

Abstract
An increase in the risk of cancer is one of the consequences of obesity. The predominant cancers associated with obesity have a hormonal basis and include breast, prostate, endometrium, colon and gall-bladder cancers. Leptin, the key player in the regulation of energy balance and body weight control also acts as a growth factor on certain organs in both normal and disease states. Therefore, it is plausible that leptin acts to promote cancer growth by acting as a mitogenic agent. However, a direct role for leptin in endometrial cancer has not been demonstrated. In this study, we analyzed the proliferative role of leptin and the mechanism(s) underlying this action in endometrial cancers which express both short and long isoforms of leptin receptors. Treatment with leptin resulted in increased proliferation of ECC1 and Ishikawa cells. The promotion of endometrial cancer cell proliferation by leptin involves activation of STAT3 and ERK2 signaling pathways. Moreover, leptin-induced phosphorylation of ERK2 and AKT was dependent on JAK/STAT activation. Therefore blocking its action at the JAK/STAT level could be a rational therapeutic strategy for endometrial carcinoma in obese patients. We also found that leptin potently induces invasion of endometrial cancer cells in a Matrigel invasion assay. Leptin-stimulated invasion was effectively blocked by pharmacological inhibitors of JAK/STAT (AG490) and phosphatidylinositol 3-kinase (LY294002). Taken together these data indicate that leptin promotes endometrial cancer growth and invasiveness and implicate the JAK/STAT and AKT pathways as critical mediators of leptin action. Our findings have potential clinical implications for endometrial cancer progression in obese patients.
Introduction
Obesity is considered as an important risk factor for many serious medical conditions. It impacts on the risk and prognosis of some of the more common forms of cancer, providing one of the few preventive interventions capable of making a significant impact on cancer (Calle et al. 2003). The management of normal body weight is regulated by adipocytokines that act on the brain to regulate food intake. The adipocytokines are biologically active polypeptides that are produced exclusively or substantially by white adipose tissue pre-adipocytes and mature adipocytes and act by endocrine, paracrine and auto-crine mechanisms (Matsuzawa et al. 1999, Rose et al. 2004). Leptin, a product of the obese (ob) gene is a neuroendocrine hormone that has attracted attention since its identification in 1995 (Halaas et al.1995, MacDougald et al. 1995). It is a multifunctional peptide hormone with wide-ranging biological activities including appetite regulation, bone formation, reproductive function and angiogenesis (Bouloumie et al. 1998, Sierra-Honigmann et al. 1998, Huang & Li 2000). These biological activities suggest an important role in cancer proliferation, invasion and metastasis (Somasunder et al. 2004a,b).
Leptin circulates as a 16 kDa protein partially bound to plasma proteins (Houseknecht et al. 1996, Sinha et al. 1996) and exerts its actions through its specific receptors present in a variety of tissues localized to the cell membrane (Bjorback et al. 1997). Leptin receptor belongs to a family of class I cytokine receptors, which typically contain a cytokine receptor homologous domain in the extra cellular region (Tartaglia 1997). All six isoforms have a similar extracellular ligand-binding domain at the amino terminus but differ at the intracellular carboxy-terminal domain. While all five short isoforms have transmembrane domains, only the long form has the intracellular motifs necessary for activation of signaling pathways (Tartaglia 1997). As with other class I cytokine receptors, the leptin signaling is thought to be transmitted mainly by the JAK/STAT pathway (Bahrenberg et al. 2002, Ahima & Osei 2004). JAKs associate constitutively with conserved box 1 and 2 motifs in the intracellular domain of Ob-Rb (long isoform leptin receptor). Binding of leptin to Ob-Rb results in autophosphorylation of JAK1 and JAK2 as well as phosphorylation of the cytoplasmic domain of Ob-Rb and the downstream transcription factors STATs (Ahima & Osei 2004). The leptin signal is terminated by induction of SOCS-3 (suppressor of cytokine signaling), a member of a family of proteins which inhibits the JAK/STAT signaling cascade (Bjorbaek et al. 1999, Emilsson et al. 1999). SOCS proteins have a variable amino-terminal domain, a central SH2 domain and a carboxy-terminal domain, termed the SOCS-box motif. They are induced by cytokines and act in a negative feedback loop to inhibit the receptor. Thus overexpression of SOCS-3 inhibits leptin-mediated tyrosine phosphorylation of JAK2 (Bjorbaek et al. 1998, 1999, Emilsson et al. 1999). Whether activation of the above pathways by leptin occurs in endometrial cancer cells remains unknown.
Endometrial cancer is the most common gynecological malignancy in developed countries, with approximately 40 000 new diagnoses each year in the US alone, where obesity is also a major health concern (Abu-Abid et al. 2002, Kaaks et al. 2002, Mueck & Seeger 2004). Therefore, the effects of obesity on human endometrial cancer represent a critical intersection between these two important health problems. A case control study of endometrial cancers in Greece showed that leptin has a strong positive association with the incidence of endometrial cancer (Petridou et al. 2002). However, whether there is a direct relationship between leptin and endometrial cancer cannot be conclusively stated, as increased leptin and endometrial cancer may both be secondary consequences of obesity. Recently, circulating levels of leptin and its mRNA were measured in patients and a possible involvement of the leptin receptor in pathogenesis of endometrial cancer was proposed (Yuan et al. 2004). Considering the fundamental role of a-dipocytokines in endocrine-related cancer progression, the growth regulation of endometrial cancer cells by leptin might affect their malignant progression. Therefore, in the present study, we examined the expression of leptin receptors using endometrial cancer cell lines. We further investigated the effects of leptin on endometrial cancer cell proliferation and invasion.
Materials and methods
Antibodies
Antibodies for phosphorylated AKT (Phospho-AKT-Ser473), AKT, phosphorylated ERK (Phospho-p44/42 MAPK-Thr202/Tyr204), ERK, phosphorylated STAT (Phospho-STAT-Tyr705) and STAT were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies for short and long forms of leptin receptors Ob-R (C-20), Ob-R (B-3) and Ob-R (H-300) were purchased from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA.
Cell culture, reagents and treatments
Ishikawa and ECC1, both human endometrial adenocarcinoma cell lines, are the most widely used human endometrial-derived cell culture models (for review, see Vollmer 2003). Most importantly, both ECC1 and Ishikawa cells form tumors with glandular structures if inoculated into athymic nude mice (Satyaswaroop & Tabibzadeh 1991). ECC1 and Ishikawa cells were grown in DMEM supplemented with 5% fetal bovine serum (Gemini Bioproducts, Woodland, CA, USA) and 2 μM l-glutamine (Invitrogen). For treatment, cells were seeded at a density of 1 × 106/100 mm tissue culture dish. After 16 h of serum starvation, the culture media were changed to serum-free media containing treatments as indicated. Cultures were treated with human recombinant leptin (Sigma) at 100 ng/ml. In other sets of experiments, cells were treated for the indicated durations with JAK/STAT inhibitor AG490 (Calbiochem) at 100 μM, phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 (Cell Signaling) at 10 μM, or MAPK inhibitor PD098059 (Sigma) at 10 μM.
RNA isolation and RT-PCR
Total cellular RNA was extracted using an RNeasy Mini Kit (Qiagen) and quantified by UV absorption. RNA integrity was confirmed by using formaldehyde agarose gel electrophoresis and ethidium bromide staining. cDNA was synthesized from 2.5 μg total RNA by reverse transcription at 42 °C for 1 h using a first-strand cDNA synthesis kit (Invitrogen). The synthesized cDNA was used as a template for PCR amplification. A semiquantitative PCR amplification was carried out using specific primers designed to amplify leptin receptors, Ob-Rb (long isoform) and Ob-Rt (short isoform). The primers were as follows: Ob-Rb sense 5′-TCA CCC AGT GAT TAC AAG CT-3′; Ob-Rb antisense 5′-CTG GAG AAC TCT GAT GTC CG-3′; Ob-Rt sense 5′-CAT TTT ATC CCC ATT GAG AAG TA-3′; Ob-Rt antisense 5′-CTG AAA ATT AAG TCC TTG TGC CCA G-3′. PCR generated 1071 and a 273 bp fragments of the Ob-Rb and Ob-Rt genes respectively. To ensure that amplification of these genes was within the exponential range, different numbers of cycles (25–40) were run. Finally, 30 cycles of PCR amplification was chosen. PCR conditions were 95 °C for 5 min (denaturation), 30 cycles of 95 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min followed by 72 °C for 5 min. In addition, specific primers for 18S RNA were used as control. The primers were sense 5′-GAG GGA GCC TGA GAA ACG G-3′ and antisense 5′-GTC GGG AGT GGG TAA TTT GC-3′. PCR products were resolved by 1.5% agarose gel electrophoresis and visualized by ethidium bromide staining.
Cell viability assay
Cell viability assay was performed by estimating reduction of XTT (2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxyanilide), using a commercially available kit (Roche) following the manufacturer’s instructions. ECC1 and Ishikawa cells were plated in 96-well plates at an initial density of 4 × 103 cells/well for 24 h. After 16 h of serum starvation, the culture media were changed to serum-free media containing treatments as indicated. XTT labeling reagent was added to each culture well to attain a final concentration of 0.3 mg/ml. After 4 h exposure at 37 °C, absorbance was measured at 450 and 690 nm using a 96-well plate reader (SPECTRAmax PLUS; Molecular Devices, CA, USA). Pilot experiments verified that the cell densities used in experiments performed were within the linear range of the XTT assay. A standard curve was prepared using cell densities from 1 × 103 to 1 × 106, and the results were calculated with respect to the number of cells.
Immunoprecipitation of Ob-Rb and Ob-Rt
For immunoprecipitation of the long and short forms of leptin receptor, 1 mg whole-cell lysates from ECC1 and Ishikawa cells were incubated with either Ob-R (C-20) (specific for the long form of leptin receptor) or Ob-R (H-300) (for both long and short forms of leptin receptor), and the mixture rotated slowly at 4 °C for 16 h. IgG was added as negative control. A total of 20 μl packed protein A/G agarose beads were added and mixture was incubated at 4 °C for 1 h with rotation. The beads were collected by gentle centrifugation and washed twice with 1.5 ml ice-cold buffer (50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% Na-deoxycholate, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM Na3VO4 and 1 mM NaF). After the final wash, the precipitated protein–beads complexes were resuspended in SDS sample-loading buffer, fractionated by SDS-PAGE, and transferred to nitrocellulose membranes. Immunodetection was performed by first blocking the membranes for 1 h in TBS buffer (20 mM Tris–HCl (pH 7.5), 137 mM NaCl, 0.05% Tween-20) containing 5% powdered milk followed by addition of the mouse monoclonal Ob-R (B-3) antibody (specific for both long and short forms of leptin receptor) in TBS and incubating for 2 h at room temperature. Specifically bound primary antibodies were detected with peroxidase-coupled secondary antibodies and developed by enhanced chemiluminescence (ECL system; Amersham Pharmacia Biotech, Arlington Heights, IL, USA) according to the manufacturer’s instructions.
Western blotting
Whole-cell lysates were prepared by scraping cells into 250 μl ice-cold modified RIPA buffer (50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% Na-deoxycholate, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM Na3VO4 and 1 mM NaF). The lysate was rotated 360° for 1 h at 4 °C followed by centrifugation at 12 000 g for 10 min at 4 °C to clear the cellular debris. Proteins were quantified using a Bradford Protein Assay Kit (Biorad). Equal amounts of proteins were resolved on SDS-polyacrylamide gels and transferred to nitrocellulose membranes, and Western blot analyses were performed using the previously described antibodies. Immunodetection was performed using the Amersham ECL system.
Tumor cell invasion assay
For an in vitro model system for metastasis, we performed a Matrigel invasion assay by using a Matrigel invasion chamber from BD Biocoat Cell-ware (San Jose, CA, USA). Cells were seeded at a density of 1 × 105 per insert and cultured overnight. After 16 h of serum starvation, the culture media were changed to serum-free media containing treatments as indicated. Triplicate wells were used for each treatment. Cells were treated with human recombinant leptin (Sigma) at 100 ng/ml. In other sets of experiments, cells were treated with JAK/STAT inhibitor AG490 (Calbiochem) at 100 μM and PI3K inhibitor LY294002 (Cell Signaling) at 10 μM along with leptin. After 24 h incubation, cells remaining above the insert membrane were removed by gentle scraping with a sterile cotton swab. Cells that had invaded through the Matrigel to the bottom of the insert were fixed in methanol for 10 min. After being washed in PBS, the cells were stained with hematoxylin–eosin. The insert was then washed in PBS and briefly air-dried and mounted. The slides were coded to prevent counting bias, and the number of invaded cells on a representative section of each membrane were counted under a light microscope. The number of invaded cells for each experimental sample represents the average of triplicate wells.
Statistical analysis
All experiments were independently performed three times in triplicate. Data were analyzed using a paired Student’s t-test. Data were considered to be statistically significant at P < 0.05. Data are expressed as means±s.e.m.
Results
Effect of leptin on the proliferation of endometrial cancer cells
Leptin exerts its biological functions through binding to its receptors, which mediates a downstream signal by activating multiple signaling pathways (Ahima & Osei 2004). We first examined the expression of leptin receptors in ECC1 and Ishikawa cells. The expression of leptin receptor mRNA and protein was examined using RT-PCR and Western blot analysis. A predicted PCR product of Ob-Rb (long isoform) was obtained as 1071 bp and Ob-Rt (short isoform) as 273 bp by specific primers (Fig. 1A) in both ECC1 and Ishikawa cells. Immunoprecipitation was performed using specific antibodies Ob-R (C-20) (recognizes only the long form of the leptin receptor) and Ob-R (H-300) (recognizes both long and short forms of leptin receptor) followed by western blot analysis using mouse monoclonal Ob-R (B-3) (recognizes both long and short forms of leptin receptor). Immunoprecipitates with specific antibodies showed the presence of both long and short forms of leptin receptor in ECC1 and Ishikawa cells whereas IgG controls did not (Fig. 1B). We next examined the effect of leptin on cell proliferation. For these experiments, ECC1 and Ishikawa cells were serum starved for 16 h followed by treatment with various concentrations of recombinant human leptin for different periods of time. The effect of leptin treatment on cell proliferation was examined by the XTT assay. Leptin stimulated the growth of ECC1 and Ishikawa cells in a time- and dose-dependent manner (Fig. 1C and D). Leptin was found to be mitogenic for both ECC1 and Ishikawa cells and the effect was maximum at 100 ng/ml for 24 h of treatment. Higher concentrations of leptin (150 and 200 ng/ml) showed no further increase for 24 h post-leptin treatments. A lower concentration, 50 ng/ml, resulted in increased proliferation after 24 h in ECC1 cells and only after 48 h in Ishikawa cells.
Figure 1.

Expression of leptin receptor and effect of leptin on the proliferation of endometrial cancer cells. (A) Total RNA was extracted from ECC1 and Ishikawa cells and analyzed by RT-PCR using specific primers for long isoform (Ob-Rb) and short isoform (Ob-Rt) leptin receptors. A primer set for 18S RNA was used as a control. (B) Total protein was isolated from ECC1 and Ishikawa cells and equal amounts of proteins were subjected to immunoprecipitation (IP) using specific antibodies for long and short forms of the leptin receptor. Immunoprecipitation with IgG was included as a negative control and 25 μg of COLO 320DM cell lysate (Santa Cruz Biotechnology) were included as a positive control. Immunoprecipitates were resolved by SDS-PAGE and subjected to immunoblot analysis using a mouse monoclonal antibody against both long and short forms of leptin receptor. Ob-Rb and Ob-Rt were found to be present in ECC1 and Ishikawa cells. (C) ECC1 cells and (D) Ishikawa cells were serum starved for 16 h followed by treatment with 50–200 ng/ml leptin for 12, 24 and 48 h. XTT assays were then performed as described in ‘Materials and methods’. Leptin treatment increased proliferation of ECC1 and Ishikawa cells in a time- and dose-dependent manner. *P < 0.01, for different times and doses compared with respective untreated cells. The data represent mean values±s.e.m. and are the results of three independent experiments performed in triplicates.
Activation of the JAK/STAT signaling pathway in growth stimulation of endometrial cancer cells by leptin
To gain insight into the mechanism underlying the proliferative effect of leptin on endometrial cancer cells, we next examined the changes in STAT3 phosphorylation after the treatment of cells with leptin. Previous studies have demonstrated that leptin activates JAK, which in turn phosphorylates and activates STATs in other systems (Bahrenberg et al. 2002, Ahima & Osei 2004). Total cellular proteins were extracted from cells treated with 100 ng/ml leptin for various time periods and lysates were immunoblotted with a specific phospho-tyrosine STAT3 antibody. STAT3 phosphorylation was stimulated by leptin in a time-dependent manner resulting in an increase in STAT3 phosphorylation within 15 min of treatment (Fig. 2A). Immunoblots were reprobed with antibodies against STAT3 showing that the increase in STAT3 phosphorylation was not due to the increased STAT3 protein expression (Fig. 2A). Additional studies performed to determine whether leptin treatment induced increased phosphorylation of STAT1, STAT5, p38, JNK and SAPKs showed no significant changes (data not shown).
Figure 2.

Leptin stimulates the phosphorylation of STAT3. (A) ECC1 were cultured, serum starved for 16 h and treated with leptin (100 ng/ml) for various intervals of time. Time zero represents the absence of leptin or untreated cells (‘U’). Cell lysates were prepared and quantified for protein content. A total of 100 μg protein were resolved on 10% SDS-PAGE, followed by immunoblot analysis with specific antibodies against total or phosphorylated (p) forms of STAT3. The representative histogram is the densitometric analysis of bands demonstrating fold increase in levels of pSTAT3 with respect to STAT3, at various intervals of leptin treatment. These data are representative of multiple independent experiments with a representative immunoblot for pSTAT3 and STAT3. The histogram represents densitometric mean values±s.e.m. of band intensities. *P < 0.05, compared with untreated (‘U’) control cells. (B) ECC1 cells were serum starved and stimulated with 100 ng/ml leptin (‘L’) or 100 μM AG490 (‘A’). For combined treatment, cells were pretreated with 100 μM AG490 for 45 min followed by leptin treatment (‘L + A’). Untreated controls are designated as ‘U’. Total proteins were immunoblotted with specific antibody against total or the phosphorylated form of STAT3. The representative histogram is the densitometric analysis of bands demonstrating fold increase in levels of pSTAT3 with respect to STAT3. *P < 0.01, compared with untreated (‘U’) control cells; #P < 0.05, compared with leptin (‘L’) treatment. (C) Leptin fails to induce proliferation of ECC1 cells in the absence of JAK/STAT3 phosphorylation. Cells were treated with leptin and AG490 as described earlier and XTT assays were performed as described in ‘Materials and methods’. The data represent experiments performed three times in triplicates. *P < 0.001, compared with untreated (‘U’) control cells; #P < 0.005, compared with leptin (‘L’) treatment.
To investigate whether activation of the STAT3 signaling pathway is directly involved in the cell proliferative effect of leptin, we used pharmacological inhibitor of JAK/STAT to block STAT3 phosphorylation. Treatment of cells with AG490 decreased the phosphorylation of STAT3 protein significantly without affecting the expression of total STAT3 protein. Importantly, leptin failed to induce STAT3 phosphorylation in the presence of AG490 (Fig. 2B). Examination of cell proliferation in these treatment conditions clearly showed that blocking STAT3 phosphorylation significantly reduced the growth stimulation of endometrial cancer cells by leptin (Fig. 2C), indicating that the activation of STAT3 is essential for the cell proliferative effect of leptin in endometrial carcinoma.
Effect of leptin on the activation of ERK and AKT pathways
In an effort to determine the intracellular signaling mechanisms responsible for the proliferative effect of leptin, we examined the phosphorylation of ERK and AKT after stimulating the cells with an optimum concentration of leptin for various intervals of time. An increased phosphorylation of ERK and AKT was observed within 5–15 min after leptin treatment followed by a decline (Fig. 3A and B). Leptin had no effect on total ERK and AKT protein expression levels.
Figure 3.

Leptin induces the phosphorylation of ERK and AKT. Endometrial cancer cells were serum starved for 16 h and treated with leptin (100 ng/ml) for various intervals of time. Time zero represents the absence of leptin (‘U’). Equal amounts of total protein lysates were immunoblotted with specific antibodies against total or phosphorylated (p) forms of ERK (A) and AKT (B). The representative histograms are the densitometric analysis of bands demonstrating fold increase in levels of phosphorylated forms of ERK and AKT with respect to total ERK and AKT respectively, at various intervals of leptin treatment. These data are representative of multiple independent experiments (means±s.e.m.) *P < 0.01, compared with untreated (‘U’) ECC1 cells.
To confirm if activation of MAPK and AKT pathways is involved in leptin-induced proliferation of endometrial cancer cells, we studied the effect of the ERK inhibitor PD098059 and PI3K inhibitor LY294002 on leptin-induced stimulation of proliferation. As demonstrated in Fig. 4A and B, both agents specifically inhibited the phosphorylation of ERK and AKT respectively, without affecting the expression of total protein. Significantly, leptin failed to induce ERK and AKT phosphorylation in the presence of their specific inhibitors. Examination of cell proliferation in these treatment conditions clearly showed that blocking ERK and AKT phosphorylation significantly reduced the growth stimulation of endometrial cancer cells by leptin (Fig. 4C and D). These data indicate that the activation of ERK and AKT signaling pathways is directly involved in mediating the cell proliferative effect of leptin in endometrial carcinoma.
Figure 4.

Leptin fails to stimulate proliferation of endometrial cancer cells in the presence of either ERK or AKT phosphorylation inhibitor. (A) Endometrial cancer cells were serum starved and stimulated with 100 ng/ml leptin (‘L’) or 10 μM PD098059 (‘P’). For combined treatment, cells were pretreated with 10 μM PD098059 for 45 min followed by leptin treatment (‘L+P’). Untreated controls are designated as ‘U’. Total proteins were immunoblotted with specific antibody against total or phosphorylated (p) forms of ERK. The representative histogram is the densitometric analysis of bands demonstrating fold increase in levels of phosphorylated ERK with respect to ERK. *P < 0.01, compared with untreated (‘U’) control cells; #P < 0.01, compared with leptin (‘L’) treatment. (B) Cells were treated with 100 ng/ml leptin (‘L’) or 10 μM LY294002 (‘LY’). For combined treatment, cells were pretreated with 10 μM LY294002 for 45 min followed by leptin treatment (‘L+Y’). Untreated controls are designated as ‘U’. Lysates were subjected to western blot analysis with specific antibody against total or the phosphorylated form of AKT. The representative histogram is the densitometric analysis of bands demonstrating fold increase in levels of phosphorylated AKT with respect to AKT. *P < 0.01, compared with untreated (‘U’) control cells; #P < 0.01, compared with leptin (‘L’) treatment. (C, D) Cells were treated with leptin, PD098059 (C), LY294002 (D) and combinations as described earlier and XTT assays were performed as described in ‘Materials and methods’. The data represent experiments performed in triplicates three times (means±s.e.m.). *P < 0.001, compared with untreated (U) control cells; #P < 0.005, compared with leptin (‘L’) treatment.
Effect of inhibition of JAK/STAT on activation of ERK and AKT pathways and proliferation
The previous experiments indicated that inhibition of the JAK/STAT pathway blocked leptin-mediated induction of cell proliferation (Fig. 2B and C). Also, inhibition of ERK or AKT resulted in significant reduction in endometrial cancer cell proliferation (Fig. 4). To probe the hierarchy of these events we sought to determine if specific inhibition of JAK/STAT affects the phosphorylation of ERK and AKT involved in uterine cancer cell proliferation. Treatment with the JAK/STAT inhibitor AG490 blocked the leptin-induced hyperphosphorylation of both ERK and AKT (Fig. 5A and B). Most importantly, simultaneous treatment with leptin and AG490 could not restore the level of phosphorylation as achieved by treatment with leptin alone. These data suggest that activation of JAK/STAT is upstream of the activation of ERK and AKT pathways.
Figure 5.
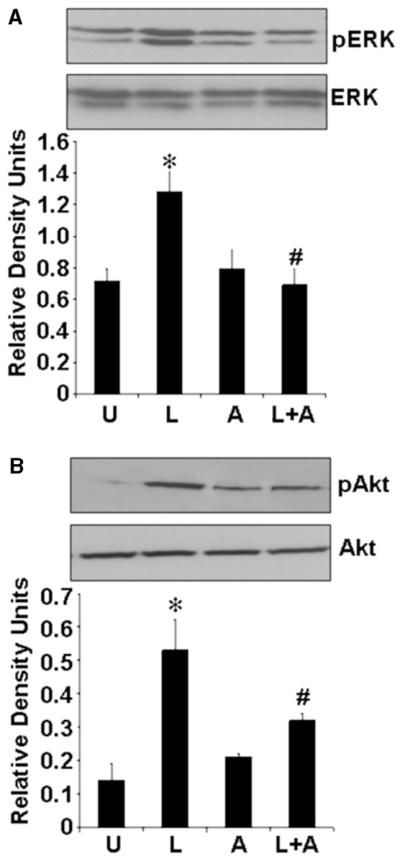
Leptin was incapable of inducing phosphorylation of ERK and AKT in the absence of JAK/STAT activation. ECC1 were cultured, serum starved for 16 h and stimulated with 100 ng/ml leptin (‘L’) or 100 μM AG490 (A). For combined treatment, cells were pretreated with 100 μM AG490 for 45 min followed by leptin treatment (‘L+A’). Untreated controls are designated as ‘U’. Total proteins were immunoblotted with specific antibody against total or phosphorylated (p) forms of ERK (A) and AKT (B). The representative histograms are the densitometric analysis of bands demonstrating fold increases in levels of phosphorylated ERK and phosphorylated AKT with respect to total ERK and AKT respectively. *P < 0.01, compared with untreated (‘U’) control cells; #P < 0.01, compared with leptin (‘L’) treatment.
Promotion of invasiveness by leptin
As Ob-Rb receptors are connected with several signaling pathways involved in cell proliferation, apoptosis and cancer progression, we addressed the question of whether leptin receptors may participate in the regulation of invasion in endometrial cancer progression. As shown in Fig. 6, ECC1 cells exhibited a remarkable invasion through Matrigel-coated inserts in response to 100 ng/ml leptin. In view of the critical role of PI3K in tumor invasion (Mareel & Leroy 2003), we examined the contribution of this lipid/protein kinase in the regulation of invasiveness by leptin. Treatment with PI3K inhibitor, LY294002, significantly inhibited the invasiveness induced by 100 ng/ml leptin in endometrial cancer cells. We next examined the possible contribution of the JAK/STAT tyrosine kinase as a signaling component of the Ob-Rb receptor, in the leptin-induced invasiveness of endometrial cancer cells. Similarly, the JAK/STAT inhibitor, AG490, also inhibited leptin-induced invasiveness. Our demonstration that PI3K and JAK/STAT inhibitors abrogate the leptin-induced invasion of Matrigel confirmed that the activity of these pathways is indeed a crucial component of the signaling machinery used by the leptin receptor in promoting invasiveness in endometrial carcinoma.
Figure 6.
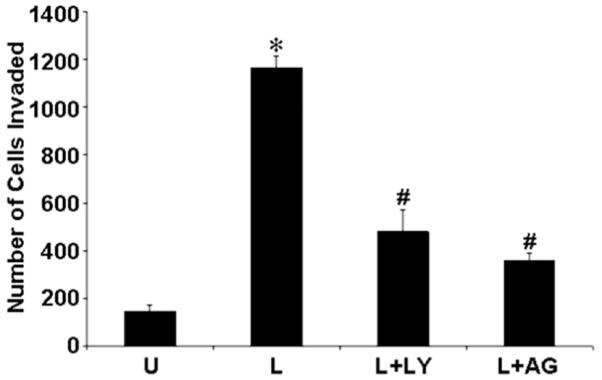
Leptin induces endometrial cancer cell invasion via AKT and JAK/STAT pathways. ECC1 were cultured in Matrigel invasion chambers in serum-free media containing 100 ng/ml leptin (‘L’). For combined treatment, cells were pretreated with 10 μM LY294002 for 45 min followed by leptin treatment (‘L+LY’) and 100 μM AG490 for 45 min followed by leptin treatment (‘L+AG’) for 24 h, and the number of cells that invaded through the Matrigel were counted. Each bar represents a Matrigel invasion assay performed in triplicate twice (means±s.e.m.). *P < 0.01, compared with untreated (‘U’) control cells; #P < 0.01, compared with leptin (‘L’) treatment.
Discussion
Obesity has been associated with various disease states, and increasing epidemiological data in humans, and many in vitro investigative reports, have suggested a strong link between leptin and cancer growth. Previous reports have described a growth-stimulatory effect of leptin on gastric (Pai et al. 2005), breast (Hu et al. 2002, Stephenson & Rose 2003, Rose et al. 2004), ovarian (Choi et al. 2005) and prostate (Somasundar et al. 2003) cancer cells. However, it inhibits the growth of pancreatic carcinoma (Somasundar et al. 2003), suggesting a differential response of various cancer cells to leptin treatment. Leptin has been shown to have a strong positive association with endometrial cancer in a case control study of incident endometrial cancers in Greece (Petridou et al. 2002), and an association between circulating levels of leptin and possible involvement of the leptin receptor in pathogenesis of endometrial cancer (Kaaks et al. 2002, Mueck & Seeger 2004, Yuan et al. 2004) has been proposed.
However, the direct role of leptin in endometrial cancer progression and the elucidation of signaling pathways involved have never been deciphered. Thus, in the present study, the expression of leptin receptor in ECC1 and Ishikawa cells was investigated. Both short and long isoforms of leptin receptors were observed in endometrial cancer cells, suggesting that leptin may be involved in endometrial cancer. In a recent report it was shown that not all ovarian cancer cell lines respond to leptin treatment, despite the presence of its receptors (Choi et al. 2005).
In a first attempt to investigate the signaling pathways involved in leptin-mediated induction of cell growth in endometrial cancer cells, we examined the effect of leptin on the activation of the JAK/STAT pathway. Our experiments clearly showed that leptin rapidly stimulates the JAK/STAT pathway and induces the phosphorylation of ERK and AKT, thus activating two key signal-transduction pathways associated with cell growth. Inhibition of these pathways with specific chemical inhibitors not only prevents the phosphorylation of respective key signal-transduction elements, it also directly blocks the endometrial cancer cell proliferation. Also, prevention of leptin-induced activation of the JAK/STAT pathway by specific chemical inhibition in turn significantly reduced the activation of ERK and AKT pathways. Thus, we deciphered in this report the signaling pathways directly involved in the cell proliferative response to leptin in endometrial carcinoma. Also, in the present study it became clear that leptin can trigger invasion by endometrial cancer cells via a pathway involving both PI3K and JAK/STAT pathways, as pharmacological inhibition of these pathways abolished leptin-induced invasiveness significantly.
These studies represent the first steps towards understanding the molecular pathway of leptin in endometrial carcinoma. AKT provides a survival signal protecting cells from apoptosis induced by various stresses (Franke et al. 1997, Kulik et al. 1997) by multiple mechanisms such as the phosphorylation of Bad, glycogen synthase-3, forkhead transcription factor and caspase-9 (Del Peso et al. 1997, Cardone et al. 1998, Pap & Cooper 1998, Brunet et al. 1999). Phosphorylation of these proteins results in inactivation of their apoptotic functions. As shown in our report, AKT phosphorylation was increased in leptin-treated human endometrial cancer cells and inhibition of PI3K with LY294002 abolished leptin-induced proliferation. LY294002 has been tested in an ectopic skin and orthotopic brain tumor model and shown to inhibit glioma tumor growth (Su et al. 2003). It has also shown efficacy against ovarian carcinoma (Hu et al. 2000). Also, more potent AKT inhibitors such as the small molecule inhibitor API-59CJ-OMe (9-methoxy-2-methylellipticinium acetate) (Jin et al. 2004) are being developed.
Our studies not only implicate the AKT pathway in endometrial cancer cell proliferation and invasion, but importantly, we report that inhibition of the JAK/STAT pathway significantly reduced the phosphorylation of AKT and ERK and proliferation mediated via these pathways, suggesting that JAK/STAT acts upstream of ERK and AKT. The main domains of STAT3 protein include the tetramerization and leucine zipper at the amino terminus, the DNA-binding domain, and the SH2 transactivation domain at the carboxy terminus. The SH2 region is responsible for the binding of STAT3 to the tyrosine phosphorylated receptors and for the dimerization necessary for DNA binding and gene expression (Bowman et al. 2000). STAT3 is activated by phosphorylation at tyrosine residue 705 leading to dimerization, nuclear translocation, recognition of STAT3-specific DNA-binding elements and activation of target gene transcription. STAT3 is frequently found to be either constitutively activated or activated in response to specific stimuli. We found that leptin activates STAT3 in endometrial cancer cells and its inhibition not only abolishes the phosphorylation of major components of the survival pathway, AKT and ERK, but also inhibits leptin-induced endometrial cancer cell proliferation. Multiple strategies have been used to block STAT activation including indirect and direct approaches. Tyrphostin AG490 has been implicated in inhibition of proliferation of human acute lymphocytic leukemia (Meydan et al. 1996) and human and mouse myeloma cells (Catlett-Falcone et al. 1999, Burdelya et al. 2002). A recent study reported that introduction of anti-sense STAT3 oligodeoxynucleotide specifically blocked expression of STAT3 mRNA in human head and neck squamous carcinoma cell lines, inhibiting proliferation (Grandis et al. 1998). Also, a small molecule inhibitor, STA21, discovered through virtual database screening, was found to inhibit human breast cancer cells expressing constitutively active STAT3 (Song et al. 2005).
In summary, our data have, for the first time, deciphered the molecular mechanisms responsible for leptin-mediated endometrial cancer cell proliferation, establishing a direct association between obesity and endometrial carcinogenesis and showing involvement of key molecules of multiple signaling pathways.
Acknowledgements
This work was supported by grant W81WXH-04-BC-030963 from The US Army Medical Research and Material Command and a grant BCTR 0503526 from The Susan G Komen Breast Cancer Research Foundation to D S and National Institutes of Health grants AA 12933 and DK 062092 awarded to F A A. It is hereby declared that there is no conflict of interest that would prejudice this research’s impartiality.
References
- Abu-Abid S, Szold A, Klausner J. Obesity and cancer. Journal of Medicine. 2002;33:73–86. [PubMed] [Google Scholar]
- Ahima RS, Osei SY. Leptin signaling. Physiology and Behavior. 2004;81:223–241. doi: 10.1016/j.physbeh.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Bahrenberg G, Behrmann I, Barthel A, Hekerman P, Heinrich PC, Joost HG, Becker W. Identification of the critical sequence elements in the cytoplasmic domain of leptin receptor isoforms required for janus kinase/signal transducer and activator of transcription activation by receptor heterodimers. Molecular Endocrinology. 2002;16:859–872. doi: 10.1210/mend.16.4.0800. [DOI] [PubMed] [Google Scholar]
- Bjorbaek C, Uotani S, da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. Journal of Biological Chemistry. 1997;272:32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Molecular Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. Journal of Biological Chemistry. 1999;274:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- Bouloumie A, Drexler HC, Lafontan M, Busse R. Leptin, the product of Ob gene, promotes angiogenesis. Circulation Research. 1998;83:1059–1066. doi: 10.1161/01.res.83.10.1059. [DOI] [PubMed] [Google Scholar]
- Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Burdelya L, Catlett-Falcone R, Levitzki A, Cheng F, Mora LB, Sotomayor E, Coppola D, Sun J, Sebti S, Dalton WS, et al. Combination therapy with AG-490 and interleukin 12 achieves greater antitumor effects than either agent alone. Molecular Cancer Therapy. 2002;1:893–899. [PubMed] [Google Scholar]
- Calle EE, Rodriquez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. New England Journal of Medicine. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- Choi JH, Park SH, Leung PC, Choi KC. Expression of leptin receptors and potential effects of leptin on the cell growth and activation of mitogen-activated protein kinases in ovarian cancer cells. Journal of Clinical Endocrinology and Metabolism. 2005;90:207–210. doi: 10.1210/jc.2004-0297. [DOI] [PubMed] [Google Scholar]
- Del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Emilsson V, Arch JR, de Groot RP, Lister CA, Cawthorne MA. Leptin treatment increases suppressors of cytokine signaling in central and peripheral tissues. FEBS Letters. 1999;455:170–174. doi: 10.1016/s0014-5793(99)00874-1. [DOI] [PubMed] [Google Scholar]
- Franke T, Kaplan D, Cantley L. PI3K: downstream AKT blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, Pitt AS, Tweardy DJ. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. Journal of Clinical Investigation. 1998;102:1385–1392. doi: 10.1172/JCI3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Houseknecht KL, Mantzoros CS, Kuliawat R, Hadro E, Flier JS, Kahn BB. Evidence for leptin binding to proteins in serum of rodents and humans: modulation with obesity. Diabetes. 1996;45:1638–1643. doi: 10.2337/diab.45.11.1638. [DOI] [PubMed] [Google Scholar]
- Hu L, Zaloudek C, Mills GB, Gray J, Jaffe RB. In vivo and in vitro ovarian carcinoma growth inhibition by a phosphatidylinositol 3′-kinase inhibitor (LY294002) Clinical Cancer Research. 2000;6:880–886. [PubMed] [Google Scholar]
- Hu X, Juneja SC, Maihle NJ, Cleary MP. Leptin-A growth factor in normal and malignant breast cells and for normal mammary gland development. Journal of the National Cancer Institute. 2002;94:1704–1711. doi: 10.1093/jnci/94.22.1704. [DOI] [PubMed] [Google Scholar]
- Huang L, Li C. Leptin: a multifunctional hormone. Cell Research. 2000;10:81–92. doi: 10.1038/sj.cr.7290038. [DOI] [PubMed] [Google Scholar]
- Jin X, Gossett DR, Wang S, Yang D, Cao Y, Chen J, Guo R, Reynolds RK, Lin J. Inhibition of AKT survival pathway by a small molecule inhibitor in human endometrial cancer cells. British Journal of Cancer. 2004;91:1808–1812. doi: 10.1038/sj.bjc.6602214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaaks R, Lukanova A, Kurzer MS. Obesity, endogenous hormones and endometrial cancer risk: A synthetic review. Cancer Epidemiology, Biomarkers and Prevention. 2002;11:1531–1543. [PubMed] [Google Scholar]
- Kulik G, Klippel A, Weber MJ. Antiapoptotic signaling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Molecular and Cellular Biology. 1997;17:1595–1606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougald OA, Hwang CS, Fan H, Lane MD. Regulated expression of the obese gene product (leptin) in white adipose tissue and 3T3-L1 adipocytes. PNAS. 1995;92:9034–9037. doi: 10.1073/pnas.92.20.9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareel M, Leroy A. Clinical, cellular and molecular aspects of cancer invasion. Physiology Reviews. 2003;83:337–376. doi: 10.1152/physrev.00024.2002. [DOI] [PubMed] [Google Scholar]
- Matsuzawa Y, Funahashi T, Nakamura T. Molecular mechanism of metabolic syndrome X: contribution of adipocytokines adipocyte-derived bioactive substances. Annals of the New York Academy of Sciences. 1999;892:146–154. doi: 10.1111/j.1749-6632.1999.tb07793.x. [DOI] [PubMed] [Google Scholar]
- Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, Leeder JS, Freedman M, Cohen A, Gazit A, et al. Inhibition of acute lymphoblastic leukemia by a Jak-2 inhibitor. Nature. 1996;379:645–648. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- Mueck AO, Seeger H. Hormone therapy after endometrial cancer. Endocrine-Related cancer. 2004;11:305–314. doi: 10.1677/erc.0.0110305. [DOI] [PubMed] [Google Scholar]
- Pai R, Lin C, Tran T, Tarnawski A. Leptin activates STAT and ERK2 pathways and induces gastric cancer cell proliferation. Biochemical and Biophysical Research Communications. 2005;331:984–992. doi: 10.1016/j.bbrc.2005.03.236. [DOI] [PubMed] [Google Scholar]
- Pap M, Cooper G. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/akt cell survival pathway. Journal of Biological Chemistry. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- Petridou E, Belechri M, Dessypris N, Koukoulomatis P, Diakomanolis E, Spanos E, Trichopoulos D. Leptin and body mass index in relation to endometrial cancer risk. Annals of Nutrition and Metabolism. 2002;46:147–151. doi: 10.1159/000063081. [DOI] [PubMed] [Google Scholar]
- Rose DP, Komninou D, Stephenson GD. Obesity, adipocytokines, and insulin resistance in breast cancer. Obesity Reviews. 2004;5:153–165. doi: 10.1111/j.1467-789X.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- Satyaswaroop PG, Tabibzadeh SS. Extracellular matrix and the patterns of differentiation of human endometrial carcinomas in vitro and in vivo. Cancer Research. 1991;51:5661–5666. [PubMed] [Google Scholar]
- Sierra-Honigmann MR, Nath AK, Murakami C, Garcia-Cardena G, Papapetropoulos A, Sessa WC, Madge LA, Schechner JS, Schwabb MB, Polverini PJ, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281:1683–1686. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- Sinha MK, Opentanova I, Ohannesian JP, Kolaczynski JW, Heiman ML, Hale J, Becker GW, Bowsher RR, Stephens TW, Caro JF. Evidence of free and bound leptin in human circulation. Studies in lean and obese subjects and during short-term fasting. Journal of Clinical Investigation. 1996;98:1277–1282. doi: 10.1172/JCI118913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundar P, Yu AK, Vona-Davis L, McFadden DW. Differential effects of leptin on cancer in vitro. Journal of Surgical Research. 2003;113:50–55. doi: 10.1016/s0022-4804(03)00166-5. [DOI] [PubMed] [Google Scholar]
- Somasundar P, Frankenberry KA, Skinner H, Vedula G, McFadden DW, Riggs D, Jackson B, Vangilder R, Hileman SM, Vona-Davis LC. Prostate cancer cell proliferation is influenced by leptin. Journal of Surgical Research. 2004a;118:71–82. doi: 10.1016/j.jss.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Somasunder P, McFadden DW, Hileman SM, Vona-Davis L. Leptin is a growth factor in cancer. Journal of Surgical Research. 2004b;116:337–349. doi: 10.1016/j.jss.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. PNAS. 2005;102:4700–4705. doi: 10.1073/pnas.0409894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson GD, Rose DP. Breast cancer and obesity: An update. Nutrition and Cancer. 2003;45:1–16. doi: 10.1207/S15327914NC4501_1. [DOI] [PubMed] [Google Scholar]
- Su JD, Mayo LD, Donner DB, Durden DL. PTEN and phosphatidylinositol 3′-kinase inhibitors up-regulate p53 and block tumor-induced angiogenesis: Evidence for an effect on the tumor and endothelial compartment. Cancer Research. 2003;63:3585–3592. [PubMed] [Google Scholar]
- Tartaglia LA. The leptin receptor. Journal of Biological Chemistry. 1997;272:6093–6096. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- Vollmer G. Endometrial cancer: experimental models useful for studies on molecular aspects of endometrial cancer and carcinogenesis. Endocrine-Related Cancer. 2003;10:23–42. doi: 10.1677/erc.0.0100023. [DOI] [PubMed] [Google Scholar]
- Yuan SS, Tsai KB, Chung YF, Chan TF, Yeh YT, Tsai LY, Su JH. Aberrant expression and possible involvement of the leptin receptor in endometrial cancer. Gynecologic Oncology. 2004;92:769–775. doi: 10.1016/j.ygyno.2003.11.043. [DOI] [PubMed] [Google Scholar]