosa. J Biol Chem. 2004;279:45791–45802. doi: 10.1074/jbc.M408303200. [DOI] [PubMed] [Google Scholar]
- 9.Montgomery BL, Lagarias JC. Phytochrome ancestry. Sensors of bilins and light. Tr Plant Sci. 2002;7:357–366. doi: 10.1016/s1360-1385(02)02304-x. [DOI] [PubMed] [Google Scholar]
- 10.Karniol B, Wagner JR, Walker JM, Vierstra RD. Phylogenetic analysis of the phytochrome superfamily reveals distinct microbial subfamilies of photoreceptors. Biochem J. 2005;392:103–116. doi: 10.1042/BJ20050826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamola AA, Blumberg WE, McClead R, Fanaroff A. Photoisomerized bilirubin in blood from infants receiving phototherapy. Proc Natl Acad Sci USA. 1981;78:1882–1886. doi: 10.1073/pnas.78.3.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDonagh AF, Palms LA, Trull FR, Lightner DA. Phototherapy for neonatal jaundice. Configurational isomers of bilirubin. J Am Chem Soc. 1982;104:6865–6869. [Google Scholar]
- 13.Zunszain PA, Ghuman J, McDonagh AF, Curry S. Crystallographic analysis of human serum albumin complexed with 4Z,15E-bilirubin-IXalpha. J Mol Biol. 2008;381:394–406. doi: 10.1016/j.jmb.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapitulnik J. Bilirubin: an endogenous product of heme degradation with both cytotoxic and cytoprotective properties. Mol Pharmacol. 2004;66:773–779. doi: 10.1124/mol.104.002832. [DOI] [PubMed] [Google Scholar]
- 15.Glazer AN. Phycobiliproteins. Meth Enzymol. 1988;167:291–303. doi: 10.1016/0076-6879(88)67034-0. [DOI] [PubMed] [Google Scholar]
- 16.Scheer H, Zhao KH. Biliprotein maturation: the chromophore attachment. Mol Microbiol. 2008;68:263–276. doi: 10.1111/j.1365-2958.2008.06160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rockwell NC, Su YS, Lagarias JC. Phytochrome structure and signaling mechanisms. Ann Rev Plant Biol. 2006;57:837–858. doi: 10.1146/annurev.arplant.56.032604.144208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giraud E, Vermeglio A. Bacteriophytochromes in anoxygenic photosynthetic bacteria. Photosynth Res. 2008;97:141–153. doi: 10.1007/s11120-008-9323-0. [DOI] [PubMed] [Google Scholar]
- 19.Rockwell NC, Lagarias JC. A Brief History of Phytochromes. Chemphyschem. 2010;11:1172–1180. doi: 10.1002/cphc.200900894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scheerer P, Michael N, Park JH, Nagano S, Choe HW, Inomata K, Borucki B, Krauss N, Lamparter T. Light-induced conformational changes of the chromophore and the protein in phytochromes: bacterial phytochromes as model systems. Chemphyschem. 2010;11:1090–1105. doi: 10.1002/cphc.200900913. [DOI] [PubMed] [Google Scholar]
- 21.Murphy JT, Lagarias JC. The Phytofluors: A new class of fluorescent protein probes. Curr Biol. 1997;7:870–876. doi: 10.1016/s0960-9822(06)00375-7. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu-Sato S, Huq E, Tepperman JM, Quail PH. A light-switchable gene promoter system. Nat Biotech. 2002;20:1041–1044. doi: 10.1038/nbt734. [DOI] [PubMed] [Google Scholar]
- 23.Fischer AJ, Lagarias JC. Harnessing phytochrome’s glowing potential. Proc Natl Acad Sci USA. 2004;101:17334–17339. doi: 10.1073/pnas.0407645101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su YS, Lagarias JC. Light independent phytochrome signaling mediated by dominant GAF-domain tyrosine mutants of Arabidopsis phytochromes in transgenic plants. Plant Cell. 2007;19:2124–2139. doi: 10.1105/tpc.107.051516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leung DW, Otomo C, Chory J, Rosen MK. Genetically encoded photoswitching of actin assembly through the Cdc42-WASP-Arp2/3 complex pathway. Proc Natl Acad Sci USA. 2008;105:12797–12802. doi: 10.1073/pnas.0801232105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shu X, Royant A, Lin MZ, Aguilera TA, Lev-Ram V, Steinbach PA, Tsien RY. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science. 2009;324:804–807. doi: 10.1126/science.1168683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischer AJ, Rockwell NC, Jang AY, Ernst LA, Waggoner AS, Duan Y, Lei H, Lagarias JC. Multiple roles of a conserved GAF domain tyrosine residue in cyanobacterial and plant phytochromes. Biochem (ACS) 2005;44:15203–15215. doi: 10.1021/bi051633z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rockwell NC, Njuguna SL, Roberts L, Castillo E, Parson VL, Dwojak S, Lagarias JC, Spiller SC. A second conserved GAF domain cysteine is required for the blue/green photoreversibility of cyanobacteriochrome Tlr0924 from Thermosynechococcus elongatus. Biochem (ACS) 2008;47:7304–7316. doi: 10.1021/bi800088t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner JR, Zhang J, von Stetten D, Gunther M, Murgida DH, Mroginski MA, Walker JM, Forest KT, Hildebrandt P, Vierstra RD. Mutational analysis of Deinococcus radiodurans bacteriophytochrome reveals key amino acids necessary for the photochromicity and proton exchange cycle of phytochromes. J Biol Chem. 2008;283:12212–12226. doi: 10.1074/jbc.M709355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ulijasz AT, Cornilescu G, von Stetten D, Cornilescu C, Velazquez Escobar F, Zhang J, Stankey RJ, Rivera M, Hildebrandt P, Vierstra RD. Cyanochromes are blue/green light photoreversible photoreceptors defined by a stable double cysteine linkage to a phycoviolobilin-type chromophore. J Biol Chem. 2009;284:29757–29772. doi: 10.1074/jbc.M109.038513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hahn J, Strauss HM, Landgraf FT, Gimenez HF, Lochnit G, Schmieder P, Hughes J. Probing protein-chromophore interactions in Cph1 phytochrome by mutagenesis. FEBS J. 2006;273:1415–1429. doi: 10.1111/j.1742-4658.2006.05164.x. [DOI] [PubMed] [Google Scholar]
- 33.von Stetten D, Seibeck S, Michael N, Scheerer P, Mroginski MA, Murgida DH, Krauss N, Heyn MP, Hildebrandt P, Borucki B, Lamparter T. Highly conserved residues Asp-197 and His-250 in Agp1 phytochrome control the proton affinity of the chromophore and Pfr formation. J Biol Chem. 2007;282:2116–2123. doi: 10.1074/jbc.M608878200. [DOI] [PubMed] [Google Scholar]
- 34.Yang X, Stojkovic EA, Kuk J, Moffat K. Crystal structure of the chromophore binding domain of an unusual bacteriophytochrome, RpBphP3, reveals residues that modulate photoconversion. Proc Natl Acad Sci USA. 2007;104:12571–12576. doi: 10.1073/pnas.0701737104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ikeuchi M, Ishizuka T. Cyanobacteriochromes: a new superfamily of tetrapyrrole-binding photoreceptors in cyanobacteria. Photochem Photobiol Sci. 2008;7:1159–1167. doi: 10.1039/b802660m. [DOI] [PubMed] [Google Scholar]
- 36.Hanzawa H, Inomata K, Kinoshita H, Kakiuchi T, Jayasundera KP, Sawamoto D, Ohta A, Uchida K, Wada K, Furuya M. In vitro assembly of phytochrome B apoprotein with synthetic analogs of the phytochrome chromophore. Proc Natl Acad Sci USA. 2001;98:3612–3617. doi: 10.1073/pnas.051629698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanzawa H, Shinomura T, Inomata K, Kakiuchi T, Kinoshita H, Wada K, Furuya M. Structural requirement of bilin chromophore for the photosensory specificity of phytochromes A and B. Proc Natl Acad Sci USA. 2002;99:4725–4729. doi: 10.1073/pnas.062713399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inomata K. Studies on the structure and function of phytochromes as photoreceptors based on synthetic organic chemistry. Bull Chem Soc Japan. 2008;81:25–59. [Google Scholar]
- 39.Bongards C, Gärtner W. The Role of the Chromophore in the Biological Photoreceptor Phytochrome: An Approach Using Chemically Synthesized Tetrapyrroles. Acc Chem Res. 2010;43:485–495. doi: 10.1021/ar800133x. [DOI] [PubMed] [Google Scholar]
- 40.Elich TD, Lagarias JC. Formation of a photoreversible phycocyanobilin-apophytochrome adduct in vitro. J Biol Chem. 1989;264:12902–12908. [PubMed] [Google Scholar]
- 41.Elich TD, McDonagh AF, Palma LA, Lagarias JC. Phytochrome chromophore biosynthesis. Treatment of tetrapyrrole-deficient Avena explants with natural and non-natural bilatrienes leads to formation of spectrally active holoproteins. J Biol Chem. 1989;264:183–189. [PubMed] [Google Scholar]
- 42.Li L, Lagarias JC. Phytochrome assembly - Defining chromophore structural requirements for covalent attachment and photoreversibility. J Biol Chem. 1992;267:19204–19210. [PubMed] [Google Scholar]
- 43.Bhoo SH, Hirano T, Jeong HY, Lee JG, Furuya M, Song PS. Phytochrome photochromism probed by site-directed mutations and chromophore esterification. J Am Chem Soc. 1997;119:11717–11718. [Google Scholar]
- 44.Kami C, Mukougawa K, Muramoto T, Yokota A, Shinomura T, Lagarias JC, Kohchi T. Complementation of phytochrome chromophore-deficient Arabidopsis by expression of phycocyanobilin:ferredoxin oxidoreductase. Proc Natl Acad Sci USA. 2004;101:1099–1104. doi: 10.1073/pnas.0307615100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rockwell NC, Shang L, Martin SS, Lagarias JC. Distinct classes of red/far-red photochemistry within the phytochrome superfamily. Proc Natl Acad Sci USA. 2009;106:6123–6127. doi: 10.1073/pnas.0902370106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma JS, Lightner DA. Facile Preparation of Symmetric Bilirubins IIIα and XIIIα from IXα. J Hetero Chem. 1984;21:1005–1008. [Google Scholar]
- 47.McDonagh AF. Bile Pigments: Bilatrienes and 5,15-Biladienes. In: Dolphin D, editor. The Porphyrins. Academic Press; New York: 1979. pp. 293–491. [Google Scholar]
- 48.Smith PK, Krohn RI, Hemanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olsen BJ, Klenk DC. Measurement of Protein using Bicinchoninic Acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 49.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein using the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 50.Lindner I, Knipp B, Braslavsky SE, Gärtner W, Schaffner K. A novel chromophore selectively modifies the spectral properties of one of the two stable states of the plant photoreceptor phytochrome. Angew Chem Intl Ed. 1998;37:1843–1846. [Google Scholar]
- 51.Frankenberg N, Lagarias JC. Phycocyanobilin:ferredoxin oxidoreductase. Biochemical and spectroscopic characterization. J Biol Chem. 2003;278:9219–9226. doi: 10.1074/jbc.M211643200. [DOI] [PubMed] [Google Scholar]
- 52.Tu S-L, Gunn A, Toney MD, Britt RD, Lagarias JC. Biliverdin reduction by cyanobacterial phycocyanobilin:ferredoxin oxidoreductase (PcyA) proceeds via linear tetrapyrrole radical intermediates. J Am Chem Soc. 2004;126:8682–8693. doi: 10.1021/ja049280z. [DOI] [PubMed] [Google Scholar]
- 53.Schluchter WM. Department of Biochemistry and Molecular Biology. Pennsylvania State University; University Park: 1994. The Characterization of Photosystem I and Ferredoxin-NADP+ Oxidoreductase in the Cyanobacterium Synechococcus sp. PCC 7002; p. 300. [Google Scholar]
- 54.Whitby FG, Phillips JD, Hill CP, McCoubrey W, Maines MD. Crystal structure of a biliverdin IXalpha reductase enzyme-cofactor complex. J Mol Biol. 2002;319:1199–1210. doi: 10.1016/S0022-2836(02)00383-2. [DOI] [PubMed] [Google Scholar]
- 55.Murphy JT, Lagarias JC. Purification and characterization of recombinant affinity peptide-tagged oat phytochrome A. Photochem Photobiol. 1997;65:750–758. doi: 10.1111/j.1751-1097.1997.tb01920.x. [DOI] [PubMed] [Google Scholar]
- 56.McDonagh AF, Assisi F. Direct evidence for the acid-catalysed isomeric scrambling of bilirubin IXα. J Chem Soc Chem Comm. 1972;1972:117–119. [Google Scholar]
- 57.Lightner DA, Holmes DL, McDonagh AF. On the Acid Dissociation Constants of Bilirubin and Biliverdin - pKa Values from C-13 Nmr Spectroscopy. J Biol Chem. 1996;271:2397–2405. doi: 10.1074/jbc.271.5.2397. [DOI] [PubMed] [Google Scholar]
- 58.Maines MD. New insights into biliverdin reductase functions: linking heme metabolism to cell signaling. Physiol (Bethesda) 2005;20:382–389. doi: 10.1152/physiol.00029.2005. [DOI] [PubMed] [Google Scholar]
- 59.Schluchter WM, Glazer AN. Characterization of cyanobacterial biliverdin reductase - Conversion of biliverdin to bilirubin is important for normal phycobiliprotein biosynthesis. J Biol Chem. 1997;272:13562–13569. doi: 10.1074/jbc.272.21.13562. [DOI] [PubMed] [Google Scholar]
- 60.Franklin E, Browne S, Hayes J, Boland C, Dunne A, Elliot G, Mantle TJ. Activation of biliverdin-IXalpha reductase by inorganic phosphate and related anions. Biochem J. 2007;405:61–67. doi: 10.1042/BJ20061651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hayes JM, Mantle TJ. The effect of pH on the initial rate kinetics of the dimeric biliverdin-IXalpha reductase from the cyanobacterium Synechocystis PCC6803. FEBS J. 2009;276:4414–4425. doi: 10.1111/j.1742-4658.2009.07149.x. [DOI] [PubMed] [Google Scholar]
- 62.Fakhrai H, Maines MD. Expression and characterization of a cDNA for rat kidney biliverdin reductase. J Biol Chem. 1992;267:4023–4029. [PubMed] [Google Scholar]
- 63.Hagiwara Y, Sugishima M, Khawn H, Kinoshita H, Inomata K, Shang L, Lagarias JC, Takahashi Y, Fukuyama K. Structural insights into vinyl reduction regiospecificity of phycocyanobilin:ferredoxin oxidoreductase (PcyA) J Biol Chem. 2010;285:1000–1007. doi: 10.1074/jbc.M109.055632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hagiwara Y, Sugishima M, Takahashi Y, Fukuyama K. Crystal structure of phycocyanobilin:ferredoxin oxidoreductase in complex with biliverdin IX{alpha}, a key enzyme in the biosynthesis of phycocyanobilin. Proc Natl Acad Sci USA. 2006;103:27–32. doi: 10.1073/pnas.0507266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hagiwara Y, Sugishima M, Takahashi Y, Fukuyama K. Induced-fitting and electrostatic potential change of PcyA upon substrate binding demonstrated by the crystal structure of the substrate-free form. FEBS Lett. 2006;580:3823–3828. doi: 10.1016/j.febslet.2006.05.075. [DOI] [PubMed] [Google Scholar]
- 66.Tu SL, Fisher AJ, Rockwell NC, Lagarias JC. Insight into the radical mechanism of phycocyanobilin:ferredoxin oxidoreductase (PcyA) revealed by x-ray crystallography and biochemical analysis. Biochem (ACS) 2007;46:1484–1494. doi: 10.1021/bi062038f. [DOI] [PubMed] [Google Scholar]
- 67.Borucki B, Otto H, Rottwinkel G, Hughes J, Heyn MP, Lamparter T. Mechanism of Cph1 phytochrome assembly from stopped-flow kinetics and circular dichroism. Biochem (ACS) 2003;42:13684–13697. doi: 10.1021/bi035511n. [DOI] [PubMed] [Google Scholar]
- 68.Essen LO, Mailliet J, Hughes J. The structure of a complete phytochrome sensory module in the Pr ground state. Proc Natl Acad Sci USA. 2008;105:14709–14714. doi: 10.1073/pnas.0806477105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goller AH, Strehlow D, Hermann G. The excited-state chemistry of phycocyanobilin: a semiempirical study. Chemphyschem. 2005;6:1259–1268. doi: 10.1002/cphc.200400667. [DOI] [PubMed] [Google Scholar]
- 70.Wagner JR, Zhang J, Brunzelle JS, Vierstra RD, Forest KT. High resolution structure of Deinococcus bacteriophytochrome yields new insights into phytochrome architecture and evolution. J Biol Chem. 2007;282:12298–12309. doi: 10.1074/jbc.M611824200. [DOI] [PubMed] [Google Scholar]
- 71.Spillane KM, Dasgupta J, Lagarias JC, Mathies RA. Homogeneity of phytochrome Cph1 vibronic absorption revealed by resonance Raman intensity analysis. J Am Chem Soc. 2009;131:13946–13948. doi: 10.1021/ja905822m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mimuro M, Füglistaller P, Rümbeli R, Zuber H. Functional assignment of chromophores and energy transfer in C phycocyanin isolated from the thermophilic cyanobacterium Mastigocladus laminosus. Biochim Biophys Acta. 1986;848:155–166. [Google Scholar]
- 73.Zhao KH, Haessner R, Cmiel E, Scheer H. Type I Reversible Photochemistry of Phycoerythrocyanin Involves Z/E-Isomerization of Alpha-84 Phycoviolobilin Chromophore. Biochim Biophys Acta Bioenerg. 1995;1228:235–243. [Google Scholar]
- 74.Zhao KH, Scheer H. Type I and Type II Reversible Photochemistry of Phycoerythrocyanin Alpha-Subunit From Mastigocladus Laminosus Both Involve Z, E Isomerization of Phycoviolobilin Chromophore and Are Controlled By Sulfhydryls in Apoprotein. Biochim Biophys Acta Bioenerg. 1995;1228:244–253. [Google Scholar]
- 75.Borucki B, Seibeck S, Heyn MP, Lamparter T. Characterization of the covalent and noncovalent adducts of Agp1 phytochrome assembled with biliverdin and phycocyanobilin by circular dichroism and flash photolysis. Biochem (ACS) 2009;48:6305–6317. doi: 10.1021/bi900436v. [DOI] [PubMed] [Google Scholar]
- 76.Stoll S, Gunn A, Brynda M, Sughrue W, Kohler AC, Ozarowski A, Fisher AJ, Lagarias JC, Britt RD. Structure of the biliverdin radical intermediate in phycocyanobilin:ferredoxin oxidoreductase identified by high-field EPR and DFT. J Am Chem Soc. 2009;131:1986–1995. doi: 10.1021/ja808573f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nogales D, Lightner DA. On the structure of bilirubin in solution. 13C[1H] heteronuclear Overhauser effect NMR analyses in aqueous buffer and organic solvents. J Biol Chem. 1995;270:73–77. doi: 10.1074/jbc.270.1.73. [DOI] [PubMed] [Google Scholar]
- 78.Awruch J, Tomaro ML, Frydman RB, Frydman B. The specificity of biliverdin reductase. The reduction of biliverdin XIII isomers. Biochim Biophys Acta. 1984;787:146–151. doi: 10.1016/0167-4838(84)90073-6. [DOI] [PubMed] [Google Scholar]
- 79.Tomaro ML, Frydman RB, Awruch J, Valasinas A, Frydman B, Pandey RK, Smith KM. The specificity of biliverdin reductase. A study with different biliverdin types. Biochim Biophys Acta. 1984;791:350–356. doi: 10.1016/0167-4838(84)90346-7. [DOI] [PubMed] [Google Scholar]
- 80.Frydman RB, Tomaro ML, Rosenfeld J, Awruch J, Sambrotta L, Valasinas A, Frydman B. Biliverdin reductase: substrate specificity and kinetics. Biochim Biophys Acta. 1987;916:500–511. doi: 10.1016/0167-4838(87)90197-x. [DOI] [PubMed] [Google Scholar]
- 81.Frydman RB, Bari S, Tomaro ML, Frydman B. The Enzymatic and Chemical Reduction Of Extended Biliverdins. Biochem Biophys Res Comm. 1990;171:465–473. doi: 10.1016/0006-291x(90)91416-p. [DOI] [PubMed] [Google Scholar]
- 82.Terry MJ, Maines MD, Lagarias JC. Inactivation of Phytochrome-Chromophore and Phycobiliprotein-Chromophore Precursors by Rat Liver Biliverdin Reductase. J Biol Chem. 1993;268:26099–26106. [PubMed] [Google Scholar]
- 83.Cunningham O, Dunne A, Sabido P, Lightner D, Mantle TJ. Studies on the specificity of the tetrapyrrole substrate for human biliverdin-IX alpha reductase and biliverdin-IX beta reductase - Structure-activity relationships define models for both active sites. J Biol Chem. 2000;275:19009–19017. doi: 10.1074/jbc.275.25.19009. [DOI] [PubMed] [Google Scholar]
- 84.Franklin EM, Browne S, Horan AM, Inomata K, Hammam MA, Kinoshita H, Lamparter T, Golfis G, Mantle TJ. The use of synthetic linear tetrapyrroles to probe the verdin sites of human biliverdin-IXalpha reductase and human biliverdin-IXbeta reductase. FEBS J. 2009;276:4405–4413. doi: 10.1111/j.1742-4658.2009.07148.x. [DOI] [PubMed] [Google Scholar]
- 85.Litts JC, Kelly JM, Lagarias JC. Structure-function studies on phytochrome. Preliminary characterization of highly purified phytochrome from Avena sativa enriched in the 124-kilodalton species. J Biol Chem. 1983;258:11025–11031. [PubMed] [Google Scholar]
- 86.Yang X, Kuk J, Moffat K. Crystal structure of Pseudomonas aeruginosa bacteriophytochrome: photoconversion and signal transduction. Proc Natl Acad Sci USA. 2008;105:14715–14720. doi: 10.1073/pnas.0806718105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang X, Kuk J, Moffat K. Conformational differences between the Pfr and Pr states in Pseudomonas aeruginosa bacteriophytochrome. Proc Natl Acad Sci USA. 2009;106:15639–15644. doi: 10.1073/pnas.0902178106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dasgupta J, Frontiera RR, Taylor KC, Lagarias JC, Mathies RA. Ultrafast excited-state isomerization in phytochrome revealed by femtosecond stimulated Raman spectroscopy. Proc Natl Acad Sci USA. 2009;106:1784–1789. doi: 10.1073/pnas.0812056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mroginski MA, von Stetten D, Escobar FV, Strauss HM, Kaminski S, Scheerer P, Gunther M, Murgida DH, Schmieder P, Bongards C, Gartner W, Mailliet J, Hughes J, Essen LO, Hildebrandt P. Chromophore structure of cyanobacterial phytochrome Cph1 in the Pr state: reconciling structural and spectroscopic data by QM/MM calculations. Biophys J. 2009;96:4153–4163. doi: 10.1016/j.bpj.2009.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rohmer T, Lang C, Hughes J, Essen LO, Gartner W, Matysik J. Light-induced chromophore activity and signal transduction in phytochromes observed by 13C and 15N magic-angle spinning NMR. Proc Natl Acad Sci USA. 2008;105:15229–15234. doi: 10.1073/pnas.0805696105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Strauss HM, Hughes J, Schmieder P. Heteronuclear solution-state NMR studies of the chromophore in cyanobacterial phytochrome Cph1. Biochem (ACS) 2005;44:8244–8250. doi: 10.1021/bi050457r. [DOI] [PubMed] [Google Scholar]
- 92.Ulijasz AT, Cornilescu G, Cornilescu CC, Zhang J, Rivera M, Markley JL, Vierstra RD. Structural basis for the photoconversion of a phytochrome to the activated Pfr form. Nature. 2010;463:250–254. doi: 10.1038/nature08671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hughes J. Phytochrome three-dimensional structures and functions. Biochem Soc Trans. 2010;38:710–716. doi: 10.1042/BST0380710. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.