

Abstract
Usher syndrome is the leading cause of combined deaf-blindness, but the molecular mechanisms underlying the auditory and visual impairment are poorly understood. Usher I is characterized by profound congenital hearing loss, vestibular dysfunction and progressive retinitis pigmentosa beginning in early adolescence. Using the c.216G>A cryptic splice site mutation in exon 3 of the USH1C gene found in Acadian Usher I patients in Louisiana, we constructed the first mouse model that develops both deafness and retinal degeneration. The same truncated mRNA transcript found in Usher 1C patients is found in the cochleae and retinas of these knock-in mice. Absent auditory-evoked brainstem responses indicated that the mutant mice are deaf at one month of age. Cochlear histology showed disorganized hair cell rows, abnormal bundles, and loss of both inner and outer hair cells in the middle turns and at the base. Retinal dysfunction as evident by an abnormal electroretinogram was seen as early as 1 month of age, with progressive loss of rod photoreceptors between 6 and 12 months of age. This knock-in mouse reproduces the dual sensory loss of human Usher I, providing a novel resource to study the disease mechanism and the development of therapies.
Keywords: Usher syndrome, deafness, retinal degeneration, mouse model
Introduction
The combination of hearing impairment and blindness is the hallmark of Usher syndrome, with an estimated world-wide prevalence of 1:16,000 - 50,000 (Keats and Corey, 1999). Three clinical subtypes of Usher syndrome (Usher I, II, and III) have been described, based on the severity and progression of deafness and retinitis pigmentosa (RP) as well as the presence or absence of vestibular function. Usher I is the most severe form with profound congenital deafness, vestibular defects and progressive retinitis pigmentosa beginning in early adolescence. Usher I is also the most genetically heterogeneous with seven different loci, named USH1B-H. Five of these genes have been identified as MYO7A (USH1B)(Weil et al., 1995; Weston et al., 1996; Adato et al., 1997; Levy et al., 1997; Liu et al., 1997), USH1C (Bitner-Glindzicz et al., 2000; Verpy et al., 2000), CDH23 (USH1D) (Bolz et al., 2001; Bork et al., 2001), PCDH15 (USH1F) (Ahmed et al., 2001; Alagramam et al., 2001; Bork et al., 2001) and SANS (USH1G) (Weil et al., 2003). These genes encode the proteins myosin VIIa, harmonin, cadherin 23, protocadherin 15 and SANS, respectively. Usher II is the most common form and three genes have been identified: they are USH2A (Eudy et al., 1998; Dreyer et al., 2000; Weston et al., 2000; van Wijk et al., 2004; Adato et al., 2005; Aller et al., 2006), which encodes the protein usherin; VLGR1b (USH2B), which encodes VLGR1b (very large G-protein coupled receptor 1b) (Weston et al., 2004); and WHRN (USH2D) (Ebermann et al., 2007), which encodes the PDZ protein whirlin. One Usher III gene, USH3A, has been identified (Joensuu et al., 2001; Adato et al., 2002); it encodes the protein clarin-1.
Spontaneous mutations in Usher 1-related mouse homologs Myo7a, Ush1c, Cdh23, Pcdh15, and Sans all cause deafness and vestibular defects. However, none of these mouse models (shaker1 (sh1) (Gibson et al., 1995); deaf circler (dfcr) (Johnson et al., 2003); waltzer (v) (Di Palma et al., 2001; Wilson et al., 2001); Ames waltzer (av) (Alagramam et al., 2001; Wada et al., 2001); and Jackson shaker (js) (Kikkawa et al., 2003)) develop the retinitis pigmentosa found in Usher I patients. Furthermore, Ush1c knock-outs (Lefevre et al., 2008) and Ush1d chlorambucil-mutagenized models (Di Palma et al., 2001) also show deafness and vestibular defects without retinal degeneration.
Because none of the existing Usher I mouse models has retinal degeneration, they are considered to be incomplete models of the syndrome. Thus, we created a knock-in mouse containing the human USH1C c.216G>A mutation (Lentz et al., 2007) to test the hypothesis that two copies of this mutation were sufficient to develop the syndrome's dual sensory loss. This cryptic splice site mutation, found in Usher 1C Acadian patients from southwest Louisiana, introduces a new splice site at the end of exon three of the USH1C gene, and results in a frame shifted transcript with a 35 base pair deletion (Bitner-Glindzicz et al., 2000; Verpy et al., 2000; Lentz et al., 2005). The Ush1c216AA knock-in mice are homozygous for the same loss of 35 base pairs seen in patients (Lentz et al., 2007). Here we demonstrate that they also reproduce the auditory and visual defects found in Acadian Usher I patients.
Materials and Methods
Mouse Strains
All procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committees at Louisiana State University Health Sciences Center in New Orleans, Louisiana and the University of Washington in Seattle, Washington. For ERG and retinal histology, mice were of a mixed C57BL/6 and 129S6 background, maintained and expanded as a single colony through more than eight generations of sibling matings. For ABR and cochlear histology, these mice were backcrossed for at least five generations to CBA (approximately 95% CBA; 5% C57BL/6, 129S6) because recessive age-related hearing loss caused by a Cdh23 mutation is found in C57BL/6 mice (Erway et al., 1993; Noben-Trauth et al., 2003). Only mice that were homozygous for the wild type allele (Cdh23753G) were included in the study. All mutant mice were compared with appropriately age- and strain-matched control mice for each assay.
Genotyping mice for Ush1c216 and Cdh23753 alleles
Tail snip DNA was isolated, and the Ush1c216 genotypes (GG, GA, AA) were detected by PCR as reported by Lentz et al. (2007). PCR analysis was also used to detect the Cdh23753 genotypes (GG, GA, AA) (Noben-Trauth et al., 2003) with 1.25 U TaKaRa Ex Taq polymerase (Fisher Scientific, Waltham, MA) and 0.4 μM primers (CdhF 5′-ggacacccatcttcatcgtcaac-3′; CdhR 5′-gtcccttatcctggtccacagca-3′) flanking nucleotide 753 in exon 7. PCR products were gel purified and sequenced on an ABI 3100 using the fluorescent dideoxy terminator method.
Auditory-Evoked Brain Stem Response
Hearing thresholds of 1 month old mutant (Ush1c216AA) and control (Ush1c216GG) mice were measured by auditory-evoked brain stem response (ABR). Mice were anesthetized (ketamine, 100 mg/Kg; xylazine, 5 mg/Kg, i.p.), placed on an isothermal heating pad to maintain body temperature near 37°C, and positioned on a bite bar in a sound attenuating chamber. Subdermal Grass electrodes were used to record ABRs. The recording configuration included three electrodes, one inserted rostral to the left ear and positioned across the midline (vertex), a second inserted ventrolateral to the left ear, and a third reference electrode was placed subcutaneously in the left hind limb. Heart rate was observed during the recording to assist with anesthesia monitoring.
Sound stimuli were generated, and ABRs were digitized by computer using custom software and an audio card (Delta 410, M-Audio). A reference microphone (Larson-Davis #2530) and an acoustic calibrator (Cal200, Larson-Davis Acoustic Calibrator) were used to calibrate the system. The sound stimuli were amplified (Ramsa WP1200, Panasonic) and delivered by a free-field speaker (Vifa D25AG-05, DST). Three msec tone pip stimuli were repeated at 75 ms intervals with alternating polarity. ABRs were produced by averaging at least 350 responses, which were amplified (1000×) and filtered (0.3-3 kHz) by a pre-amplifier (P55; Grass-Telefactor). The threshold was defined using VDT (visual detection threshold) to determine the lowest sound pressure level (SPL, in 5 dB increments) in which a recognizable waveform was present and repeatable. Thresholds were determined at nine frequencies; 3.5, 4.0, 5.6, 8.0, 11.3, 16.0, 22.6, 32.0, and 40.0 kHz.
Tissue Preparation
Three different methods were used to study the cochleae of one month old mutant and control mice: (1) fluorescent labeling of microdissected whole-mount preparations of the organ of Corti; (2) fluorescent labeling of the intact cochlea followed by embedding in epoxy resin (Hardie et al., 2004); and (3) scanning electron microscopy (SEM). For experiments involving immunofluorescence processing (Methods 1 and 2), cochleae were isolated from the auditory bulla, the stapes was removed from the oval window, and a small opening was created in the apex. The cochleae were gently perfused with 4% paraformaldehyde in 0.1M phosphate buffer, pH 7.4, and post-fixed by overnight immersion in the same fixative at 4°C. Tissues were washed with PBS following fixation. For Method 1, segments (half turns) of the organ of Corti were carefully dissected free from the cochlea. The stria vascularis was pulled off or trimmed down, and the tectorial membrane was lifted free with fine forceps and discarded. For Method 2, an opening was created in the bone overlying the basal turn scala tympani. The cochleae were then decalcified in 0.5 M EDTA for four days, washed with PBS, and processed for immunohistochemistry as described below.
Antibodies
A mouse monoclonal anti-parvalbumin antibody (parv19, Cat. No. P3088, Sigma-Aldrich, St. Louis MO, 1:500; Sage et al., 2000) was used to label cochlear hair cells. A mouse monoclonal anti-neurofilament 200 kDa antibody (Cat. No. N0142, Sigma-Aldrich) was used at a dilution of 1:500 to label nerve fibers (Hardie et al., 2004).
Immunofluorescent Labeling
Isolated segments of organ of Corti (Method 1) were treated for 30 minutes with 0.1% saponin/0.1% Tween 20 (Sigma-Aldrich) in phosphate-buffered saline (PBS) in order to make membranes more permeable to antibodies. Nonspecific binding of the primary antibody was reduced by incubating tissues for 1 hour in a blocking solution consisting of 10% normal donkey serum /0.03% saponin/0.1% Triton X-100 (Sigma-Aldrich) in PBS. Primary antibody incubations were performed for 1 day at 4°C in PBS containing 0.03% saponin, 3% normal donkey serum (Vector Laboratories, Burlingame, CA), 2 mg/ml bovine serum albumin (Sigma-Aldrich), and 0.1% Triton x-100. Secondary antibodies derived from donkey and conjugated to Alexa 488, Alexa 568 (Invitrogen/Molecular Probes, Carlsbad, CA), or Cy5 (Jackson Immunoresearch, West Grove, PA) were used at a dilution of 1:200 in the same buffer for 2-4 hours (at room temperature) or overnight (at 4°C). For mouse antibodies against parvalbumin, the M.O.M. kit was used as specified by the manufacturer (Vector Laboratories). Tissues were washed after each antibody incubation (3 times for 10-15 min. each) in 0.1% Tween-20 in PBS. After counterstaining nuclei for 15 minutes in DAPI (Cat. No. D9542, Sigma-Aldrich, 1 μg/ml), specimens were mounted in Fluoromount-G™ (Cat. No. 0100-01, Southern Biotech, Birmingham AL), coverslipped, and examined with either epifluorescence or confocal fluorescence microscopy.
For Method 2, whole cochleae were processed in microcentrifuge tubes at 4 °C, with rotation, as follows. Tissues were incubated for 2 hours in a blocking solution consisting of 0.5% BSA, 10% normal donkey serum, and 0.1% Triton X-100 in PBS. Cochleae were then exposed to primary antibodies diluted 1:500 in blocking solution for 3 days. After extensive washing for 1 day, cochleae were incubated for 3 days in secondary antibodies from donkey and conjugated to Alexa 488, Alexa 568, Alexa 594 (Invitrogen/Molecular Probes) or Cy5 (Jackson Immunoresearch) diluted 1:500 in blocking solution. Following washes in PBS and PBS plus 0.1% Triton X-100, cochleae were dehydrated in a graded ethanol series and embedded in Spurr's epoxy resin (Polysciences, Warrington PA). Using a diamond wheel saw, the polymerized cochleae were hemisectioned in a plane through the modiolus to create two slabs, each containing the half turns of one side of the cochlea. The slabs were placed facedown into a thin film of immersion oil on a # 1.5 coverslip for imaging with confocal fluorescence microscopy.
Controls to verify the immunofluorescent labeling method and antibody specificity consisted of substituting non-immune sera for the primary antibody and using a series of dilutions of the primary antibody. In double-labeling experiments, antibodies raised in different species were used to avoid cross reactivity among secondary antibodies.
Immunofluorescent Imaging
Whole-mount and plastic embedded organ of Corti preparations were examined with a laser scanning confocal microscopic (Olympus FV-1000) attached to an IX-81 inverted microscope and equipped with a 405 nm diode laser, a multi-line argon laser (457 nm, 488 nm and 514 nm), a 561 nm solid state laser, and a 637 nm helium neon laser. The confocal microscope was controlled by Fluoview acquisition software, version 1.6a. Sequential image acquisition was performed to avoid bleed-through between channels. Files were imported into ImageJ and/or Adobe Photoshop for processing and analysis.
Scanning Electron Microscopy (SEM)
For Method 3, an intra-labyrinthine perfusion was performed with 2.5% glutaraldehyde/1% paraformaldehyde in 0.12 M phosphate buffer (pH 7.2–7.4) containing1.5% sucrose. Cochleae were post-fixed by immersion for 3 days in the same fixative at 4°C with gentle rotation. Tissues were washed three times in PBS for 30 minutes and post-fixed in 1% OsO4 in PBS for 40 minutes. After additional 30 minute washes in PBS, specimens were serially dehydrated in ethanol, dried in a critical point drier (Autosamdri – 814, Tousinis Research Corporation, Rockville, MD), and mounted on aluminum stubs. The bony capsule of the cochlea, spiral ligament, stria vascularis and Reissner's membrane were removed with a temporal bone drill and fine dissecting instruments. After exposure of the entire organ of Corti, specimens were coated in gold/palladium with a Hummer VIA sputter coater (Anatech Ltd., Alexandria, VA) and viewed on a JEOL JSM 6300 F scanning electron microscope. At least three individual animals representative of each experimental paradigm were analyzed for each method and each genotype.
Estimation of hair cell number in whole-mount preparations
Whole-mount cochlear preparations of fluorescently labeled Ush1c216AA mutant (n=5) and wild type (n=5) littermates were examined with the FV-1000 confocal microscope. Confocal image stacks were collected through the sensory epithelium in a plane parallel to the basilar lamina using 1.0 μm steps with a 20X/NA.75 Plan-Fluor objective lens, zoom = 5. Three-to-five confocal z-series were collected from each organ of Corti tissue segment. Segments were 125 μm in length and their location was measured in degrees from the extreme apex through the middle turn, including the apex, 90°, 180°, 240°, 300° and 360° from the extreme apex (Fig 3). The number of hair cells was counted and mean values were determined for each segment. Analysis of variance was carried out using the two-way Anova command in Origin (Origin v8.0).
Figure 3.
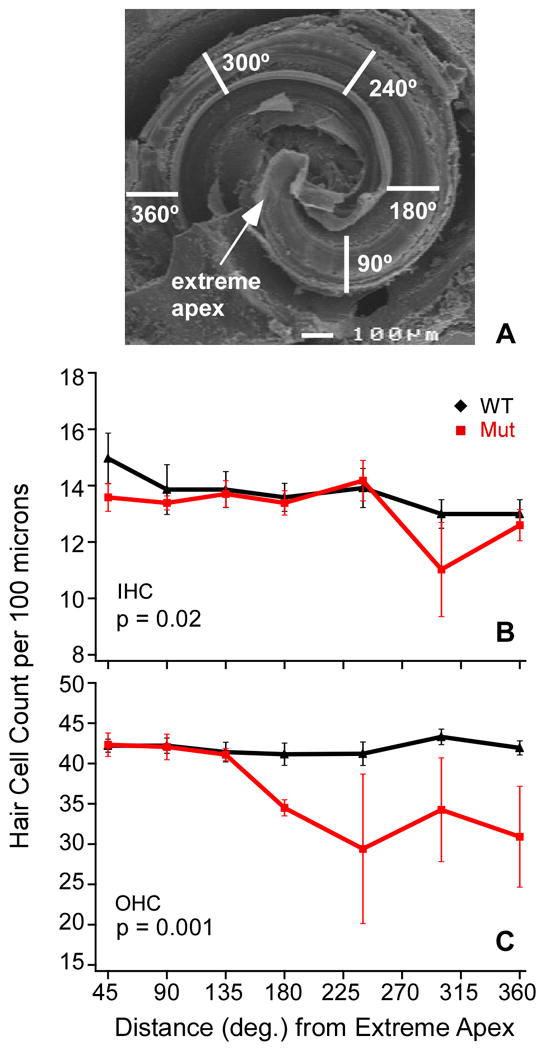
Average number of inner and outer hair cells per 100 μm longitudinal section of organ of Corti for wild type and Ush1c216AA mutant mice at P30. A Illustration of the region where cochlear hair cells were counted, scale bar = 100 μm. B,C Graphs show significantly fewer IHCs (B, p=.02) and OHCs (C, p=.001) in more basally located regions in Ush1c216AA mutant mice as compared with their wild type littermates (two-way Anova, Origin 8.0). (IHC, inner hair cells, OHC, outer hair cells) Error bars represent the standard error.
Electroretinography
Electroretinograms were recorded in Ush1c216AA mutant mice and their age-matched wild type littermate controls at 1 (n=12 wild type, n=4 mutants), 3 (n=12 wild type, n=6 mutant), 6.5 (n=18 wild type, n=12 mutants) and 12 months of age (n=9 wild type, n=5 mutants). Prior to ERG recording, mice were dark adapted overnight (12-16 hours) and anesthetized with an I.P. injection of 100 mg/kg ketamine and 6 mg/kg xylazine. Pupil dilation was accomplished with topical 1.0% atropine. Body temperature was maintained near 38°C with a heat pad (Harvard Apparatus). ERG's were recorded with a silver-silver chloride wire (0.03 o.d.) placed on the cornea; the subcutaneous reference and ground electrodes were placed in the forehead. A drop of 1% methylcellulose was placed on the cornea to prevent corneal desiccation and provide improved electrical contact.
Scotopic ERG responses were elicited with either short duration LED flashes or a Xenon strobe delivered in a Ganzfield dome (Espion, Diagnosys, LLC) with interstimulus intervals of 0.5 – 2 minutes, depending on the stimulus intensity. For flash intensities that elicited an a-wave response, flash duration was no longer than 50 μsec. At lower intensities that elicited the positive scotopic threshold response (pSTR) and the beginnings of the b-wave, flash duration was no longer than 1 msec. ERG responses were recorded from a series of flash intensities, which ranged from 0.0005 – 5000 cd-s/m2 in ¼ - ½ log steps. Two to four responses were averaged for each step depending on the stimulus intensity. ERG responses were filtered using low-pass (0.15 Hz) and high-pass (100 Hz) filters and digitized for later analysis. The a-wave amplitude was measured from the pre-stimulus baseline to the peak of the first trough while the b-wave was measured from the pre-stimulus baseline or the peak of the a-wave trough (when present) to the maximum positive value.
Intensity-response amplitude data were displayed on log-linear coordinates. To evaluate changes in ERG amplitude and sensitivity, scotopic a- and b-wave amplitudes were iteratively fit using a non-linear sigmoid function (Origin v8.0 software). Means and standard errors were plotted over the fit. The maximum response (Vmax) was calculated from the mean amplitude response across all intensity stimuli. One-way Anova (Origin v8.0) was used to compare response amplitudes at the intensity stimulus that gave Vmax. The a- and b-wave implicit times were measure from the onset of stimulus to either the peak of the first trough (a-wave time to response) or maximum positive value (b-wave time to response).
Retinal Histology
Following ERG analysis, eyes were collected from each animal, corneas slit, and whole eyes placed in fixative (2% glutaraldehyde and 2% paraformaldehyde in 0.135 M sodium cacodylate) overnight at 4 °C. Each eye was cut in half through the optic nerve along the superior-inferior meridian, the lens removed, and the superior cornea notched for later orientation of the light microscope sections. After returning to fixative for at least one additional hour, these hemisected eyes were placed in 6.8% sucrose overnight at room temperature on a rotator, and then dehydrated through a 20% stepped ethanol series to acetone, infiltrated, and embedded in Technovit 8100 methacrylate (Electron Microscopy Sciences, Hatfield, PA). Thick sections (1 μm) were stained with toluidine blue, viewed with bright-field microscopy (Nikon Optiphot-2), and images collected with Metamorph software (Universal Imaging Corp, Downingtown, PA).
These sections were also used to construct the retinal profiles in wild type (n=3-4) and mutant (n=3-4) mice at 6.5 months and 1 year of age. The number of photoreceptor nuclei was counted in four regions (marginal, peripheral, central and medial) along each vertical section of the superior and inferior meridians (Fig 7A). All nuclei within a 130 μm wide section (22 fields per retina) were counted; 3 marginal, 3 peripheral, 3 central and 2 medial, in both the superior and inferior retina. Orientation was verified in all sections by the notch in the superior cornea.
Figure 7.
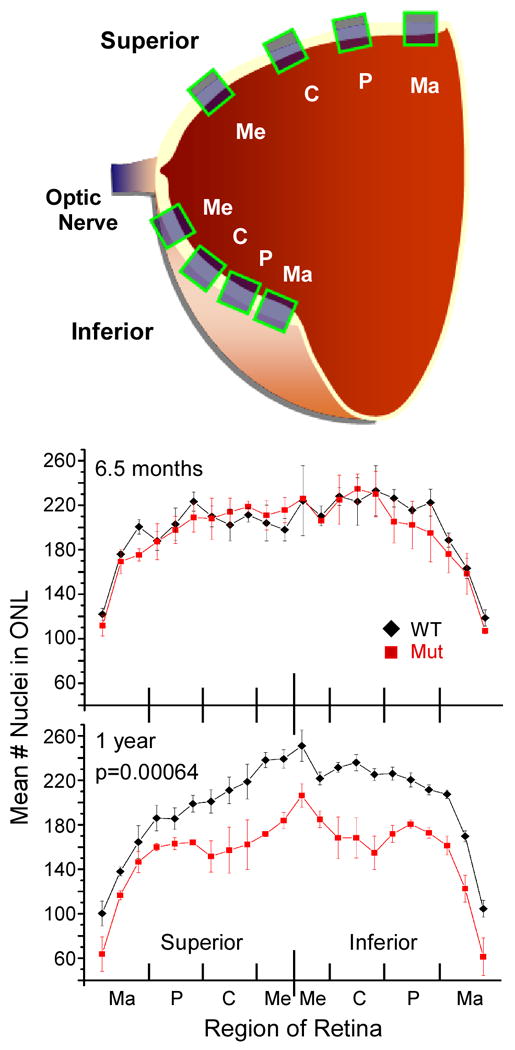
Progressive retinal degeneration in Ush1c216AA mutant mice from 6.5 months (6.5 mo) to 1 year of age (1 yr). A Diagram of hemisected retina and regions (marginal, peripheral, central, medial) where number of rods were counted. B Average number of nuclei in the ONL of the superior and inferior meridians of 6.5 month old wild type (black line) and Ush1c216AA mutant (red line) mice. C Average number of nuclei in the ONL of the superior and inferior meridians of 1 year old wild type (black line) and Ush1c216AA mutant (red line) mice. There are significantly fewer ROD nuclei across the entire retina in mutants compared with age-matched littermates (p=0.00035, one-way Anova, Origin 8.0). Error bars represent the standard error.
Results
Auditory-evoked brain stem response (ABR) analysis
Initial evaluation of the Ush1c216AA mutant mice showed that they did not exhibit an acoustic startle response at 3 weeks of age and showed the characteristic circling and head-tossing behavior seen in mice with cochlear defects. To investigate these observations further, ABR thresholds were done in 3 one-month old Ush1c216AA mice and compared with 10 age-matched CBA (Ush1c216GG) wild type controls. No response was elicited with 110 dB SPL stimuli from the mutant mice at any of the nine frequencies (3.5 kHz to 40.0 kHz) tested, while the CBA controls showed the expected thresholds for mice with normal hearing (Figs 1A & 1B).
Figure 1.
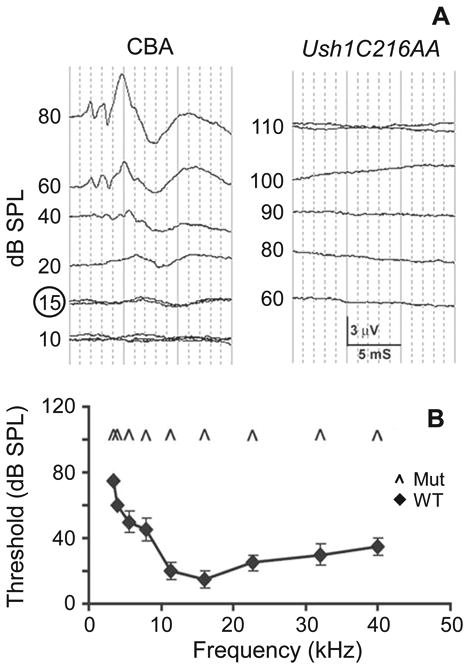
Representative audiograms at 16 kHz (A) of P30 wild type CBA controls (left panel) and Ush1c216AA mutant mice (right panel). CBA control mice hearing thresholds at 16 kHz (15 dB SPL) is circled. B Average ABR thresholds (dB SPL) to pure tones ranging in frequency from 3.5 to 40 kHz in Ush1c216AA mutant mice and CBA controls at P30. Error bars represent standard deviation. Caret marks indicate no detectable response to 110 dB SPL tones.
Cochlear Histology
The deafness and circling behavior of mutant mice suggested the presence of inner ear pathology. Therefore, cochleae from mutant and control mice were examined using whole-mount light microscopic immunocytochemically stained preparations and scanning electron microscopy (SEM) preparations. The microdissected organs of Corti labeled with DAPI (blue), parvalbumin (red), and neurofilament (green) showed disorganized inner and outer hair cell rows in the P30 mutant mice as compared with the well organized pattern in control mice. Degeneration of both inner and outer hair cells of the mutant cochlea was pronounced in the basal turn relative to control mice (Figs 2D & 2E). In this region, virtually all the hair cells were gone in all the preparations examined in the mutant mice. Disorganization and degeneration was also evident in the middle turn, and less so at the apex (Figs 2A-C). The extent of hair cell degeneration was quantified by counting the hair cells in seven regions spanning from the apex through the middle turn (count comparing at the base was not needed because there were so few hair cells in this region of mutant cochlea) in three mutant mice and compared with the same region in three control mice (Fig 3A). By one month of age, significantly fewer inner (p<0.02, Fig 3B) and outer hair cells (p<0.001, Fig 3C) were observed in Ush1c216AA mutant mice. These differences were most apparent in the middle turn (at 240°, 300°, and 360° from the apex) and basal region of the cochlea (Fig 2E). The large error size in the number of hair cells found in the mutant cochleae at 1 month of age is likely due to variable age of onset and rate of degeneration. In addition, scanning electron microscopy (SEM) analysis showed that the outer hair cell stereocilia patterning, height, and orientation were highly abnormal in the mutants relative to the controls (Figs 4A-D).
Figure 2.
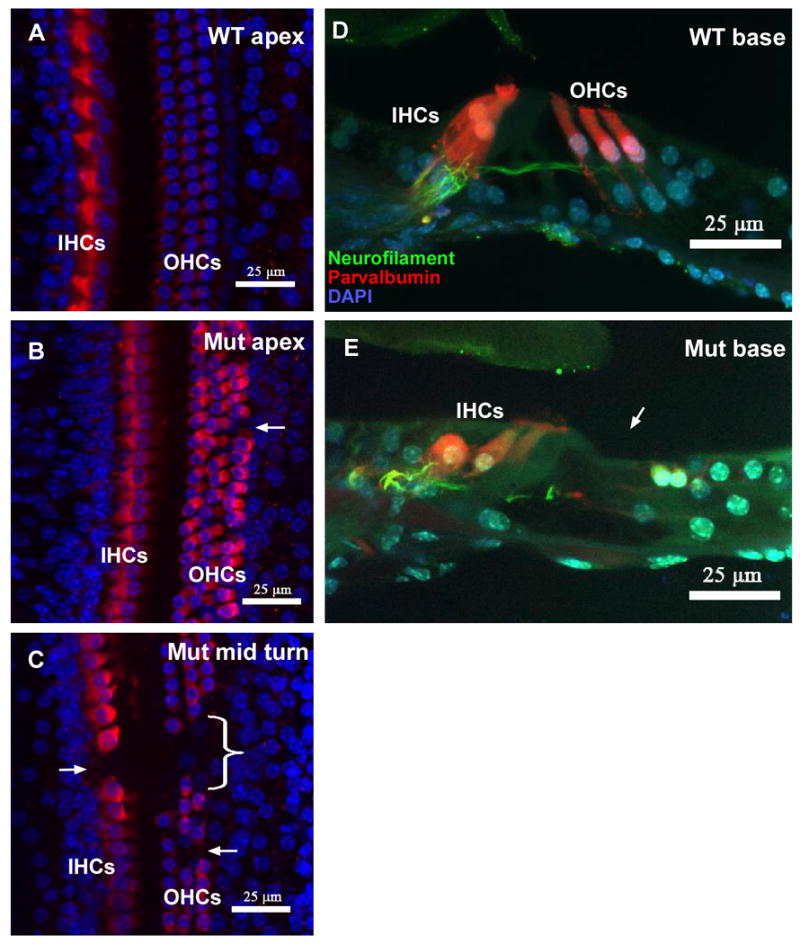
Cochlear histology of wild type and Ush1c216AA mutant mice at P30. Figures A-C show whole-mount preparations of the organ of Corti at the apex of wild type (A) and the apex (B) and middle turns (C) of mutant mice. All images are brightest point projections from a confocal Z series spanning the full depth of the sensory epithelium. The cytoplasm of inner (IHCs) and outer (OHCs) hair cells is immunolabeled with parvalbumin (red) and nuclei are counterstained with DAPI (blue). White arrows and bracket indicate missing OHCs. Figures D, E show cross sections of the basal turn of the organ of Corti from plastic embedded cochleae from wild type (D) and mutant (E) mice. Both images are brightest point projections from a confocal Z series spanning the depth of 2-3 outer hair cells. Nerve fibers (Neurofilament, green) are visible innervating IHCs and OHCs in the wild type mice, while OHCs are missing entirely from this region of the cochlea in mutant mice. Additionally, IHCs appear abnormal, and few nerve fibers cross the tunnel of Corti. Scale bars in all figures are 25 μm.
Figure 4.
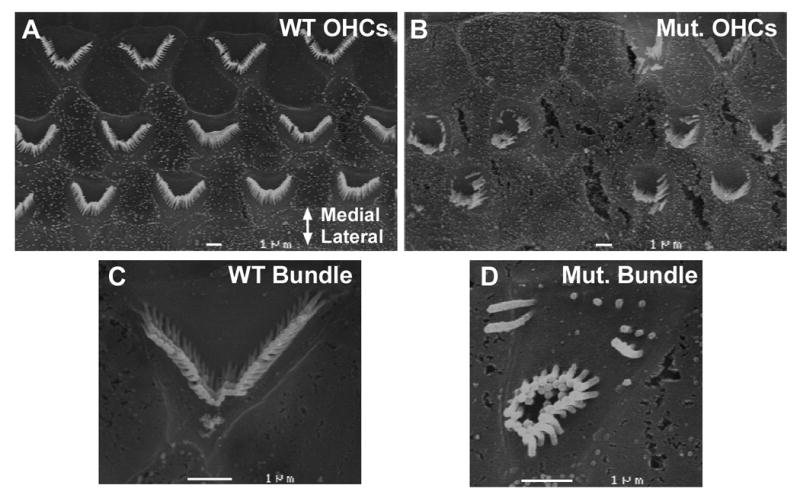
Scanning electron micrographs of cochleae from Ush1c216AA mutant and CBA controls at P30. A OHCs in the middle turn of the cochlea from a wild type mouse. The most medial and lateral rows of OHCs are at the top and base of the figure, respectively. B Disorganized OHC rows with missing hair cells in the middle turn from a mutant mouse. Hair cell stereociliary bundles are abnormal. C Higher magnification (15,000× original magnification) of an OHC stereociliary bundle from a wild type mouse. D Higher magnification of an abnormal OHC stereociliary bundle from a mutant mouse (15,000× original magnification). Scale bars = 1 μm.
Electroretinography analysis
To determine if the c.216G>A mutation affects retinal function in the mouse, electroretinography (ERG), which measures the summed electrical activity of all retinal cells, was performed. ERGs were recorded in dark adapted (scotopic) wild type and Ush1c216AA mutant littermates at 1, 3, 6.5, and 12 months of age. The maximum amplitude and time to response of both major components of the scotopic ERG, the a-wave, which reflects the functional integrity of the photoreceptors, and the b-wave, which is the response of the neural retina post-synaptic to the photoreceptors, were analyzed. At all time points tested, the general shape of the mutant ERG was similar to that of their wild type age-matched littermate controls, but the maximum amplitudes of both the a- and b-waves were significantly reduced. The mutant a- and b-waves were significantly attenuated as early as 1 month of age by 30% and 34%, respectively (p=0.02 and p=0.03, respectively, one-way Anova; Figs 5C,D). Another difference observed as early as 1 month was the a-wave implicit time, measured from the onset of stimulus to the peak of the a-wave. At the two brightest light intensities, the implicit time was faster in the mutants compared with age-matched control mice (data not shown).
Figure 5.
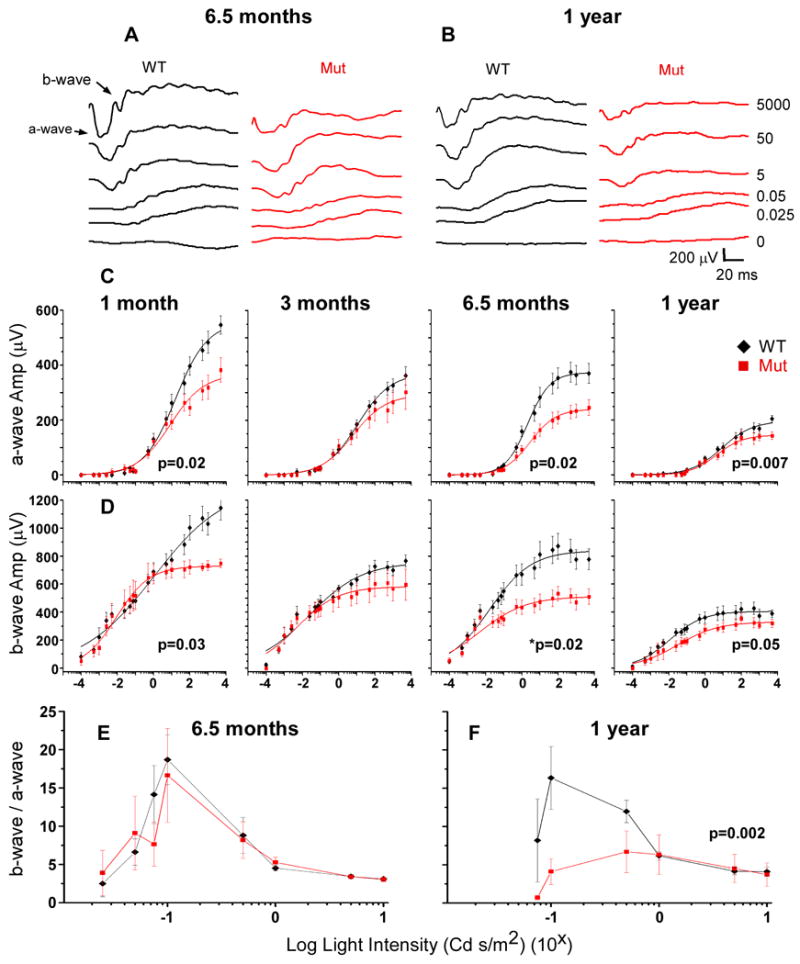
ERG analysis of wild type and Ush1c216AA mutant mice at 1, 3, 6.5 and 12 months of age. A,B Representative ERGs of mutant (Ush1c216AA) and control (wild type) at 6.5 and 12 months of age. C,D Averaged a- and b-wave amplitudes at 1, 3, 6.5 and 12 months of age of control mice (black line) and mutant mice (red line). At all ages tested, mutant a- and b-wave maximum amplitudes are lower (1 month: a-wave Vmax=382.48 μV, b-wave Vmax=747.48 μV; 3 month: a-wave Vmax=302.6, b-wave Vmax=608.62 μV; 6.5 month: a-wave Vmax=304.56 μV, b-wave Vmax=676.58 μV; 12 months: a-wave Vmax=136.36 μV, b-wave Vmax=324.14 μV) than controls (1 month: a-wave Vmax=546.68, b-wave Vmax=1143.73 μV; 3 month a-wave Vmax=362.13 μV, b-wave Vmax=765.87 μV; 6.5 month: a-wave Vmax=374.54 μV, b-wave Vmax=870.96 μV; 12 month: a-wave Vmax=199.72 μV, b-wave Vmax=415.89 μV). By 12 months of age, mutant a- and b-wave thresholds are higher compared to controls (WT a-wave threshold=0.005 Cd s/m2 vs. Mut a-wave threshold=0.0075; WT b-wave threshold=0.0001 Cd s/m2 vs. Mut b-wave threshold=0.0005). Error bars represent the standard error. E,F b/a-wave ratio of wild type (black line) and Ush1c216AA mutant mice (red line) at 6.5 (left graph) and 12 months (right graph) of age. Graphs show that at one year of age mutant and wild type mice have significantly different b/a wave ratios at lower light intensities (p=0.002, two-way Anova, Origin 8.0). Light intensity is shown in log scale with x=log cycle number. Error bars represent the standard error.
Interestingly, at 3 and 6.5 months of age, the mutant a-wave and b-wave maximum responses were less attenuated than at 1 month. At 3 months, mutant a-wave maximum responses were 16% lower, while the b-wave maximum response was 21% lower than controls, although not statistically significant. At 6.5 months of age, the mutant a-wave and b-wave maximum amplitudes were reduced by 19 and 22%, relative to controls, respectively (p=0.02 for both a- and b-wave reduction, one-way Anova; Figs 5A-D). The implicit time of the mutant a-wave at 3 and 6.5 months of age was similar to that of controls.
By 1 year of age, the mutant a-wave and b-wave maximum responses were 32% and 22%, respectively, lower than the control responses (p=0.007 and p=0.05, respectively; one-way Anova; Figs 5B-D). Furthermore, by one year of age, the light intensities at threshold for both the a- and b-waves were higher (Figs 5C,D). Additionally, the mutant and control b-to-a-wave ratios are significantly different (p=0.002, two-way Anova; Fig 5F). The implicit time of the mutant b-wave was faster at the highest light intensities, although not significantly, than that of wild type controls at 6.5 months and one year of age.
Rod photoreceptor degeneration
To look for rod photoreceptor degeneration characteristic of Usher-associated retinitis pigmentosa, the number of rod photoreceptor nuclei in the outer nuclear layer of Ush1c216AA mutant mice was compared with control littermates at 1, 4, 6.5 and 12 months of age. Six 130 μm wide sections at the periphery (3 marginal and 3 peripheral), three in the central region (central), and two near the optic nerve (medial) of both the superior and inferior meridians, were counted and analyzed independently. Figure 6 shows representative 6.5 month (A, B) and 1 year old (C, D) wild type and mutant retinal sections from the superior meridian. The average number of photoreceptor nuclei counted along the superior and inferior meridians of wild type and mutant retinas at these ages was calculated (Fig 7). Up to 6.5 months of age, wild type and mutant retinas appear morphologically similar at the light microscopy level (Figs 6A,B). At 6.5 months, mutant retinas show a slight, but reproducible, decrease (6-8%) at the periphery (both superior and inferior) compared to wild type littermates (Fig 7B). However, at 1 year of age, wild type and mutant retinas (Fig 6C and D, respectively) show major differences in the thickness of the outer nuclear layer in all regions, but more significantly at the superior and inferior marginal and central regions. Section 4× of figure 6D shows the severe loss of the outer nuclear layer (black arrow) from the periphery of a severely affected mutant superior marginal section. In contrast, the outer nuclear layer from the same region of a wild type littermate (section 3× of Fig 6C) is apparent to the outer edge of the retina. Section 6× of figure 6D shows another severely affected section in the superior central region of a mutant retina, compared to the same region of a representative wild type littermate (3× and 5×, respectively). As shown, the outer nuclear layer from the mutant retina is only 4 – 5 nuclei in thickness compared to 8 – 9 nuclei in the wild type control. These results demonstrate that by 1 year of age, there are significantly fewer rods (21-26%) in Ush1c216AA mutant mice compared with age-matched controls across the entire retina (p=0.00035, one-way Anova, Origin v8.0).
Figure 6.
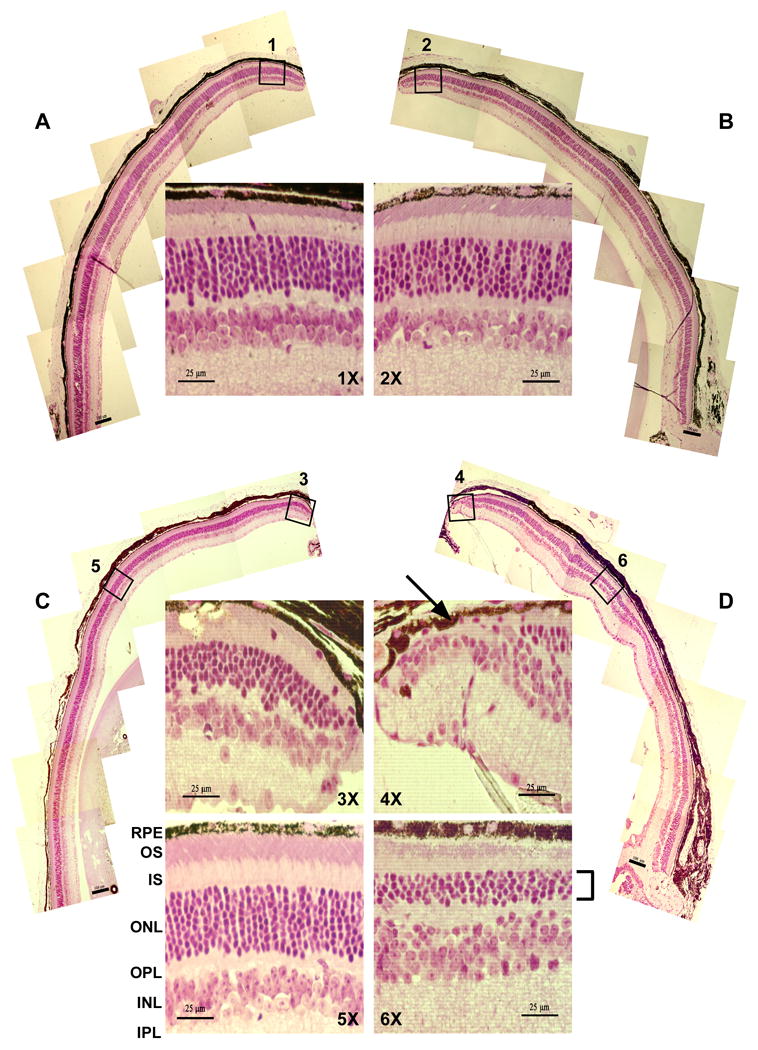
Histology (100× and 400× original magnification) of superior peripheral retina of wild type and Ush1c216AA mutant mice at 6.5 months and 1 year of age. Figures A,B show 6.5 month old wild type (A) and mutant (B) sections from the margin of the superior retina. Figures C-F show sections from the margin (C,D) and central (E,F) regions of the superior retina of wild type and mutant 1 year old mice. (RPE, retinal pigment epithelium; OS, outer segments; IS, inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer.) Scale bars in superior meridians (100× magnification) are 100 μm, inside panels (400× magnification) are 25 μm.
Discussion
Usher I patients have profound deafness and vestibular dysfunction at birth and progressive retinal degeneration beginning in early adolescence. Retinal degeneration in patients is apparent by fundus examination and the progressive reduction in ERG amplitudes over the course of the disorder. Previous mouse models for each Usher I type have shown the characteristic congenital deafness and vestibular defects, but they all lack retinal degeneration. Spontaneous mutations in the mouse Ush1 homologs Myo7a, Ush1c, Cdh23, Pcdh15 and Sans have been shown to be responsible for the deafness and circling and head-tossing behavior in the shaker 1 (sh1), deaf-circler (dfcr), waltzer (v), Ames waltzer (av), and Jackson shaker (js) mice, respectively (Gibson et al., 1995; Alagramam et al., 2001; Di Palma et al., 2001; Johnson et al., 2003; Kikkawa et al., 2003). Some of these mutants (sh1, v, av) showed ERG anomalies (Libby and Steel, 2001), and a defective retinal pigment epithelium has been reported in sh1 mice (Gibbs et al., 2003; Gibbs et al., 2004). However, retinal degeneration has not been observed in any of these mouse models. In contrast, the tubby mouse is a spontaneous mouse model that does have combined retinal and cochlear degeneration with late-onset obesity. Unlike Usher syndrome type 1, the tubby mouse has early onset retinal degeneration and later onset hearing loss, with maturity onset obesity and insulin resistance. Tub knock-out mice share the same phenotype as tubby mice, suggesting the mechanism involves a loss of function of the tub protein (Stubdal et al., 2000). This contrasts with the Ush1c models in which knock-outs produce only a partial phenotype with cochlear degeneration alone, while our Ush1c216AA knock-in mice develop both cochlear and retinal degeneration.
In humans, the mechanisms responsible for the combined deafness and blindness characteristic of Usher syndrome are poorly understood. The USH1C gene is organized into 28 exons, eight of which are alternatively spliced, generating three different classes of the protein harmonin. Classes a, b and c differ in the number of protein-protein interaction domains (PDZ, post-synaptic density/disc-large/zonal occludens 1), coiled-coiled domains (CC), and the presence of a proline-serine-threonine (PST) rich domain (Verpy, et al., 2000). With at least 12 known RNA transcripts and three protein classes, harmonin is likely to have multiple functions that are tissue specific. In mice, it has been localized to both the sensory hair cells of the cochlea and photoreceptors of the retina (Boeda et al., 2002; Reiners et al., 2005; Reiners et al., 2005); however its function remains unclear. Harmonin is expressed in the cuticular plate and stereociliary tip region in both developing and mature hair cell bundles (Verpy et al., 2000; Boeda et al., 2002; Lefevre et al., 2008; Grillet et al., 2009). Hair bundle morphogenesis is abnormal in mice lacking harmonin, as well as in mice with a missense mutation in the PDZ2 domain, which is required for binding with cadherins CDH23 and PCDH15 (Boeda et al., 2002; Siemens et al., 2002; Adato et al., 2005; Reiners and Wolfrum, 2006; Kazmierczak et al., 2007). Deletion of the PST and coiled-coiled domains does not appear to affect bundle formation, but does affect localization in functionally mature hair cells and mechanotransduction (Grillet et al., 2009). In the adult mouse retina, harmonin has been immunolocalized to the photoreceptor inner and outer segments, the outer plexiform layer and ganglion cell layer. Electron microscopy shows harmonin associated with photoreceptor outer segment discs, cytoplasm of the inner segments and both the pre- and post-synaptic regions (Reiners et al., 2003; Williams et al., 2009).
The Ush1c216AA mice are hyperactive and display circling and head-tossing behavior by three weeks of age, and their ABR thresholds are greatly increased at all frequencies tested, indicating profound deafness and vestibular dysfunction. Abnormal hair cell bundle morphology is apparent in all regions of the cochlea. The number and height of the stereocilia vary irregularly, suggesting an essential role for the actin-binding and PDZ-domain containing protein harmonin in hair bundle development. By one month of age, the sensory cells have begun to degenerate. The basal region of the cochlea appears to be affected first, with more severe inner and outer hair cell loss than in the middle and apical regions. Cochleae from mice older than one month were not examined, but the extent of the degeneration would be expected to increase with age. Although, the abnormal stereocilia morphology and hair cell degeneration observed in Ush1c216AA knock-in mice is similar to that reported in all USH1 mouse models (Gibson et al., 1995; Alagramam et al., 2001; Di Palma et al., 2001; Wilson et al., 2001; Johnson et al., 2003; Kikkawa et al., 2003; Pawlowski et al., 2006; Lefevre et al., 2008), our work provides the first Usher I mouse model with the combined progressive retinal degeneration and deafness and vestibular dysfunction characteristic of patients. The fact that the Ush1c216AA mouse is homozygous for the human mutation that is found in Acadian Usher 1C patients may be an important factor in explaining why this Usher I mouse model has retinal degeneration while others do not.
Progressive retinal degeneration was observed in our mice beginning with a decline in retinal function as early as one month of age. The wild type ERG development progressed as expected (Li et al., 2001; Libby and Steel, 2001), with a- and b- wave maximum amplitudes decreasing with age by approximately 50% from 1 month to 1 year, suggesting the deficit in the mutant is due to the presence of the c.216G>A mutation. ERG a- and b-wave maximum amplitudes of Ush1c216AA mutant mice were attenuated at all ages tested compared with control littermates. Additionally, by one year of age, an increase in threshold was observed in the mutants, indicating that their retinas are less sensitive. Because the a-wave of the ERG is the result of the rod photoreceptors' response to light, a reduction in a-wave amplitudes suggests Ush1c216AA mutant mice have abnormal photoreceptor function. The implicit time of the mutant b-wave was faster than controls as early as 1 month of age, and at 6.5 and 12 months of age. This faster mutant b-wave latency, as well as a change in the b-to-a wave ratio observed at one year of age, suggests specific defects in the kinetics of the b-wave.
Subsequent to the decline in retinal function, progressive degeneration of rod photoreceptors was observed in mutant retinas between 6.5 months and 1 year of age. The periphery of the retina appears to be affected first. At 6.5 months of age, there are 6-8% fewer rods from the margin through the peripheral region, and this reduction increases to 16-23% by one year of age. Degeneration then follows in the central and medial retinas, where patches of severe rod loss were observed. Ush1c216AA mutant mice exhibit a loss of visual function prior to photoreceptor cell disappearance, meaning that functional changes have occurred without observable cell loss, as in the case of normal animals that show ERG decline with age without cell loss (Libby and Steel, 2001). This suggests that when the photoreceptor cells begin to disappear, the rate of loss is not reflected in as great a decline in ERG as might be anticipated, as was found in the mutant mice. The precise mechanisms that underlie the relationship between amplitude decline and photoreceptor cell loss remain to be elucidated.
With respect to the molecular mechanism of this mutation, the c.216G>A mutation is a single base pair change from a guanine (G) to an adenine (A) at position 216 in the USH1C gene. This change shifts the splice donor site upstream of the wild type splice donor, resulting in the loss of 35 base pairs at the end of exon 3. In Acadian Usher 1C patients, a truncated mutant splice variant is detected (Lentz et al., 2005). The same truncated transcript is found in the retina and cochlea of our knock-in mouse. Rt-pcr was used to investigate the splice variants in patients and mutant mice. Under competitive conditions, only a mutant mRNA transcript was found in patients and mutant mice, and not in unaffected family members and wild type littermates, suggesting the mutant splice variant is much more abundant (Lentz et al., 2007). Computational translation (DNA Star Software) of the mutant mRNA transcript found in our mice, as well as Usher IC patients, predicts a premature stop codon resulting in a severely truncated protein of about 135 amino acids (aa), as compared to wild type harmonin classes a (544-548 aa), b (852-910 aa) or c (403-476 aa). This mutant protein would not contain the characteristic PDZ, CC or PST domains believed to play important roles in the function of harmonin. The predicted mutant protein would, however, retain one of two Src homology 3 (SH3) domains (PLIPLK site), and contain a new SH3 domain (PSAPRR site) that is not present in wild type harmonin (SH3-Hunter). SH3 domains are protein-protein interaction domains that have been found to participate in nearly all cellular functions including cell growth, differentiation, motility, polarity and apoptosis (Li, 2005). It would seem reasonable to hypothesize that this truncated protein is present, but highly dysfunctional in its protein-protein interactions. Ush1c knock-out mice show a similar phenotype in the ear, with deafness and abnormal hair bundle formation, suggesting this splice-site mutation acts as a loss of function in the hair cells. The fact that knock-outs do not, however, have a retinal phenotype suggests the possibility that mutant protein is present in the c.216G>A knock-in mouse photoreceptor cells and contributes to their degeneration. With the retention of one wild type SH3 domain, this mutant protein may continue to interact in a non-functional manner with harmonin binding partners rendering them unable to function properly. A gain of function could also be hypothesized, in which this mutant protein takes on a new function through interactions with the SH3 domain that is not present in wild type harmonin. Future studies of molecular mechanisms and protein functions, as well as localization and interactions, will clarify the reasons that abnormalities are observed in both the cochlea and the retina of the USH1C c.216G>A knock-in mouse.
In conclusion, our data demonstrate that two copies of the human USH1C c.216G>A mutation are sufficient to create the human Usher I phenotype in the mouse. This c.216G>A knock-in mouse model of Usher I with combined deafness, vestibular defects and retinal degeneration thus provides a valuable resource which will contribute to the understanding of the mechanisms associated with sensory development in general, and dual sensory loss in particular, and facilitate the development of therapies that prevent the retinal degeneration in these patients.
Acknowledgments
The authors thank Dr. Richard Bobbin for his technical help with ABRs, Dr. Juan Sagredo (LSUHSC-NC) for his expert histological preparation of the retinas, and Dr. Jeffrey Jamison (Alcon Laboratories, Inc.) for his technical advice with the ERGs. The authors also thank Dr. Prescott Deininger for his critical comments on the manuscript. This work was supported by grants from the Foundation Fighting Blindness, NIH 5P20RR016816 (NGB and HEF), NIH P30 DC004661 (EWR), NIH R01 DC002739 (BLT), NIH RO1 DC03944 (EO), and NICHHD P30 HD002274.
References
- Adato A, Lefevre G, Delprat B, Michel V, Michalski N, Chardenoux S, Weil D, El-Amraoui A, Petit C. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum Mol Genet. 2005;14:3921–3932. doi: 10.1093/hmg/ddi416. [DOI] [PubMed] [Google Scholar]
- Adato A, Michel V, Kikkawa Y, Reiners J, Alagramam KN, Weil D, Yonekawa H, Wolfrum U, El-Amraoui A, Petit C. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005;14:347–356. doi: 10.1093/hmg/ddi031. [DOI] [PubMed] [Google Scholar]
- Adato A, Vreugde S, Joensuu T, Avidan N, Hamalainen R, Belenkiy O, Olender T, Bonne-Tamir B, Ben-Asher E, Espinos C, Millan JM, Lehesjoki AE, Flannery JG, Avraham KB, Pietrokovski S, Sankila EM, Beckmann JS, Lancet D. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur J Hum Genet. 2002;10:339–350. doi: 10.1038/sj.ejhg.5200831. [DOI] [PubMed] [Google Scholar]
- Adato A, Weil D, Kalinski H, Pel-Or Y, Ayadi H, Petit C, Korostishevsky M, Bonne-Tamir B. Mutation profile of all 49 exons of the human myosin VIIA gene, and haplotype analysis, in Usher 1B families from diverse origins. Am J Hum Genet. 1997;61:813–821. doi: 10.1086/514899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman TB, Wilcox ER. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet. 2001;69:25–34. doi: 10.1086/321277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP. The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet. 2001;27:99–102. doi: 10.1038/83837. [DOI] [PubMed] [Google Scholar]
- Alagramam KN, Yuan H, Kuehn MH, Murcia CL, Wayne S, Srisailpathy CR, Lowry RB, Knaus R, Van Laer L, Bernier FP, Schwartz S, Lee C, Morton CC, Mullins RF, Ramesh A, Van Camp G, Hageman GS, Woychik RP, Smith RJ. Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum Mol Genet. 2001;10:1709–1718. doi: 10.1093/hmg/10.16.1709. [DOI] [PubMed] [Google Scholar]
- Aller E, Jaijo T, Beneyto M, Najera C, Oltra S, Ayuso C, Baiget M, Carballo M, Antinolo G, Valverde D, Moreno F, Vilela C, Collado D, Perez-Garrigues H, Navea A, Millan JM. Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J Med Genet. 2006;43:e55. doi: 10.1136/jmg.2006.041764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, Hussain K, Furth-Lavi J, Cosgrove KE, Shepherd RM, Barnes PD, O'Brien RE, Farndon PA, Sowden J, Liu XZ, Scanlan MJ, Malcolm S, Dunne MJ, Aynsley-Green A, Glaser B. A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nat Genet. 2000;26:56–60. doi: 10.1038/79178. [DOI] [PubMed] [Google Scholar]
- Boeda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J, Houdusse A, Legrain P, Wolfrum U, Richardson G, Petit C. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21:6689–6699. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG, Seeliger M, del CSCM, Vila MC, Molina OP, Gal A, Kubisch C. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet. 2001;27:108–112. doi: 10.1038/83667. [DOI] [PubMed] [Google Scholar]
- Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, Wayne S, Bellman S, Desmukh D, Ahmed Z, Khan SN, Kaloustian VM, Li XC, Lalwani A, Bitner-Glindzicz M, Nance WE, Liu XZ, Wistow G, Smith RJ, Griffith AJ, Wilcox ER, Friedman TB, Morell RJ. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001;68:26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001;27:103–107. doi: 10.1038/83660. [DOI] [PubMed] [Google Scholar]
- Dreyer B, Tranebjaerg L, Rosenberg T, Weston MD, Kimberling WJ, Nilssen O. Identification of novel USH2A mutations: implications for the structure of USH2A protein. Eur J Hum Genet. 2000;8:500–506. doi: 10.1038/sj.ejhg.5200491. [DOI] [PubMed] [Google Scholar]
- Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, Millan JM, Aller E, Mitter D, Bolz H. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet. 2007;121:203–211. doi: 10.1007/s00439-006-0304-0. [DOI] [PubMed] [Google Scholar]
- Erway LC, Willott JF, Archer JR, Harrison DE. Genetics of age-related hearing loss in mice: I. Inbred and F1 hybrid strains. Hear Res. 1993;65:125–132. doi: 10.1016/0378-5955(93)90207-h. [DOI] [PubMed] [Google Scholar]
- Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C, Cremers C, Davenport S, Moller C, Talmadge CB, Beisel KW, Tamayo M, Morton CC, Swaroop A, Kimberling WJ, Sumegi J. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998;280:1753–1757. doi: 10.1126/science.280.5370.1753. [DOI] [PubMed] [Google Scholar]
- Gibbs D, Azarian SM, Lillo C, Kitamoto J, Klomp AE, Steel KP, Libby RT, Williams DS. Role of myosin VIIa and Rab27a in the motility and localization of RPE melanosomes. J Cell Sci. 2004;117:6473–6483. doi: 10.1242/jcs.01580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs D, Kitamoto J, Williams DS. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc Natl Acad Sci U S A. 2003;100:6481–6486. doi: 10.1073/pnas.1130432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson F, Walsh J, Mburu P, Varela A, Brown KA, Antonio M, Beisel KW, Steel KP, Brown SD. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature. 1995;374:62–64. doi: 10.1038/374062a0. [DOI] [PubMed] [Google Scholar]
- Grillet N, Xiong W, Reynolds A, Kazmierczak P, Sato T, Lillo C, Dumont RA, Hintermann E, Sczaniecka A, Schwander M, Williams D, Kachar B, Gillespie PG, Muller U. Harmonin mutations cause mechanotransduction defects in cochlear hair cells. Neuron. 2009;62:375–387. doi: 10.1016/j.neuron.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie NA, MacDonald G, Rubel EW. A new method for imaging and 3D reconstruction of mammalian cochlea by fluorescent confocal microscopy. Brain Res. 2004;1000:200–210. doi: 10.1016/j.brainres.2003.10.071. [DOI] [PubMed] [Google Scholar]
- Joensuu T, Hamalainen R, Yuan B, Johnson C, Tegelberg S, Gasparini P, Zelante L, Pirvola U, Pakarinen L, Lehesjoki AE, de la Chapelle A, Sankila EM. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet. 2001;69:673–684. doi: 10.1086/323610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Gagnon LH, Webb LS, Peters LL, Hawes NL, Chang B, Zheng QY. Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum Mol Genet. 2003;12:3075–3086. doi: 10.1093/hmg/ddg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak P, Sakaguchi H, Tokita J, Wilson-Kubalek EM, Milligan RA, Muller U, Kachar B. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature. 2007;449:87–91. doi: 10.1038/nature06091. [DOI] [PubMed] [Google Scholar]
- Keats BJ, Corey DP. The usher syndromes. Am J Med Genet. 1999;89:158–166. [PubMed] [Google Scholar]
- Kikkawa Y, Shitara H, Wakana S, Kohara Y, Takada T, Okamoto M, Taya C, Kamiya K, Yoshikawa Y, Tokano H, Kitamura K, Shimizu K, Wakabayashi Y, Shiroishi T, Kominami R, Yonekawa H. Mutations in a new scaffold protein Sans cause deafness in Jackson shaker mice. Hum Mol Genet. 2003;12:453–461. doi: 10.1093/hmg/ddg042. [DOI] [PubMed] [Google Scholar]
- Lefevre G, Michel V, Weil D, Lepelletier L, Bizard E, Wolfrum U, Hardelin JP, Petit C. A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development. 2008;135:1427–1437. doi: 10.1242/dev.012922. [DOI] [PubMed] [Google Scholar]
- Lentz J, Pan F, Ng SS, Deininger P, Keats B. Ush1c216A knock-in mouse survives Katrina. Mutat Res. 2007;616:139–144. doi: 10.1016/j.mrfmmm.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Lentz J, Savas S, Ng SS, Athas G, Deininger P, Keats B. The USH1C 216G-->A splice-site mutation results in a 35-base-pair deletion. Hum Genet. 2005;116:225–227. doi: 10.1007/s00439-004-1217-4. [DOI] [PubMed] [Google Scholar]
- Levy G, Levi-Acobas F, Blanchard S, Gerber S, Larget-Piet D, Chenal V, Liu XZ, Newton V, Steel KP, Brown SD, Munnich A, Kaplan J, Petit C, Weil D. Myosin VIIA gene: heterogeneity of the mutations responsible for Usher syndrome type IB. Hum Mol Genet. 1997;6:111–116. doi: 10.1093/hmg/6.1.111. [DOI] [PubMed] [Google Scholar]
- Li C, Cheng M, Yang H, Peachey NS, Naash MI. Age-related changes in the mouse outer retina. Optom Vis Sci. 2001;78:425–430. doi: 10.1097/00006324-200106000-00015. [DOI] [PubMed] [Google Scholar]
- Li SS. Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem J. 2005;390:641–653. doi: 10.1042/BJ20050411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby RT, Steel KP. Electroretinographic anomalies in mice with mutations in Myo7a, the gene involved in human Usher syndrome type 1B. Invest Ophthalmol Vis Sci. 2001;42:770–778. [PubMed] [Google Scholar]
- Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet. 1997;16:188–190. doi: 10.1038/ng0697-188. [DOI] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet. 2003;35:21–23. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlowski KS, Kikkawa YS, Wright CG, Alagramam KN. Progression of inner ear pathology in Ames waltzer mice and the role of protocadherin 15 in hair cell development. J Assoc Res Otolaryngol. 2006;7:83–94. doi: 10.1007/s10162-005-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiners J, Marker T, Jurgens K, Reidel B, Wolfrum U. Photoreceptor expression of the Usher syndrome type 1 protein protocadherin 15 (USH1F) and its interaction with the scaffold protein harmonin (USH1C) Mol Vis. 2005;11:347–355. [PubMed] [Google Scholar]
- Reiners J, Reidel B, El-Amraoui A, Boeda B, Huber I, Petit C, Wolfrum U. Differential distribution of harmonin isoforms and their possible role in Usher-1 protein complexes in mammalian photoreceptor cells. Invest Ophthalmol Vis Sci. 2003;44:5006–5015. doi: 10.1167/iovs.03-0483. [DOI] [PubMed] [Google Scholar]
- Reiners J, van Wijk E, Marker T, Zimmermann U, Jurgens K, te Brinke H, Overlack N, Roepman R, Knipper M, Kremer H, Wolfrum U. Scaffold protein harmonin (USH1C) provides molecular links between Usher syndrome type 1 and type 2. Hum Mol Genet. 2005;14:3933–3943. doi: 10.1093/hmg/ddi417. [DOI] [PubMed] [Google Scholar]
- Reiners J, Wolfrum U. Molecular analysis of the supramolecular usher protein complex in the retina. Harmonin as the key protein of the Usher syndrome. Adv Exp Med Biol. 2006;572:349–353. doi: 10.1007/0-387-32442-9_49. [DOI] [PubMed] [Google Scholar]
- Siemens J, Kazmierczak P, Reynolds A, Sticker M, Littlewood-Evans A, Muller U. The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions. Proc Natl Acad Sci U S A. 2002;99:14946–14951. doi: 10.1073/pnas.232579599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubdal H, Lynch CA, Moriarty A, Fang Q, Chickering T, Deeds JD, Fairchild-Huntress V, Charlat O, Dunmore JH, Kleyn P, Huszar D, Kapeller R. Targeted deletion of the tub mouse obesity gene reveals that tubby is a loss-of-function mutation. Mol Cell Biol. 2000;20:878–882. doi: 10.1128/mcb.20.3.878-882.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk E, Pennings RJ, te Brinke H, Claassen A, Yntema HG, Hoefsloot LH, Cremers FP, Cremers CW, Kremer H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am J Hum Genet. 2004;74:738–744. doi: 10.1086/383096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, Slim R, Petit C. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet. 2000;26:51–55. doi: 10.1038/79171. [DOI] [PubMed] [Google Scholar]
- Wada T, Wakabayashi Y, Takahashi S, Ushiki T, Kikkawa Y, Yonekawa H, Kominami R. A point mutation in a cadherin gene, Cdh23, causes deafness in a novel mutant, Waltzer mouse niigata. Biochem Biophys Res Commun. 2001;283:113–117. doi: 10.1006/bbrc.2001.4724. [DOI] [PubMed] [Google Scholar]
- Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995;374:60–61. doi: 10.1038/374060a0. [DOI] [PubMed] [Google Scholar]
- Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, Delmaghani S, Adato A, Nadifi S, Zina ZB, Hamel C, Gal A, Ayadi H, Yonekawa H, Petit C. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum Mol Genet. 2003;12:463–471. doi: 10.1093/hmg/ddg051. [DOI] [PubMed] [Google Scholar]
- Weston MD, Eudy JD, Fujita S, Yao S, Usami S, Cremers C, Greenberg J, Ramesar R, Martini A, Moller C, Smith RJ, Sumegi J, Kimberling WJ. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am J Hum Genet. 2000;66:1199–1210. doi: 10.1086/302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston MD, Kelley PM, Overbeck LD, Wagenaar M, Orten DJ, Hasson T, Chen ZY, Corey D, Mooseker M, Sumegi J, Cremers C, Moller C, Jacobson SG, Gorin MB, Kimberling WJ. Myosin VIIA mutation screening in 189 Usher syndrome type 1 patients. Am J Hum Genet. 1996;59:1074–1083. [PMC free article] [PubMed] [Google Scholar]
- Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet. 2004;74:357–366. doi: 10.1086/381685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D, Aleman T, Lillo C, Lopes VS, Hughes LC, Stone EM, Jacobson SG. Harmonin in the murine retina and the retinal phenotypes of Ush1c-mutant mice and human USH1C. Invest Ophthalmol Vis Sci. 2009:iovs.08–3358. doi: 10.1167/iovs.08-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SM, Householder DB, Coppola V, Tessarollo L, Fritzsch B, Lee EC, Goss D, Carlson GA, Copeland NG, Jenkins NA. Mutations in Cdh23 cause nonsyndromic hearing loss in waltzer mice. Genomics. 2001;74:228–233. doi: 10.1006/geno.2001.6554. [DOI] [PubMed] [Google Scholar]