

Abstract
A convergent method to access the fused indoline ring system present in a multitude of bioactive molecules has been developed. The strategy involves the condensation of hydrazines with latent aldehydes to ultimately deliver indoline-containing products by way of an interrupted Fischer indolization sequence. The method is convergent, mild, operationally simple, broad in scope, and can be used to access enantioenriched products. In addition, our approach is amenable to the synthesis of furoindoline and pyrrolidinoindoline natural products as demonstrated by the concise formal total syntheses of physovenine and debromoflustramine B. The strategy will likely enable the synthesis of more complex targets such as the communesin alkaloids.
Keywords: indoline, natural products, Fischer indole synthesis, total synthesis, heterocycles
1. Introduction
The discovery of efficient methods to synthesize complex bioactive molecules continues to be a vital area of research.i A subset of compounds that have received substantial interest due to their medicinal properties and impressive structures are those that possess a fused indoline motif, of the type 1 (Figure 1 and Scheme 1). The simplest of these compounds are the acetylcholinesterase inhibitors physovenine (2) and physostigmine (3),ii,iii which are composed of basic furo- and pyrrolidinoindoline motifs, respectively (Figure 1). Numerous relatives of pyrrolidinoindoline 3 have been isolated, including bis(prenylated) derivatives,iv dimeric structures,v and compounds possessing a heteroatom substituent at C3 (e.g., 4– 7, respectively).vi,vii Beyond these compounds, a variety of more architecturally complex indoline containing natural products are known, such as the akuammiline alkaloids (e.g., 8–11),viii,ix perophoramidine (12),x the communesins (e.g., 13),xi diazonamide A (14),xii and bipleiophylline (15).xiii Many of these molecules possess interesting biological properties, which further enhance their appeal as targets for total synthesis.
Figure 1.

Parent indoline 1 and representative natural products 2–15
Scheme 1.

Approach to indoline 1.
The importance of indoline-containing compounds has prompted the development of a number of methods to access such motifs, with numerous studies particularly in the area of pyrrolidinoindoline synthesis. In most cases, the fused indoline ring systems 1 are constructed by cyclization of precursors of the type 16, which in turn are derived from substituted indolexiv or oxindolexv intermediates (Scheme 1). Herein, we report the development of a powerful cascade reaction that allows direct access to 1 (via 16) from the coupling of two simple fragments.xvi The transformation is convergent, broad in scope, proceeds under mild reaction conditions, and can be used to synthesize a variety of natural product scaffolds.
Our approach to the indoline scaffold 1 of compounds 2– 15 is inspired by the classic Fischer indole synthesis,xvii,xviii and is presented in Scheme 2. We envisioned that an arylhydrazine 17 and an α-disubstituted aldehyde 18 would react in the presence of acid to afford enamine intermediate 19. Subsequent [3,3]-sigmatropic rearrangement and re-aromatization would provide aniline 20, which in turn would cyclize with loss of NH3 to furnish transient indolenine 16. Intramolecular attack by a proximal heteroatom substituent (X=NR or O) would deliver the desired product 1. This interrupted Fischer indolization process would allow for the formation of three new bonds, two heterocyclic rings and two stereogenic centers, one of which is quaternary (C3).
Scheme 2.

Proposed fragment coupling / cyclization cascade to access indoline 1.
A key feature of our approach to 1 is the ready availability of starting materials 17 and 18. The arylhydrazine coupling partners 17 could be easily prepared or accessed from commercial sources.xix Although the required α-branched aldehyde fragments 18 would not be obtainable commercially, isomeric lactols and hemiaminals 21 could likely serve as suitable aldehyde surrogates in the desired transformation (Scheme 3).xx,xxi In turn, lactols and hemiaminals 21 could be accessed by reduction of readily available lactones or lactams 22.xxii
Scheme 3.

Lactols and hemiaminals as latent aldehydes.
Only scattered examples of the interrupted Fischer indolization process have been reported over the past fifty years.xxiii,xxiv,xxv Most notable are the seminal studies by Grandberg summarized in Figure 2.xxiii In 1967, C2 substituted pyrrolidinoindoline 25 was prepared by reacting phenylhydrazine (23) and 5-chloro-3-methylpentan-2-one (24).xxiiia However, this method is not applicable to the synthesis of furoindolines, or to the more complex ring systems encountered in numerous natural products. It was later demonstrated that furoindolines could be accessed by the acid-promoted reaction of phenylhydrazines with α-disubstituted lactones.xxiiib For example, reaction of hydrazine 26 and lactone 27 in HCl/iPrOH afforded furoindoline 28 in 22% yield. This method bears limitations, such as the modest yields of products, the use of strongly acidic conditions, and the constraint to furoindoline ring systems. Despite these laudable efforts, and those of others,xxiv,xxv a general and mild method to access 1 using the interrupted Fischer indolization strategy outlined in Scheme 2 has remained elusive. Moreover, with the exception of our studies,xvi the notion that such a method could be used to prepare the indoline scaffold present in a multitude of complex biologically important compounds has not been realized.
Figure 2.

Grandberg’s syntheses of pyrrolidinoindoline 25 and furoindoline 28
2.Results and discussion
2.1. Synthesis of furoindolines
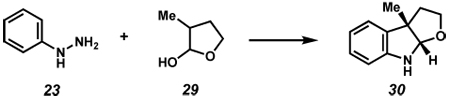
The feasibility of the proposed cascade reaction sequence of Scheme 2 was established in the context of furoindoline synthesis. Thus, the reaction between commercially available phenylhydrazine (23) and latent aldehyde 29 (1 equiv) was carried out under a variety of acidic conditions (Table 1). Lewis acids were examined and found to be ineffective (entries 1 and 2). However, use of p-toluenesulfonic acid, trifluoroacetic acid, or HCl each afforded the desired product 30 in modest yield (entries 3–5). Sulfuric acid-mediated reaction conditions were also explored, and ultimately provided the desired product in 87% yield (entry 6). Recognizing that a milder acid source would be more generally useful, acetic acid was examined. Although the use of glacial acetic acid afforded modest product yields (entry 7), employment of a 1:1 mixture of acetic acid and water at 60 °C furnished indoline 30 in 89% isolated yield (entry 8).xxvi
Table 1.
Survey of acids to promote furoindoline formation.
 | |||
|---|---|---|---|
| entry | acid source | conditions | yielda |
| 1 | PCl3 | benzene, 60 °C | < 5% |
| 2 | ZnCl2 | EtOH, 100 °C | < 5% |
| 3 | TsOH | EtOH, H2O, 60 °C | 51% |
| 4 | TFA | CH3CN, 60 °C | 64% |
| 5 | 5% HCl | CH3CN, 60 °C | 70% |
| 6 | 4% H2SO4 | CH3CN, 60 °C | 87% |
| 7 | AcOH | AcOH, 60 °C | 52% |
| 8 | AcOH | 1:1 AcOH/H2O, 60 °C | 89%b |
Unless otherwise noted, yields determined by 1H NMR analysis.
Isolated yield.
As shown in Table 2, a number of arylhydrazines bearing N-substitution were examined in the interrupted Fischer indolization reaction. In addition to parent arylhydrazine 23 (entry 1), N-methyl,xxvii N-benzyl, and N-allyl substituted hydrazines were deemed competent coupling partners (entries 2–4). Interestingly, the use of N-acetyl and N-Boc phenylhydrazines (entries 5 and 6) led predominantly to the recovery of unreacted starting materials.xxviii,xxix
Table 2.
Variation of the hydrazine N-substituent.
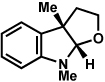
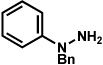
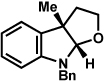
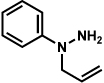
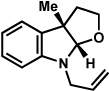
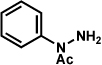
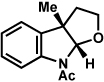
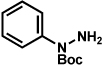
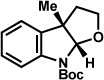
Conditions unless otherwise noted: lactol 29 (1 equiv), 1:1 AcOH/H2O, 60 °C.
AcOH as solvent.
Isolated yield.
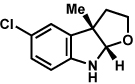
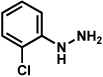
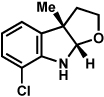
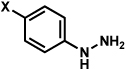
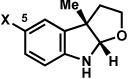
Substitution on the aryl ring of the hydrazine component was also investigated (Table 3). It was found that para, meta, and ortho substituents were tolerated under the reaction conditions (entries 1–6). Importantly, use of chlorohydrazines furnished haloindolines (entries 4 and 5), which could be further functionalized by transition metal-catalyzed cross-coupling chemistry. The transformation proceeded smoothly with p-methoxyphenylhydrazine as a substrate, thus affording C5-oxygenated products in good yields (entry 6). However, use of p-(trifluoromethyl)phenylhydrazine as a substrate led to low yields of product (entry 7).
Table 3.
Variation of the hydrazine aryl substituents.
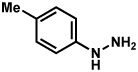
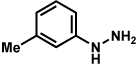
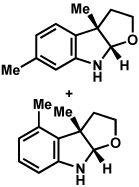
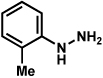
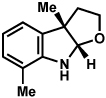
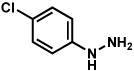
Conditions unless otherwise noted: lactol 29 (1 equiv), 1:1 AcOH/H2O, 60 °C.
Isolated yield.
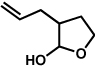
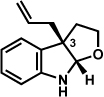
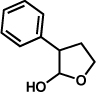
The scope of the lactol component for furoindoline synthesis was examined in the interrupted Fischer indolization process (Table 4). Allyl and phenyl substituents were tolerated, thus providing fused indolines with alternate C3 substitution (entries 1 and 2). It should be noted, however, that substrates bearing either a t-butyl or a methyl ester substituent led only to trace amounts of product formation (entries 3 and 4). Nonetheless, a 6-membered homologue of the furoindoline framework was accessible using this methodology using our standard reaction conditions (entry 5).
Table 4.
Variation of the lactol component.
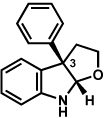
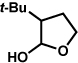
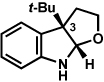
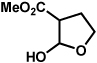
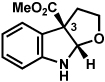
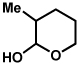
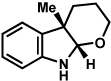
Conditions: hydrazine 23 (1 equiv), 1:1 AcOH/H2O, 60 °C.
Isolated yield.
2.2. Synthesis of pyrrolidinoindolines
Having established the viability of the interrupted Fischer indolization approach for the synthesis of furoindolines, we sought to develop the corresponding transformation that would enable the synthesis of pyrrolidinoindolines and related derivatives. Thus, hemiaminal 31 was prepared from the corresponding lactam following a known procedure, and then subjected to phenylhydrazine (23) in the presence of 1:1 H2O/AcOH (Scheme 4). To our delight, the interrupted Fischer indolization reaction proceeded smoothly at 100 °C and delivered the desired indoline 32 in 88% yield.
Scheme 4.

Synthesis of pyrrolidinoindoline 32.
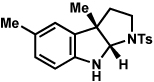
Analogous to our studies in the area of furoindoline synthesis, the interrupted Fischer indolization reaction was found to be an effective means to access a range of pyrrolidinoindolines. As shown in Table 5, a variety of arylhydrazines were tolerated in the transformation. Reactions of N-substituted arylhydrazines furnished the desired indoline products in good yield (entries 1–3), whereas a range of arylhydrazines bearing benzenoid substitution were deemed competent coupling partners (entries 4–9). Similar to the results obtained in our furoindoline studies, use of p-(trifluoromethyl)phenylhydrazine as a substrate led to low yields of product (entry 10).
Table 5.
Variation of the hydrazine component.
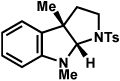
| entrya | hydrazine | product | yieldd |
|---|---|---|---|
| 1b | 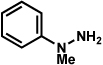 |
 |
81% |
| 2 | 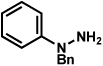 |
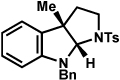 |
83% |
| 3 | 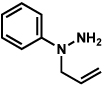 |
 |
70% |
| 4 | 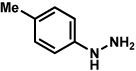 |
 |
71% |
| 5 | 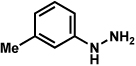 |
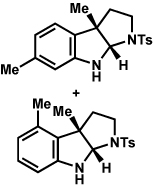 |
55% (4 : 3) |
| 6 | 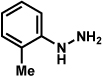 |
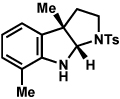 |
73% |
| 7 | 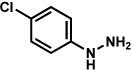 |
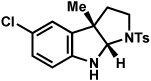 |
77% |
| 8 | 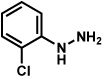 |
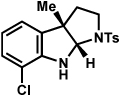 |
84% |
| 9c | 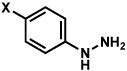 |
 |
X = OMe; 70% |
| 10 | X = CF3; < 5% |
Conditions unless otherwise noted: hemiaminal 31 (1 equiv), 1:1 AcOH/H2O, 100 °C.
23 °C, AcOH as solvent.
75 °C.
Isolated yield.
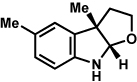
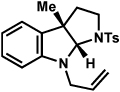
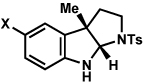
The scope of the hemiaminal component was also investigated (Table 6). C3-allylated and -phenylated pyrrolidinoindolines could be accessed without difficulty (entries 1 and 2). Of note, these pyrrolidinoindoline motifs are present in an array of medicinally important compounds, such as debromoflustramine B (5, Figure 1)iva and the hodgkinsine alkaloids.xxx Furthermore, a 6-membered homologue was prepared in 81% yield (entry 3) reminiscent of the communesin and perophoramidine core structures. Finally, it was determined that a carbamylated hemiaminal could be employed in place of a sulfonamide (entry 4).
Table 6.
Variation of the hemiaminal component.
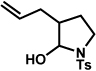
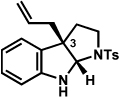
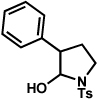
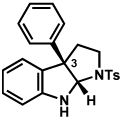
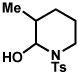
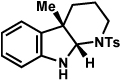
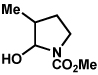
Conditions: hydrazine 23 (1 equiv), 1:1 AcOH/H2O, 100 °C.
Isolated yield.
As shown in Figure 3, the N-substituents of our pyrrolidinoindoline products can easily be manipulated. The sulfonamide group of 33 was removed upon treatment with Mg and NH4Cl in MeOHxxxi to provide pyrrolidinoindoline 34 in 79% yield.xxxii Additionally, carbamate 35 was converted to the corresponding N-methylated product 36 when reacted with Red-Al. The latter of these results is particularly notable given that many pyrrolidinoindoline natural products possess this N-methylated substitution pattern (e.g, Figure 1, 3–7).
Figure 3.

Manipulation of the N-substituent.
2.3. Formal total syntheses of physovenine and debromoflustramine B, and assembly of the communesin indoline scaffold
Having developed a powerful means to synthesize fused indoline ring systems, we examined the scope and limitations of our methodology in more complex settings. As shown in Scheme 5, the newly discovered transformation has been utilized to achieve a concise formal total synthesis of the furoindoline natural product physovenine (2).xxxiii Reaction of hydrazine 37xxxiv with lactol 29 in AcOH furnished furoindoline 38 in 77% yield, which has previously been converted to physovenine (2) in two additional steps.xva Although asymmetric routes to intermediate 38 have previously been reported, our single step route to (±)-38 is substantially shorter (one step compared to 7,xva or 18xxxiiig steps). Furthermore physovenine (2) can be optically resolved, on preparative scale, using column chromatography with cellulose triacetate.xxxiiio
Scheme 5.

Formal total synthesis of physovenine (2).
The interrupted Fischer indolization reaction could also be used to complete a formal total synthesis of the pyrrolidinoindoline natural product debromoflustramine B (5).xxxv Pyrrolidinone 39xxxvi was elaborated to hemiaminal 40 using a standard two-step sequence. Treatment of 40 with 1-allyl-1-phenylhydrazine in H2O/AcOH at 100 °C facilitated the key condensation/sigmatropic rearrangement to deliver bis(allylated)pyrrolidinoindoline 41. In turn, 41 was reacted with 2-methyl-2-butene in the presence of Grubbs’ second generation catalyst to afford bis(prenylated) derivative 42,xxxvii which was converted to 5 by reduction with Red-Al.
Finally, we explored the scope and limitations of our methodology in the context of the communesin natural products (Scheme 7).xxxviii Known sulfonamide 43xxxix was reacted with 1-ethoxypropene in the presence of Cs2CO3 to afford hetero-Diels–Alder product 44, following the general procedure described by Corey.xxxix Exposure of 44 to N-methyl phenylhydrazine (26) in 1:1 AcOH/H2O delivered indoline 45, which possesses the tetracyclic 6,5,6,6-ring system of the communesin alkaloids.xl As noted earlier, the previously described [3,3]-sigmatropic rearrangement strategies for the synthesis of fused indoline ring systems are not amenable to this complex scaffold.
Scheme 7.

Synthesis of communesin indoline scaffold 45.
2.4. Access to enantioenriched indoline products
Having demonstrated that the interrupted Fischer indolization reaction provides an effective means to access indoline scaffolds, we hoped to uncover a variant that would give access to enantioenriched indoline products. The most appealing scenario to achieve this goal would involve asymmetric catalysis. Thus, efforts were put forth to carry out the interrupted Fischer indolization reaction in the presence of chiral non-racemic phosphoric acids.xli
As shown in Scheme 8, this asymmetric transformation proved challenging. Despite an extensive survey of reaction conditions (e.g., variations in substrates, phosphoric acid promoter, stoichiometry, solvent, and temperature), only modest levels of enantioselectivity could be obtained. For example, reaction of hydrazine 23 and lactol 29 in the presence of 1.2 equivalents of phosphoric acid 46 (prepared from (R)-BINOL)xlii in benzene at 40 °C provided furoindoline 30 in 62% yield and 28% ee.xliii Similar results were obtained when hemiaminal substrates were employed in place of lactols.
Scheme 8.

Furoindoline synthesis using phosphoric acid promoter 46.
Given the difficulty in achieving a reagent or catalyst-controlled asymmetric interrupted Fischer indolization, we turned to an auxiliary-based approach.xliv Thus, Nishida’s enantioenriched arylhydrazine 50 was prepared using the sequence shown in Scheme 9.xxve,xlv,xlvi Bromobenzene (47) was coupled with commercially available enantioenriched amine (–)-48 under Pd catalysis to provide aniline 49. Using a standard protocol, aniline 49 was converted to the targeted hydrazine 50 in 81% yield over two steps.
Scheme 9.

Synthesis of arylhydrazine 50.
The utility of arylhydrazine 50 in our interrupted Fischer indolization process was evaluated in the context of furoindoline synthesis (Figure 4). Gratifyingly, the reaction of 50 and lactol 29 proceeded smoothly under a variety of acidic conditions. When the reaction was carried out in the presence of 3 equivalents of chloroacetic acid in benzene at 40 °C, an 80% yield of diastereomeric indoline products 51 and 52 was obtained (d.r.=2.4:1).xlvii The isomers were easily separable using conventional flash column chromatography on silica gel. The major isomer 51 was treated with Pd(OH)2 and 1,4-cyclohexadiene in EtOH to remove the auxiliary and deliver optically enriched indoline 30.xlviii,xlix The ee of 30 was found to be 97%,xxii thus demonstrating that our methodology can be utilized to access enantioenriched products. The absolute configuration of 30 was determined based on correlation to known data,xxxiiic and was found to be as depicted in Figure 4.
Figure 4.

Synthesis of enantioenriched 30.
3. Conclusions
In summary, we have developed an efficient method to access the fused indoline ring systems present in a variety of natural products. Our interrupted Fischer indolization strategy involves the condensation of readily available hydrazines with latent aldehydes to deliver indoline-containing products by way of a tandem [3,3]-sigmatropic rearrangement / cyclization cascade sequence. The method is convergent, mild, operationally simple, broad in scope, and can be used to access enantioenriched products. In addition, our approach is amenable to the synthesis of furoindoline and pyrrolidinoindoline natural products as demonstrated by the concise formal total syntheses of physovenine and debromoflustramine B. We expect that the interrupted Fischer indolization strategy will enable the synthesis of more complex targets such as the communesins and akuammiline alkaloids. Such studies in the realm of natural product synthesis are currently underway in our laboratory.
4. Experimental
4.1 Representative experimental procedure for furoindoline synthesis (Table 2, entry 1)
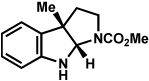
Lactol 29 (126 mg, 1.22 mmol) was dissolved in a 1:1 mixture of acetic acid and water (6 mL). Phenylhydrazine (23) (0.121 mL, 1.23 mmol) was added to the resulting mixture. The reaction was heated to 60 °C for 4.5 h, then cooled to 23 °C, and quenched with a solution of sat. aq. NaHCO3 (15 mL). The resulting mixture was extracted with EtOAc (3 × 15 mL). The combined organic layers were dried over MgSO4. Evaporation of the solvent under reduced pressure afforded the crude product. Purification by flash chromatography (7:1 hexanes:EtOAc) afforded indoline 30 (196 mg, 89% yield). Rf 0.7 (1:1 EtOAc:hexanes); 1H NMR (300 MHz, CDCl3): δ 7.08 (d, J = 7.2, 1H), 7.05 (t, J = 7.5, 1H), 6.76 (t, J = 7.5, 1H), 6.59 (d, J = 7.8, 1H), 5.28 (s, 1H), 3.96 (ddd, J = 8.4, 7.2, 1.8, 1H),3.56 (ddd, J = 10.8, 8.4, 5.1, 1H), 2.13 (ddd, J = 11.7, 5.4, 1.5, 1H), 2.07 (ddd, J = 11.7, 7.2, 4.2, 1H), 1.47 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 148.8, 133.9, 127.9, 122.9, 118.8, 108.8, 99.5, 67.3, 53.8, 41.4, 24.7; IR (film): 2967, 2845, 1611, 1486, 1265, 1055 cm−1; HRMS-ESI (m/z) [M + H]+ calcd for C11H14NO, 176.1075; found 176.1078.
4.2. Representative experimental procedure for pyrrolidinoindoline synthesis (Table 6, entry 1)
3-Allyl-1-tosylpyrrolidin-2-olxxii (105 mg, 0.37 mmol) was dissolved in a 1:1 mixture of acetic acid and water (1.8 mL). Phenylhydrazine 23 (0.036 mL, 0.36 mmol) was added to the resulting mixture. The reaction was heated to 100 °C for 1 h 40 min, cooled to 23 °C, and then diluted with Et2O (20 mL). The reaction mixture was then quenched with sat. aq. NaHCO3 (20 mL), and the layers were separated. The aqueous layer was extracted with Et2O (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over MgSO4, and concentrated in vacuo to afford the crude indoline product. Purification by flash chromatography (18:1:1 benzene:Et2O:CH2Cl2) afforded the 3-allyl-indoline (Table 6, entry 1) as a yellow oil (88.0 mg, 68% yield). Rf 0.6 (8:1:1 benzene:Et2O:CH2Cl2); 1H NMR (500 MHz, CDCl3): δ 7.75 (d, J = 8.0, 2H), 7.32 (d, J = 8.0, 2H), 7.08 (t, J = 7.5, 1H), 7.00 (d, J = 7.0, 1H), 6.75 (t, J = 7.5, 1H), 6.62 (d, J = 7.5, 1H), 5.55 (ddt, J = 16.8, 10.0, 7.5, 1H), 5.13 (s, 1H), 4.96–5.00 (m, 2H), 4.84 (s, 1H), 3.43 (ddd, J = 10.0, 8.0, 2.0, 1H), 3.13 (ddd, J = 10.5, 10.5, 6.0, 1H), 2.44 (s, 3H), 2.31 (ddd J = 19.5, 13.5, 7.5, 2H), 2.07 (ddd, J = 6.,5, 6.0, 2.0, 1H), 1.84 (ddd, J = 10.5, 8.0, 6.5, 1H); 13C NMR (125 MHz, CDCl3): δ 149.1, 143.7, 136.4, 133.5, 131.4, 130.0 128.8, 127.3 123.2, 119.3, 118.8, 109.7, 82.7, 58.0, 47.6, 42.3, 36.2, 21.7; IR (neat): 3391, 3076, 1611, 1485, 1337, 1160 cm−1; HRMS-ESI (m/z) [M+Na]+ calcd for C20H22N2O2SNa, 377.1300; found, 377.1298.
4.3 Synthesis of indoline diastereomers 51 and 52
To a mixture of aryl hydrazine 50 (52.4 mg, 0.20 mmol), lactol 29 (20.6 mg, 0.20 mmol), and benzene (1 mL) was added chloroacetic acid (56.7 mg, 0.60 mmol). The resulting mixture was heated at 40 °C for 24 h. The reaction mixture was cooled to 23 °C, diluted with CH2Cl2 (20 mL), washed with sat. aq. NaHCO3 (5 mL) and extracted with CH2Cl2 (10 mL). The combined organic layers were dried over MgSO4 and evaporated to dryness. Purification by flash chromatography (15:1 →10:1 hexanes:EtOAc) afforded a mixture of diastereomers as an orange solid (53.0 mg, 80% yield, 2.4:1 dr). To separate the diastereomers the mixture was repurified by flash chromatography, under the same conditions. The stereochemical configurations of 51 and 52 were inferred after the conversion of 51 to (+)-30. Indoline 51: Rf 0.5 (4:1 hexanes:EtOAc); 1H NMR (500 MHz, CDCl3) δ 8.23 (d, J = 8.0, 1H), 7.96 (d, J = 7.5, 1H), 7.82 (t, J = 8.0, 2H), 7.60 (t, J = 7.0, 1H), 7.56 (t, J = 7.5, 1H), 7.49 (t, J = 8.0, 1H), 7.11 (d, J = 7.5, 1H), 6.91 (t, J = 7.5, 1H), 6.03 (d, J = 7.5, 1H), 5.65 (s, 1H), 5.56 (q, J = 7.0, 1H), 4.03 (t, J = 8.0, 1H), 3.70–3.75 (m, 1H), 2.28 (dd, J = 11.5, 4.5, 1H), 1.86 (d, J = 6.5, 3H), 1.58 (s, 3H); 13C NMR (125 MHz, CDCl3) δ149.1, 139.4, 134.5, 133.9, 130.8, 129.1, 127.8, 127.5, 125.9, 125.8, 125.2, 123.6, 122.5, 122.4, 117.3, 106.0, 101.4, 66.7, 52.3, 51.7, 41.9, 25.8, 19.0; IR (neat): 3046, 2963, 2852, 1606, 1594, 1487, 1459, 1395, 1298, 1236, 1013 cm−1; HRMS-ESI (m/z) [M + Na]+ calcd for C23H23NONa, 352.1677; found 352.1686; [α]D24.4 + 125.4 (c 0.01, CHCl3). Indoline 52: Rf 0.5 (4:1 hexanes:EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.95 (d, J = 8.5, 1H), 7.90 (d, J = 8.0, 1H), 7.84 (d, J = 8.5, 1H), 7.74 (d, J = 7.5, 1H), 7.45–7.54 (m, 3H), 7.14 (t, J = 8.0, 1H), 7.10 (d, J = 7.0, 1H), 6.75 (d, J = 7.0, 1H), 6.51 (d, J = 7.5, 1H), 5.52 (q, J = 7.0, 1H), 4.69 (s, 1H), 3.93 (t, J = 7.5, 1H), 3.53 (ddd, J = 13.0, 8.5, 4.5, 1H), 2.18 (dd, J = 12.0, 4.5, 1H), 1.98 (ddd, J = 11.5, 11.5, 7.0, 1H), 1.90 (d, J = 6.5, 3H), 1.24 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 148.8, 136.3, 134.7, 133.8, 131.7, 128.7, 128.1, 128.0, 126.2, 125.5, 125.4, 124.2, 123.2, 122.7, 117.3, 105.0, 101.0, 67.0, 52.2, 49.3, 41.2, 24.7, 18.1; IR (film): 3681, 2973, 2845, 1605, 1487, 1215, 1059 cm−1; HRMS-ESI (m/z) [M + Na]+ calcd for C23H23NONa, 352.1677; found 352.1680; [α]D24.2 –54.6 (c 0.01, CHCl3).
4.4 Synthesis of indoline (+)-30
A mixture of indoline 51 (32.9 mg, 0.1 mmol), 1,4-cyclohexadiene (80.0 mg, 1.0 mmol), and palladium hydroxide (20% wt on carbon, 10.0 mg) in ethanol (1 mL) was heated at 80 °C for 6 h. The reaction mixture was cooled to 23 °C, filtered through celite, washed with CH2Cl2 (10 mL), and the solvent was removed under reduced pressure. Purification by flash chromatography (5:1 hexanes:EtOAc) furnished furoindoline (+)-30 (14.0 mg, 80% yield, 97% ee). [α]D24.3 +124.5 (c 0.01, CHCl3), SFC (CHIRALPAK AS-H, CO2/MeOH = 9/10, flow 1.5 mL/min, at 23 °C, detection at 254 nm) tR 3.06 min (major) and tR 4.43 min (minor). The absolute configuration of 30 was determined based on correlation to known data.xxxiiic
Supplementary Material
Scheme 6.

Formal total synthesis of debromoflustramine B (5).
Acknowledgments
The authors are grateful to the NIH-NIGMS (R00 GM079922), the University of California, Los Angeles and Boehringer Ingelheim for financial support. We also thank Materia Inc. for the donation of chemicals, the Garcia–Garibay laboratory (UCLA) for the generous access to instrumentation, Dr. John Greaves (UC Irvine) for mass spectra, and Professors Ken Houk (UCLA), Patrick Harran (UCLA), and Jon Antilla (University of South Florida) for fruitful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This manuscript is dedicated to Professor Brian M. Stoltz on the occasion of his receipt of the Tetrahedron Young Investigator Award.
Supplementary Data
Supplementary data associated with this article, including experimental procedures, characterization data, and NMR spectra, can be found in the online version.
References and notes
- i.a) Hudlicky T, Reed JW. The Way of Synthesis. Weinheim: Wiley–VCH; 2007. [Google Scholar]; b) Wender PA, Verman VA, Paxton TJ, Pillow TH. Acc. Chem. Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- ii.For a review of these alkaloids, see: Takano S, Ogasawara K. Alkaloids. 1989;36:225–251.
- iii.Physostigmine (3) has been used to treat glaucoma, Alzheimer’s disease, and myasthenia gravis. For a pertinent review, see Triggle DJ, Mitchell JM, Filler R. CNS Drug Rev. 1998;4:87–136.
- iv.a) Carle JS, Christophersen C. J. Am. Chem. Soc. 1979;101:4012–4013. [Google Scholar]; b) Holst PB, Anthoni U, Christophersen C, Nielsen PH. J. Nat. Prod. 1994;57:997–1000. doi: 10.1021/np50109a020. [DOI] [PubMed] [Google Scholar]
- v.For a pertinent review, see: Steven A, Overman LE. Angew. Chem. Int. Ed. 2007;46:5488–5508. doi: 10.1002/anie.200700612.
- vi.a) Nakao Y, Yeung BKS, Yoshida WY, Scheuer PJ, Kelly-Borges M. J. Am. Chem. Soc. 1995;117:8271–8272. [Google Scholar]; b) Yeung BKS, Nakao Y, Kinnel RB, Carney JR, Yoshida WY, Scheuer PJ, Kelly-Borges M. J. Org. Chem. 1996;61:7168–7173. doi: 10.1021/jo960725e. [DOI] [PubMed] [Google Scholar]; c) Nakao Y, Kuo J, Yoshida WY, Kelly M, Scheuer PJ. Org. Lett. 2003;5:1387–1390. doi: 10.1021/ol026830u. [DOI] [PubMed] [Google Scholar]; d) Takayama H, Mori I, Kitajima M, Aimi N, Lajis NH. Org. Lett. 2004;6:2945–2948. doi: 10.1021/ol048971x. [DOI] [PubMed] [Google Scholar]
- vii.For synthetic studies, see: Newhouse T, Baran PS. J. Am. Chem. Soc. 2008;130:10886–10887. doi: 10.1021/ja8042307. Newhouse T, Lewis CA, Baran PS. J. Am. Chem. Soc. 2009;131:6360–6361. doi: 10.1021/ja901573x. Marsden SP, Watson EL, Raw SA. Org. Lett. 2008;10:2905–2908. doi: 10.1021/ol801028e. Toumi M, Couty F, Marrot J, Evano G. Org. Lett. 2008;10:5027–5030. doi: 10.1021/ol802155n. Espejo VR, Rainier JD. J. Am. Chem. Soc. 2008;130:12894–12895. doi: 10.1021/ja8061908. Matsuda Y, Kitajima M, Takayama H. Org. Lett. 2008;10:125–128. doi: 10.1021/ol702637r.
- viii.a) Subramaniam G, Hiraku O, Hayashi M, Koyano T, Komiyama K, Kam T-S. J. Nat. Prod. 2007;70:1783–1789. doi: 10.1021/np0703747. [DOI] [PubMed] [Google Scholar]; b) Jagetia G, Baliga MS, Venkatesh P, Ulloor JN, Mantena SK, Genebriera J, Mathuram VJ. Pharm. Pharmacol. 2005;57:1213–1219. doi: 10.1211/jpp.57.9.0017. [DOI] [PubMed] [Google Scholar]; c) Ramírez A, García-Rubio S. Curr. Med. Chem. 2003;10:1891–1915. doi: 10.2174/0929867033457016. [DOI] [PubMed] [Google Scholar]
- ix.For pertinent total syntheses, see: Dounay AB, Humphreys PG, Overman LE, Wrobleski AD. J. Am. Chem. Soc. 2008;130:5368–5377. doi: 10.1021/ja800163v. Shen L, Zhang M, Wu Y, Qin Y. Angew. Chem. Int. Ed. 2008;47:3618–3621. doi: 10.1002/anie.200800566. Jones SB, Simmons B, MacMillan DWC. J. Am. Chem. Soc. 2009;131:13606–13607. doi: 10.1021/ja906472m. Zhang M, Huang X, Shen L, Qin Y. J. Am. Chem. Soc. 2009;131:6013–6020. doi: 10.1021/ja901219v.
- x.For isolation, see: Verbitski SM, Mayne CL, Davis RA, Concepcion GP, Ireland CM. J. Org. Chem. 2002;67:7124–7126. doi: 10.1021/jo026012f. For the total synthesis of (±)-12, see: b) Fuchs JR, Funk RL. J. Am. Chem. Soc. 2004;126:5068–5069. doi: 10.1021/ja049569g.
- xi. Numata A, Takahashi C, Ito Y, Takada T, Kawai K, Usami Y, Matsumura E, Imachi M, Ito T, Hasegawa T. Tetrahedron Lett. 1993;34:2355–2358. Jadulco R, Edrada RA, Ebel R, Berg A, Schaumann K, Wray V, Steube K, Proksch P. J. Nat. Prod. 2004;67:78–81. doi: 10.1021/np030271y. For a comprehensive review, see: c) Siengalewicz P, Gaich T, Mulzer J. Angew. Chem. Int. Ed. 2008;47:8170–8176. doi: 10.1002/anie.200801735.
- xii.For biological studies, see: Williams NS, Burgett AWG, Atkins AS, Wang X, Harran PG, McKnight SL. Proc. Natl. Acad. Sci. U.S.A. 2007;104:2074–2079. doi: 10.1073/pnas.0611340104. Wang G, Shang L, Burgett AWG, Harran PG, Wang X. Proc. Natl. Acad. Sci. U.S.A. 2007;104:2068–2073. doi: 10.1073/pnas.0610832104. For a review of synthetic studies, see: c) Lachia M, Moody CJ. Nat. Prod. Rep. 2008;25:227–253. doi: 10.1039/b705663j.
- xiii.Kam T-S, Tan S-J, Ng S-K, Komiyama K. Org. Lett. 2008;10:3749–3752. doi: 10.1021/ol801354s. [DOI] [PubMed] [Google Scholar]
- xiv.For examples of pyrrolidinoindoline and furoindoline syntheses from substituted indoles, see: Depew KM, Marsden SP, Zatorska D, Zatorski A, Bornmann WG, Danishefsky SJ. J. Am. Chem. Soc. 1999;121:11953–11963. Austin JF, Kim SG, Sinz CJ, Xiao WJ, MacMillan DWC. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5482–5487. doi: 10.1073/pnas.0308177101. Movassaghi M, Schmidt MA. Angew. Chem. Int. Ed. 2007;46:3725–3728. doi: 10.1002/anie.200700705. Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238–241. doi: 10.1126/science.1170777.
- xv.For examples of pyrrolidinoindoline and furoindoline syntheses from substituted oxindoles, see: Matsuura T, Overman LE, Poon DJ. J. Am. Chem. Soc. 1998;120:6500–6503. Trost BM, Zhang Y. J. Am. Chem. Soc. 2006;128:4590–4591. doi: 10.1021/ja060560j.
- xvi.For a preliminary communication describing this chemistry, see: Boal BW, Schammel AW, Garg NK. Org. Lett. 2009;11:3458–3461. doi: 10.1021/ol901383j.
- xvii.For reviews, see: Robinson B. Chem. Rev. 1963;63:373–401. Robinson B. Chem. Rev. 1969;69:227–250. doi: 10.1021/cr60262a003.
- xviii.a) Fischer E, Jourdan F. Ber. 1883;16:2241–2245. [Google Scholar]; b) Fischer E, Hess O. Ber. 1884;17:559–568. [Google Scholar]
- xix.Hydrazines could be employed in either free-base form or as the hydrochloric acid salts.
- xx.All lactol and hemiaminal substrates were employed as an inconsequential mixture of diastereomers.
- xxi.Lactols and hemiaminals have previously been employed in modifications of the Fischer indole synthesis, see: Campos KR, Woo JCS, Lee S, Tillyer RD. Org. Lett. 2004;6:79–82. doi: 10.1021/ol036113f. Brodfuehrer PR, Chen B-C, Sattelberg TR, Smith PR, Reddy JP, Stark DR, Quinlan SL, Reid JG, Thottathil JK, Wang S. J. Org. Chem. 1997;62:9192–9202. Adachi H, Palaniappan KK, Ivanov AA, Bergman N, Gao Z-G, Jacobson KA. J. Med. Chem. 2007;50:1810–1827. doi: 10.1021/jm061278q.
- xxii.See Supplementary Data for details.
- xxiii. Grandberg II, Zuyanova TI, Afonina NI, Ivanova TA. Dokl. Akad. Nauk SSSR. 1967;176:583–585. For a synthesis of furoindolines, albeit in modest yield, see: b) Grandberg II, Tokmakov GP. Khim. Geterotsikl. Soedin. 1975:207–210.
- xxiv.Takano et al. has synthesized a pyrrolidinoindoline product using an interrupted Fischer indolization reaction; see: Takano S, Moriya M, Iwabuchi Y, Ogasawara K. Chem. Lett. 1990:109–112. Takano S, Ogasawara K, Iwabuchi R, Moriya M. 1991 JP 03112989 A 19910514.
- xxv.For other examples, see: Tsuji R, Nakagawa M, Nishida A. Heterocycles. 2002;58:587–593. Britten AZ, Bardsley WG, Hill CM. Tetrahedron. 1971;27:5631–5639. Rosenmund P, Sadri E. Liebigs Ann. Chem. 1979:927–943. Rosenmund P, Gektidis S, Brill H, Kalbe R. Tetrahedron Lett. 1989;30:61–62. Nishida A, Ushigome S, Sugimoto A, Arai S. Heterocycles. 2005;66:181–185.
- xxvi.For the use of H2O/AcOH to facilitate the Fischer indole synthesis, see: Desaty D, Keglević D. Croat. Chem. Acta. 1964;36:103–109. Keglević D, Stojanac N, Desaty D. Croat. Chem. Acta. 1961;33:83–88.
- xxvii.Interestingly, the reaction between N-methylphenylhydrazine (26) and lactol 29 in 1:1 H2O/AcOH leads to the desired product, albeit with significant (ca. 20–30%) formation of NH furoindoline 30.
-
xxviii.Attempts to generate furoindoline 30 from i and 29 using Buchwald’s methodology were unsuccessful despite an extensive survey of acidic conditions. For Buchwald’s modification of the Fischer indole synthesis, see:
Wagaw S, Yang BH, Buchwald SL. J. Am. Chem. Soc. 1998;120:6621–6622.
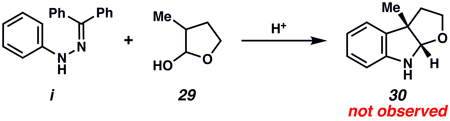
-
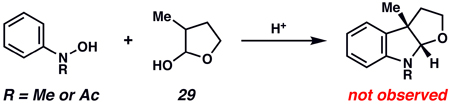
xxix.Efforts to synthesize furoindoline products from the reaction of aryl hydroxylamine derivatives and 29 were not successful.
- xxx.Guéritte-Voegelein F, S#x000E9;venet T, Pusset J, Adeline M-T, Gillet B, Beloiel J-C, Gu#x000E9;nard D, Potier P, Rasolonjanahary R, Kordon C. J. Nat. Prod. 1992;55:923–930. doi: 10.1021/np50085a012. [DOI] [PubMed] [Google Scholar]
- xxxi.May JA, Stoltz BM. Tetrahedron. 2006;62:5262–5271. [Google Scholar]
- xxxii.The N-sulfonyl group of pyrrolidinoindoline products could also be removed using Na2K–SG(1), a solid-supported alkali metal reagent developed by SiGNa Chemistry, Inc.; see: Nandi P, Redko MY, Petersen K, Dye JL, Lefenfeld M, Vogt PF, Jackson JE. Org. Lett. 2008;10:5441–5444. doi: 10.1021/ol8021337.
- xxxiii.For syntheses of 2, see ref xva and the following: Kulkarni MG, Dhondge AP, Borhade AS, Gaikwad DD, Chavhan SW, Shaikh YB, Nigdale VB, Desai MP, Birhade DR, Shinde MP. Eur. J. Org. Chem. 2009:3875–3877. doi: 10.3762/bjoc.5.4. Shao Z, Zhang H. Youji Huaxue. 2005;25:282–289. Sunazuka T, Yoshida K, Kojima N, Shirahata T, Hirose T, Handa M, Yamamoto D, Harigaya Y, Kuwajima I, Omura S. Tetrahedron Lett. 2005;46:1459–1461. Zhang TY, Zhang H. Tetrahedron Lett. 2002;43:1363–1365. Morales-Ríos MS, Santos-Sánchez NF, Joseph-Nathan P. J. Nat.Prod. 2002;65:136–141. doi: 10.1021/np010475j. Tanaka K, Taniguchi T, Ogasawara K. Tetrahedron Lett. 2001;42:1049–1052. ElAzab AS, Taniguchi T, Ogasawara K. Org. Lett. 2000;2:2757–2759. doi: 10.1021/ol006065o. Ishibashi H, Kobayashi T, Machida N, Tamura O. Tetrahedron. 2000;56:1469–1473. Node M, Hao X-J, Nishide K, Fuki K. Chem. Pharm. Bull. 1996;44:715–719. Yu QS, Lu BY. Heterocycles. 1994;39:519–525. Morales-Ríos MS, Santos-Sánchez NF, Fragoso-Vázquez MJ, Alagille D, Villagómez-Ibarra JR, Joseph-Nathan P. Tetrahedron. 2003;59:2843–2853. Clark AJ, Jones K. Tetrahedron. 1992;48:6875–6882. Horne S, Taylor N, Collins S, Rodrigo R. J. Chem. Soc. Perkin Trans. 1. 1991;12:3047–3051. Takano S, Moriya M, Ogasawara K. J. Org. Chem. 1991;56:5982–5984. Yu Q, Liu C, Brzostowska M, Chrisey L, Brossi A, Greig NH, Atack JR, Soncrant TT, Rapoport SI, Radunz H. Helv. Chim. Acta. 1991;74:761–766. Luo Y, Yu Q, Chrisey L, Brossi A. Heterocycles. 1990;31:283–287. Shishido K, Azuma T, Shibuya M. Tetrahedron Lett. 1990;31:219–220. Shishido K, Shitara E, Komatsu H, Hiroya K, Fukumoto K, Kametani T. J. Org. Chem. 1986;51:3007–3011. Tadamasa O. Tetrahedron Lett. 1971;12:4391–4392. Dale FJ, Robinson B. J. Pharm. Pharmacol. 1970;22:889–896. doi: 10.1111/j.2042-7158.1970.tb08469.x. Longmore RB, Robinson B. Chem. Commun. 1967;32:2184–2192.
- xxxiv.Srivastava S, Ruane PH, Toscano JP, Sullivan MB, Cramer CJ, Chiapperino D, Reed EC, Falvey DE. J. Am. Chem. Soc. 2000;122:8271–8278. [Google Scholar]
- xxxv.For syntheses of 5, see ref xivd and the following: Rivera-Becerril E, Joseph-Nathan P, P#x000E9;rez-Álvarez VM, Morales-Ríos MS. J. Med. Chem. 2008;51:5271–5284. doi: 10.1021/jm800277g. Cardoso ASP, Marques MMB, Srinivasan N, Prabhakar S, Lobo AM. Tetrahedron. 2007;63:10211–10225. Miyamoto H, Okawa Y, Nakazaki A, Kobayashi S. Tetrahedron Lett. 2007;48:1805–1808. López-Alvarado P, Caballero E, Avendaño C, Men#x000E9;ndez JC. Org. Lett. 2006;8:4303–4306. doi: 10.1021/ol061631m. Dix AV, Meseck CM, Lowe AJ, Mitchell MO. Bioorg. Med. Chem. Lett. 2006;16:2522–2524. doi: 10.1016/j.bmcl.2006.01.093. Morales-Ríos MS, Rivera-Becerril E, Joseph-Nathan P. Tetrahedron: Asymmetry. 2005;16:2493–2499. Tan GH, Zhu X, Ganesan A. Org. Lett. 2003;5:1801–1803. doi: 10.1021/ol034516+. Cardoso AS, Srinivasan N, Lobo AM, Prabhakar S. Tetrahedron Lett. 2001;42:6663–6666. Morales-Ríos MS, Suárez-Castillo OR, Trujillo-Serrato JJ, Joseph-Nathan P. J. Org. Chem. 2001;66:1186–1192. doi: 10.1021/jo0012647. Morales-Ríos MS, Suárez-Castillo OR, Joseph-Nathan P. J. Org. Chem. 1999;64:1086–1087. doi: 10.1021/jo0012647. Somei M, Yamada F, Izumi T, Nakajou M. Heterocycles. 1997;45:2327–2330. Jensen J, Anthoni U, Christophersen C, Nielsen PH. Acta Chem. Scand. 1995;49:68–71. Bruncko M, Crich D, Samy R. J. Org. Chem. 1994;59:5543–5549. Muthusubramanian P, Carle JS, Christophersen C. Acta Chem. Scand. Ser. B. 1983;B37:803–807.
- xxxvi.Khoukhi N, Vaultier M, Carri#x000E9; R. Tetrahedron. 1987;43:1811–1822. [Google Scholar]
- xxxvii.42 has previously been converted to debromoflustramine B (5) by reduction with LiAlH4; see ref xxxve.
- xxxviii.For the total synthesis of communesin F, see: Yang J, Wu H, Shen L, Qin Y. J. Am. Chem. Soc. 2007;129:13794–13795. doi: 10.1021/ja075705g.
- xxxix.Steinhagen H, Corey EJ. Angew. Chem. Int. Ed. 1999;38:1928–1931. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1928::AID-ANIE1928>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- xl.Indoline 45 has previously been prepared by an intermolecular aza-Diels–Alder strategy; see ref xxxi.
- xli.a) Terada M. Chem. Commun. 2008:4097–4112. doi: 10.1039/b807577h. [DOI] [PubMed] [Google Scholar]; b) Taylor MS, Jacobsen EN. Angew. Chem. Int. Ed. 2006;45:1520–1543. doi: 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]
- xlii.Akiyama T, Morita H, Itoh J, Fuchibe K. Org. Lett. 2005;7:2583–2585. doi: 10.1021/ol050695e. [DOI] [PubMed] [Google Scholar]
- xliii.The enantiomer formed in excess is believed to have the (R,R) configuration, as depicted in Scheme 8, based on correlation to known data (see ref xxxiiic).
- xliv.Takano has shown that a proximal stereogenic center can be used to induce diastereoselectivity in a Fischer indolization reaction; see ref xxiva.
- xlv.Although experimental procedures for the synthesis of 50 were not reported previously, the synthetic route shown in Scheme 9 parallels that described by Nishida (see ref xxve). Experimental details for the synthesis of 50 are included in the Supplementary Data that accompanies this manuscript.
- xlvi.Nishida has utilized 50 to synthesize an optically enriched pyrrolidinone, albeit in modest yield;
- xlvii.The stereochemical configurations of 51 and 52 were inferred after the conversion of 51 to (+)-30.
- xlviii.30 can be converted to physovenine (2) in four steps; see ref xxxiiic.
- xlix.Numerous attempts to cleave the auxiliary using H2 with various metal catalysts led to reduction of the N,O-acetal linkage, a problem that was not encountered when cleaving the auxiliary from a pyrrolidinoindoline derivative (see ref xxve).


Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.