

Abstract
Aureothin is a shikimate-polyketide hybrid metabolite from Streptomyces thioluteus with a rare nitroaryl moiety, a chiral tetrahydrofuran ring and an O-methylated pyrone ring. The antimicrobial and antitumor activities of aureothin have caught our interest in modulating its structure as well as its bioactivity profile. In an integrated approach using mutasynthesis, biotransformation and combinatorial biosynthesis, a defined library of aureothin analogues was generated. The promiscuity of the polyketide synthase assembly line towards different starter units and the plasticity of the pyrone and tetrahydrofuran ring formation were exploited. A selection of fifteen new aureothin analogues with modifications at the aryl residue, the pyrone ring, and the oxygenated backbone was produced on a preparative scale and fully characterized. Remarkably, various new aureothin derivatives are less cytotoxic than aureothin but have improved antiproliferative activities. Furthermore, we found that the THF ring is crucial for the remarkably selective activity of aureothin analogues against certain pathogenic fungi.
Introduction
Natural products are known to represent the major source for novel therapeutics, in particular in the field of antiinfectives and antitumoral agents.1,2 This is not surprising in light of the hypothesis that many of these compounds contribute towards defending the producer’s ecological niche and have undergone evolutionary selection and optimization.3,4 Nonetheless, natural products as such are not necessarily suited as therapeutics. To reduce undesired side effects, improve bioavailability issues and modulate their bioactivity profile, natural products typically undergo a chemical post-evolution through semisynthesis (i.e. derivatization) or total synthesis of analogues prior to clinical trials.5,6 During the past two decades, the portfolio of methods for structural variation has been greatly increased by the knowledge of biosynthetic pathways and their rational reprogramming. Synthetic methods can now be complemented by feeding synthetic building blocks to suitable mutant strains (mutasynthesis),7,8 using individual enzymes for biotransformations,9 and swapping biosynthesis genes in a mix-and-match fashion (combinatorial biosynthesis).10-12 These techniques have been predominantly applied to the large and diverse group of polyketide metabolites,13 mainly macrolide antibiotics.14-18 Yet in comparison, polyketide-derived pyrone compounds, which are considered as a privileged group of bioactive natural products,19-21 have been less thoroughly investigated to date. The densely functionalized shikimate-polyketide hybrid metabolite aureothin (1) is the prototype of a series of related pyrone compounds from actinomycetes. While aureothin (1) is known as a cytotoxic agent from the soil bacterium Streptomyces thioluteus,22 N-acetylaureothamine (2) and neoaureothin (spectinabilin, 3) were identified because of their potent anti-Helicobacter23 and antiviral activities, respectively.24 SNF4435C (4) and D are known as immunosuppressants25 derived from neoaureothin by a photoinduced electrocyclic rearrangement,26 (Figure 1). To gain an insight into the biological assembly lines involved in the formation of these metabolites and to harness the biosynthetic potential for the generation of novel analogues, we previously cloned and sequenced the aur and nor biosynthesis gene loci.27,28 Here, we report an integrated approach using the combination of mutasynthesis, biotransformation and combinatorial biosynthesis for the generation of a focused library of aureothin analogues with remarkably selective antifungal and antiproliferative activities.
Figure 1.
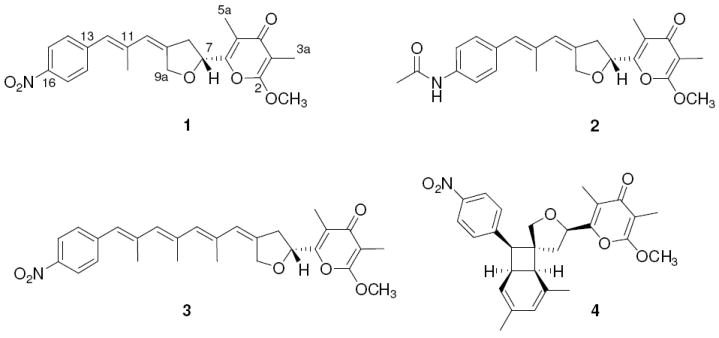
Structures of aureothin (1), N-acetylaureothamine (2), neoaureothin (spectinabilin, 3), and SNF4435C (4).
Results and Discussion
Levels of diversification in the enzymatic aureothin assembly line
According to our previous functional analyses, the aureothin biosynthetic pathway is initiated by the formation of p-aminobenzoate (PABA) and its subsequent transformation into p-nitrobenzoate (PNBA) by a novel nitro group forming N-oxygenase (AurF). 29-31 PNBA is then activated by the acyl-CoA ligase AurE and loaded onto the first module of a non-colinear modular type I polyketide synthase (AurA-C), which catalyzes five elongation cycles (Figure 2).32 After off-loading of the pentaketide with lactonization, the resulting pyrone ring is methylated by AurI,33 and finally the cytochrome P450 monooxygenase AurH introduces the tetrahydrofuran ring.34,35 The pathway thus exhibits three general levels that could be envisaged for diversification: incorporation of the starter unit building block,36 formation of the polyketide backbone, and enzymatic tailoring (i.e. alkylation and oxidative heterocyclization). As initial attempts to alter the aureothin polyketide synthase failed (data not shown) we concluded that the thiotemplate itself is not readily amenable to manipulations. In contrast, mutational analyses of the aur gene cluster suggested that both the choice of starter unit and post-PKS tailoring reactions would in principle leave some room for genetically engineering structural variants. While an aurF null mutant could be used for mutasynthesis approaches, variants lacking aurH or aurI would result in deoxy-/hydroxyl- or desmethylaureothin derivatives, respectively. Considering x possible starter units, three variants in backbone functionalization (methylene, hydroxyl, tetrahydrofuran), and three different pyrone rings (non-methylated, α-methlyated, γ-methylated), at least in principle 9 x aureothin derivatives could be generated.
Figure 2.
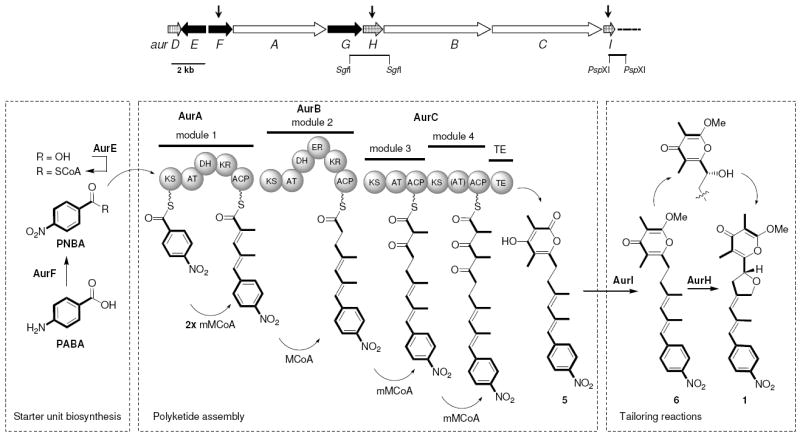
Organization of the aur biosynthesis gene cluster and model of the aureothin biosynthetic assembly line. AurG: PABA synthase, AurF: N-oxygenase, AurE: ligase, AurA-C: type I PKS, AurI: methyl transferase, AurH: Cyp450 monooxygenase.
Tolerance of the aur PKS Towards Alternative Starter Units
A mutant lacking the N-oxygenase gene aurF is suitable for the mutasynthesis of aureothin derivative aureonitrile.37 However, apart from cyanobenzoate we could not observe the incorporation of further alternative starter units. Surprisingly, the situation changed dramatically when we used another genetic construct in a different expression host. To generate deoxyaureothin derivatives, we deleted the N-oxygenase gene aurF as well as the cytochrome P450 monooxygenase gene aurH from the aur biosynthesis gene cluster. Both genes were subsequently excised from a cosmid bearing the entire aur locus by a combination of PCR-targeting and cloning strategies, respectively. The resulting plasmid, pMZ01, was introduced into the host strain S. albus, and the mutant strain was fermented in liquid production media. HPLC-MS monitoring of the extracts revealed that aureothin biosynthesis was fully abolished (Figure 3d). To complement the double knockout mutant, we supplemented the medium with PNBA and introduced a compatible expression plasmid (pMZ04), a pHJ110 derivative that contains the aurF gene downstream of the constitutive ermE promoter. Because these conditions fully restored aureothin biosynthesis, polar effects could be excluded. Using the ΔaurFH mutant supplemented with PNBA, we yielded 44.8 mg L-1 deoxyaureothin on a 50 mL scale. This result encouraged probing starter unit surrogates. For testing the substrate tolerance of the PKS, the mutant S. albus∷pMZ01 was supplemented with a series of substituted benzoates (BA, substitutions see below) in 25% aq DMSO (c = 100 μg mL-1) on a 50 mL scale. Initially, we tested the impact of chemical starter unit activation. For this purpose, we synthesized N-acetyl cysteamine (NAC) adducts that mimic CoA thioesters and readily diffuse into bacterial cells. However, when applying mutasynthons it proved to be irrelevant whether the analogues were supplied as SNAC thioester or as free acids. Apparently, the endogenous acyl-CoA ligase AurE is highly promiscuous and does not represent a bottleneck for engineering experiments.
Figure 3.

Metabolic profile of ΔaurFH null mutant (S. albus∷pMZ01) supplemented with mixtures of mutasynthons a) lacking PNBA (p-nitrobenzoate) and PCBA (p-cyanobenzoate), b) lacking PNBA, c) complete mixture; d) without supplement. (* not related to aureothin biosynthesis)
The successful incorporation of the mutasynthons was monitored by HPLC and HRMS. The expected masses and isotope patterns were recorded for all deoxyaureothin derivatives (7-11) resulting from the incorporation of all p-halogenated BAs, and the pseudohalide p-cyanobenzoate. Likewise, we observed the incorporation of p-methyl-BA and p-methoxy-substituted benzoate, yielding 12 and 13. It is remarkable that even benzoic acid is loaded onto the aur PKS and processed to a phenyl analogue of deoxyaureothin (14). The most surprising observation, however, was that even feeding 2-naphthoic acid yielded the corresponding pyrone (15). Another unexpected finding is that m-nitro-BA was tolerated as a surrogate, too, giving access to 16. However, feeding alternative PNBA surrogates equipped with ortho- or meta-substituents or multiple functionalizations were not accepted as priming units. Likewise, no polyketide production was detected when supplementing the mutant with p-hydroxy-BA, p-amino-BA, p-N-methyl-amino-BA, p-N,N’-dimethyl-amino-BA or p-N-formamido-BA. Considering the tolerated substitutions, it appears that mutasynthons with electron acceptor groups result in a higher production rate of the corresponding deoxyaureothin derivative than those with electron donor groups.
To evaluate the viability of a parallel biosynthesis of aureothin derivatives, we added mixtures of substituted benzoates to the ΔaurFH mutant (Figure 3a-c) and monitored the resulting metabolic profiles. Interestingly, mainly deoxyaureothin and deoxyaureonitrile are produced when adding the entire variety of possible starter units (Figure 3c). We next omitted PNBA and noted an increased production of deoxyaureonitrile (7) and several halogenated analogues (8-11) in low amounts (Figure 3b). Feeding a mixture lacking p-nitro-BA and p-cyano-BA led to a similar pattern, but increased the yields of halogen- and methyl-substituted analogues (Figure 3a). These results clearly demonstrate the competition between the various possible starter units and that the formation of products derived from less-favored primers is repressed in the presence of p-nitro-BA and p-cyano-BA.
The p-cyanophenyl- and naphthoate-substituted derivatives were obtained from two fermentations in liquid R2YE media (20 L), yielding 2 mg L-1 of 7 and 1.8 mg L-1 of 15, respectively. For the large scale production of p-halogenated derivatives, we selected liquid E1 medium, which proved to be superior and less expensive, providing deoxyaureothin derivatives 8-11 in higher yields (R = F: 5.4 mg L-1, R = Cl: 3.1 mg L-1, R = Br: 7.8 mg L-1, R = I: 18 mg L-1). Taken together, we have shown that the aur PKS is rather promiscuous with regard to the starter units and that besides PNBA, ten different starter units can be employed for the preparative-scale precursor-directed biosynthesis of aureothin derivatives (Figure 4).
Figure 4.
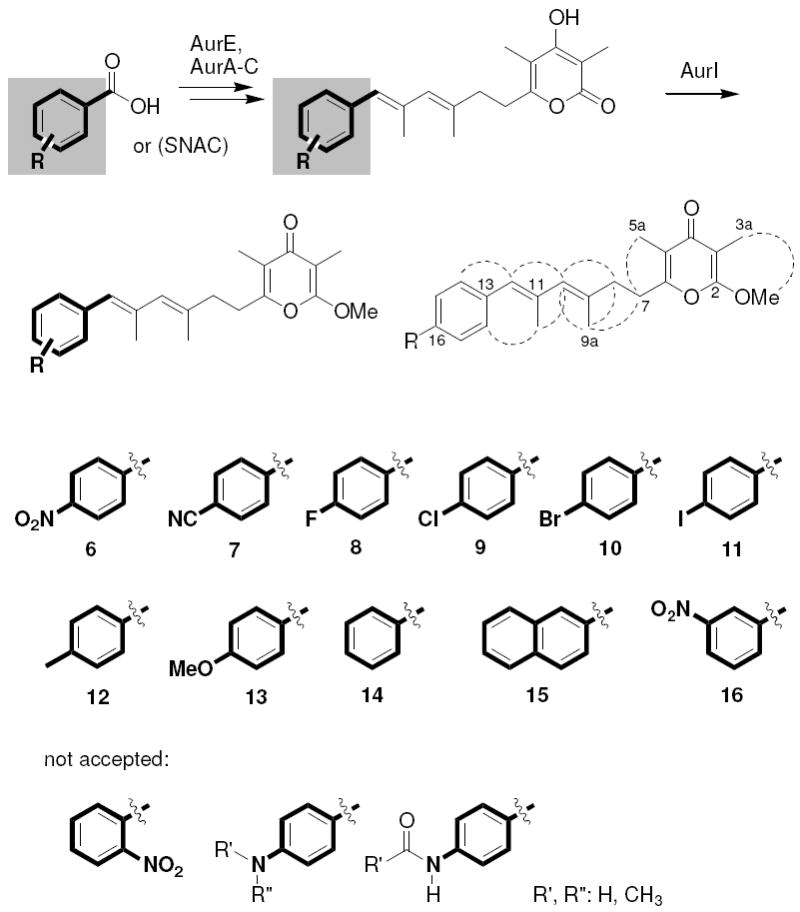
Structures of deoxyaureothin derivatives.
Exploiting plasticity in pyrone formation through mutational and combinatorial biosynthesis
At the pyrone moiety, three different substitution patterns (α-methylated, γ-methylated and non-methylated variants) are conceivable. For the production of nordeoxyaureothin derivatives, a triple mutant lacking aurF, aurH as well as the O-methyltransferase gene aurI was constructed. This was achieved by a PspXI restriction digest of cosmid pMZ01, which harbors a variant of the aur gene cluster lacking aurF and aurH. Since only two PspXI sites are present in pMZ01, one within aurI and the other downstream of the aur locus, the target gene could be excised through restriction and re-ligation. After verification of the correct construct, the resulting cosmid, pMZ02, was introduced into S. albus by conjugation. The exoconjugant S. albus∷pMZ02 was tested at a 50 mL scale for the ability to produce desmethylated analogues. While no polyketide production was observed for the mutant in liquid R2YE complex medium, supplementation with PNBA yielded nordeoxyaureothin (5), which features the more stable non-methylated α-pyrone ring.
PNBA surrogates were analogously accepted by the triple mutant, yielding analogues such as nordeoxyaureonitrile (7) with different aryl substitutions. Complementation of the triple mutant with aurI restored the formation of the methylated γ-pyrone.
In order to expand the scope of substitutions, we aimed at generating an O-metylated α-pyrone. As yet, the only known enzyme catalyzing an O-methyltransfer of a pyrone metabolite is EncK from the enterocin pathway in Streptomyces maritimus.38,39 To test whether this enzyme has a sufficiently broad specificity to methylate the aureothin pyrone ring, we planned to complement the ΔaurFHI null mutant with encK. For this purpose, encK was recovered from cosmid 1A9 as 1.8 kb EcoRI fragment and cloned downstream of the constitutive ermE promoter of pWHM4*. The resulting construct, pHJ72, was introduced together with pMZ02 into S. albus. The resulting strain (S. albus∷pMZ02/pHJ72) was cultured with apramycin and thiostreptone as selection markers and its metabolic profile monitored by HPLC/MS. In fact, supplementation of the mutant with PNBA yielded a new product (17). While 6 and 17 share the same molecular mass, they have a markedly different retention time in the HPLC profile, thus suggesting they represent two regioisomers (Figure 5). This was firmly corroborated by NMR data. The successful methylation of 5 using EncK is surprising in light of the different polyketide core structures of aureothin and enterocin (Figure 6). Notably, only encK can complement the ΔaurI mutant, but not vice versa (data not shown), indicating that EncK has broader substrate specificity than AurI. In sum, the ΔaurFHI mutant allows access to aureothin derivatives with modified pyrone moieties. Upon supplementation with PNBA or other synthetic analogues, it is possible to yield various homologues.
Figure 5.
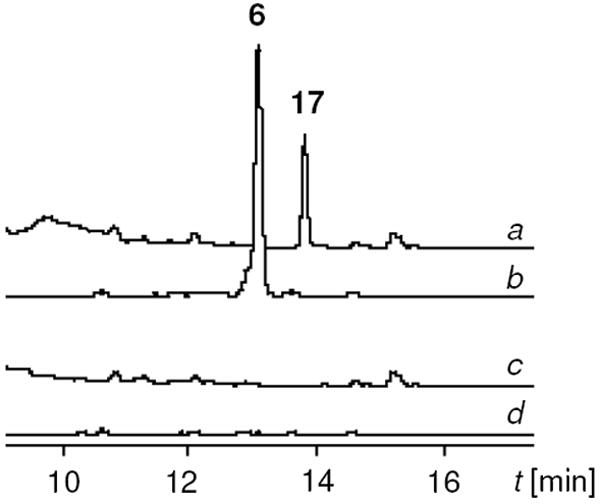
Regiospecific pyrone O-methylation by AurI and EncK. HPLC profiles of extracts from a) ΔaurFHI triple mutant (S. albus∷pMZ02), complemented with encK (S. albus∷pMZ02/pHJ72) supplemented with PNBA, b) ΔaurFH mutant (S. albus∷pMZ01) supplemented with PNBA, c) S. albus∷pMZ02/pHJ72, d) S. albus∷pMZ01.
Figure 6.
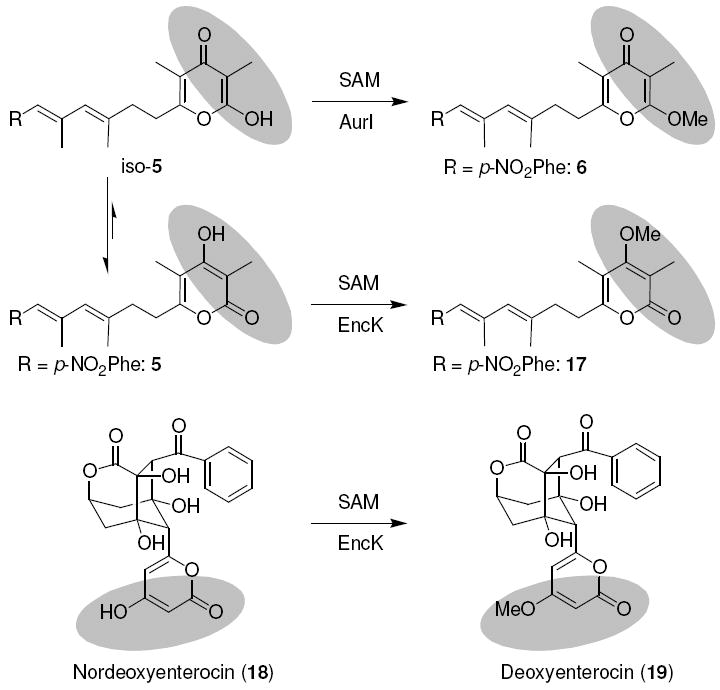
Regiospecific pyrone O-methylation by AurI and EncK.
Promiscuity of the Bifunctional Monooxygenase AurH in Polyketide Hydroxylation and Heterocyclization
The sequential, AurH-catalyzed hydroxylation-heterocyclization represents the last step in the aureothin pathway. From initial mutasynthesis experiments using the ΔaurF mutant, it appeared that various aureothin derivatives are toxic for the producer strain, thus limiting their continuous formation. For this reason, we chose to generate aureothin derivatives containing the characteristic chiral tetrahydrofuran moiety by AurH-mediated biotransformation of the deoxyaureothin derivatives. While the in vitro biotransformation with AurH is in principal viable,35 an in vivo approach using whole cells of a host expressing aurH40 proved to be more efficient on the preparative scale. For this purpose, the aureothin cytochrome P450 monooxygenase AurH was heterologously expressed in S. albus and S. lividans using expression plasmid pHJ110. HPLC-MS monitoring of 50 mL cultures of S. lividans/pHJ110 supplemented with deoxyaureothin derivatives indicated in all cases the formation of novel peaks with complete disappearance of the starting material (Figure 7). AurH proved to be remarkably tolerant towards artificial substrates. The oxygenase is capable of transforming deoxyaureonitrile (7), all four p-halogenated derivatives (8-11), and even the unusual naphthoate (15) into the corresponding aureothin analogues 21-26. However, when α-pyrones like 17 were administered, no heterocycle formation was observed. Instead, 7-hydroxy derivatives such as 20 were produced. O-Heterocyclization obviously only takes place in the presence of a methylated γ-pyrone moiety, where an exact enzyme fit is warranted. If desired, it would also be possible to introduce a 7-hydroxyl moiety into methylated γ-pyrones and circumvent THF ring formation. This is generally feasible when complementing the aurH null mutant with norH from the neoaureothin pathway, since NorH is not capable of catalyzing the full reaction sequence when an aureothin backbone is supplied. In this way, appreciable amounts of the 7-hydroxylated γ-pyrone 27 were obtained.
Figure 7.
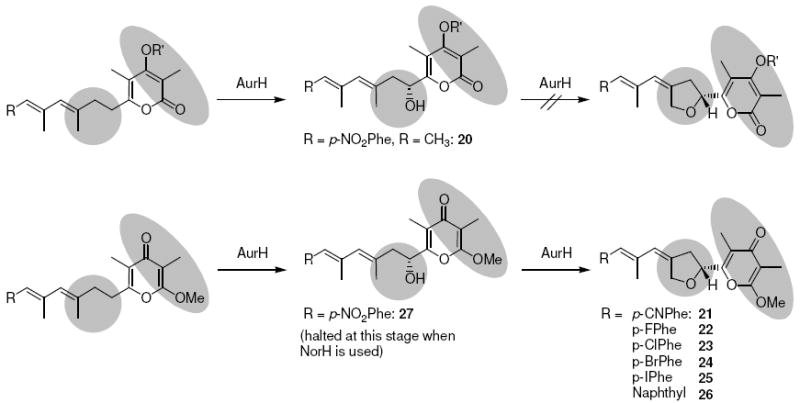
Biotransformation of α- and γ-pyrones by AurH.
The scale-up for preparative in vivo biotransformations was optimized using p-iododeoxyaureothin (11) dissolved in DMSO (6.5 μmol, c = 1 mg 25 μL-1) in 250 mL M10 media. Due to its reduced metabolic background S. albus/pHJ110 proved to be superior to S. lividans. Varying amounts of 11 (a. 6.5 μmol, b. 13 μmol, c. 26 μmol, d. 52 μmol) were tested, revealing an optimum of production when using the lowest quantity of the corresponding aureothin analogue 25 (a. 74%, b. 52%, c. 52%, d. 40%) as quantified by HPLC.
Production of a Focused Library of Aureothin Derivatives by Fermentation
The mutasynthesis experiments revealed that at least eleven different starter units are accepted by the aureothin PKS. Furthermore, three modes of backbone substitutions and three different types of pyrone moieties can be engineered through biotransformation and combinatorial biosynthesis, respectively. The only restriction is that the tetrahydrofuran ring is not readily formed in the presence of α-pyrones. Accordingly, at least in principle, 77 aureothin derivatives would be accessible through the combination of all methods presented. However, not all compounds appeared to be equally profitable; we found that non-methylated α-pyrones and hydroxyl-substituted aureothin analogues (e.g. 20, 27) are prone to acid-catalyzed degradation and rearrangement, which renders them less favorable from a pharmacological point of view. Furthermore, initial biological assays indicated that a deoxyaureothin derivative featuring an α-pyrone (17) moiety exhibit a markedly lower antiproliferative effect (Huvec GI50 >20 μg mL-1) compared to the γ-pyrone 6 (Huvec GI50 9.3 μg mL-1). Furthermore, the α-pyrone proved to be slightly more cytotoxic, which is against the desired disposition. In addition, the methylated α-pyrone compounds do not show any antifungal activity (Figure 10). We thus focused on the large scale production of all p-cyano-, p-halogen- and the naphthoate-substituted deoxyaureothin (7-11, 14, 15) and aureothin derivatives (22-26) featuring the methylated γ-pyrone group. The α-pyrone 17 and the 7-hydroxy substituted analogues 20 and 27 were also produced on a preparative scale for comparison.
Figure 10.
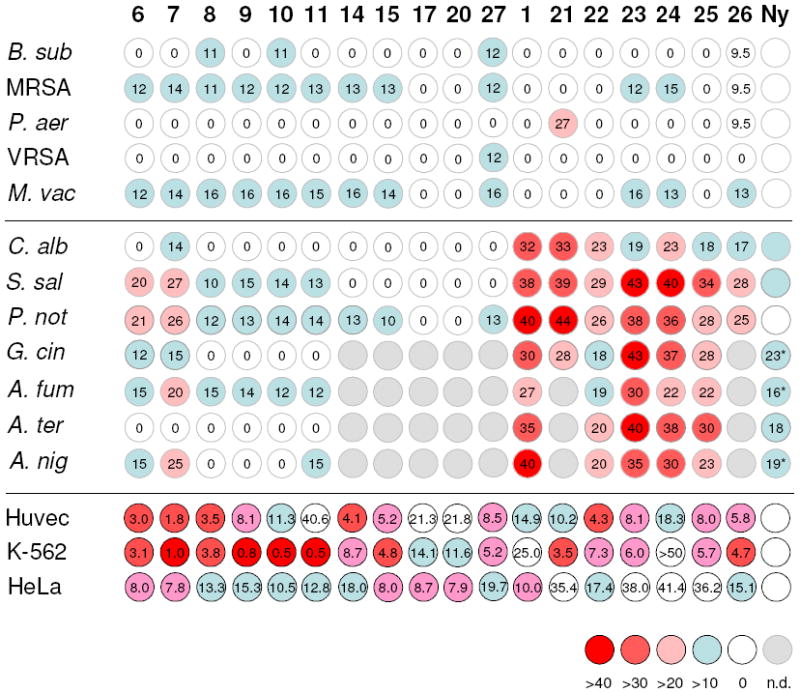
Color-coded survey on antimicrobial (inhibition diameter in mm), cytotoxic (IC50 μg mL-1) and antiproliferative (GI50 μg mL-1) activities of aureothin (1) and its derivatives. Less effective activities indicated with blue and color-staggered to dark red for the most effective ones. Test strains: Bacillus subtilis ATCC 6633; Staphylococcus aureus SG511; Pseudomonas aeruginosa K799/61; Enterococcus faecalis (VRSA) 1528; Mycobacterium vaccae 10670; Candida albicans BMSY 212; Sporobolomyces salmonicolor 549; Penicillium notatum JP36; Glomerella cingulata; Aspergillus fumigatus ATCC 46645; Aspergillus terreus DSM 826; Aspergillus niger DSM 737; Ny: nystatin.
The mutasynthesis experiments for the production of the methylated γ-pyrones were performed by multiple 20 L or 60 L fermentations. The culture broths were neutralized with 1 N HCl and mycelia and culture filtrates were individually extracted with ethyl acetate. Crude extracts were subjected to open column chromatography (silica gel, normal phase) using a CHCl3/MeOH gradient. Final purification was achieved by preparative reverse phase HPLC. For subsequent biotransformations, the deoxyaureothin derivatives were transformed into the corresponding aureothin derivatives using AurH in a whole cell approach as described above, yielding 74% conversion.
It should be highlighted that the structures of all new compounds (7-11, 14, 15, 17, 20, 22-27) were fully elucidated by 1D and 2D NMR experiments, high resolution mass spectrometry, and infrared spectroscopy. NMR shifts of the core structures were similar to those of 1 and 6, respectively. The influence of the groups exerting different I- and M-effects was clearly detected in shifts of the neighboring atoms. Signals of the fluoro-substituted analogues showed typical C-F coupling constants (ranging from 259 Hz to 3 Hz) up to 6 atom bondings in the 13C-NMR. Two-dimensional NMR as well as NOESY experiments fully support the proposed structures. HR-MS data confirmed the expected molecular masses, and chloro- and bromo-derivatives showed the typical isotopic pattern (“A+2”) in the mass spectra. By IR-spectroscopy the vibration of the nitrile group as well as the C-Hal vibrations could be clearly observed. The IR fingerprint (ca. 850-810 cm-1) indicated the para-substitution of the phenyl head group.
Identification of novel aureothin derivatives with selective antifungal and improved antiproliferative activities
The biological activities of all new aureothin derivatives were analyzed in a panel of assays with cell lines like human cervix carcinoma HeLa, mouse fibroblast cells L-929, human umbilical vein endothelial cells Huvec, as well as human K-562 leukemia cells. Notably, almost all new compounds show increased antiproliferative and cytostatic activities compared to aureothin (1), and have significantly lower cytotoxicity. For deoxyaureonitrile (7) and the halogenated deoxyaureothin derivatives (8-11) a dramatic increase in antiproliferative activities (up to 50-fold compared to 1) could be determined, with GI50 values (K-562) as low as 0.5 μg mL-1. Furthermore, the antiproliferative compounds show intriguing dose-response curves with a plateau over several orders of magnitude (Figure 9). Such a flat curve is favorable considering a broad therapeutic window. A combination of both a flat curve and low IC50 values may be beneficial for potential therapeutic applications.
Figure 9.

Antiproliferative effects of p-fluoro-aureothin (□, 22) and p-fluoro-deoxyaureothin (◇, 8) on human K-562 leukemia cells displayed in a dose-response curve; concentration vs. proliferation (% vs. control, bold line).
The most remarkable finding in our biological assays was the selective antifungal activity of the new aureothin derivatives. Using a panel of bacteria, yeasts and fungi, the novel aureothin derivatives were compared to the known substances deoxyaureothin (6), aureothin (1) and aureonitrile (21). Except for deoxyaureonitrile (7), deoxyaureothin analogues do not show any notable antibacterial activities. Compared to deoxyaureothin (6), deoxyaureonitrile (7) exhibits a higher activity against Sporobolomyces salmonicolor and Penicillium notatum. In contrast, the new aureothin derivatives 22-25 show similar activities like 1 and 21 against these test strains as well as the important pathogen Candida albicans. Due to the remarkable activity of various aureothin derivatives against the yeast Sporobolomyces salmonicolor, the facultative human pathogen Candida albicans and the mould Penicillium notatum, an extended array of fungal test strains including human pathogenic Aspergillus spp. was probed (Figure 10). Deoxyaureonitrile (7) shows selective antifungal activity against Aspergillus niger and Aspergillus fumigatus, the major cause for human aspergillosis. Compounds 22-25 are even more active against aspergilli, in particular the pathogen Aspergillus terreus. While 22 and 24 do not show any activity against Candida glabrata, they effectively inhibit the growth of Glomerella cingulata, the phytopathogenic fungus and causative agent of apple bitter rot. Among all tested compounds, p-chloroaureothin 23 proved to be the most active antifungal agent.
Comparing the activity profiles of deoxyaureothin and aureothin derivatives, it is obvious that the presence of the tetrahydrofuran unit is critical for the antifungal activity, in particular against yeasts. It should be highlighted that 22 and 24 are significantly more active against Aspergillus fumigatus, Aspergillus terreus and Aspergillus niger than the standard references nystatin and amphotericin B (not shown). The selective activities against fungal strains are striking.
Conclusion
Aureothin is a densely functionalized natural product with intriguing biological activities, yet toxic and pharmacologically unfavorable due to the nitroaryl substitutent. The aim of our study was to generate a series of aureothin analogues for structure-activity relationship (SAR) studies. Because of the relatively small size of both molecule and the corresponding biosynthesis gene cluster, this target was ideally suited as a model for combining genetic, biochemical and chemical methods to generate structural diversity. Furthermore, the aureothin pathway exhibits three principal levels that could be envisaged for diversification: the aryl starter unit, the oxygenated backbone, and the pyrone head group. In order to efficiently replace the nitro substituent we chose the concept of bioisosterism that is well known in drug design.41 This goal was achieved through mutasynthesis using an engineered ΔaurF/aurH null mutant that exhibits a broad substrate tolerance towards ten alternative PKS starter units. Furthermore, in a combinatorial biosynthesis approach, we found that the endogenous γ-regiospecific pyrone methyltransferase (AurI) can be replaced by an α-regiospecific homologue from the enterocin pathway (EncK), yielding the corresponding iso-deoxyaureothin derivatives. Finally, the aureothin cytochrome P450 monooxygenase, AurH, was used as a tool for whole cell enzymatic hydroxylation and heterocyclization, repectively, providing a convenient access to the chiral tetrahydrofuran moiety. By this highly integrative approach in total fifteen aureothin analogues were generated on a preparative scale. A biological evaluation of these compounds revealed new insights into structure-activity relationships and identified various promising candidates. Some of the new deoxyaureothin derivatives are less cytotoxic but stronger antiproliferative agents than the parent natural product. Furthermore, some of the new aureothin derivatives hold promise as highly selective antifungal, in particular anti-Candida, and it seems that the THF ring markedly increases antifungal activity. Our results demonstrate that exploiting enzymatic promiscuity may well complement synthetic approaches to generate structural diversity. Thus, our results are another illustration for the viability and potential of biosynthetic engineering.
Supplementary Material
Figure 8.
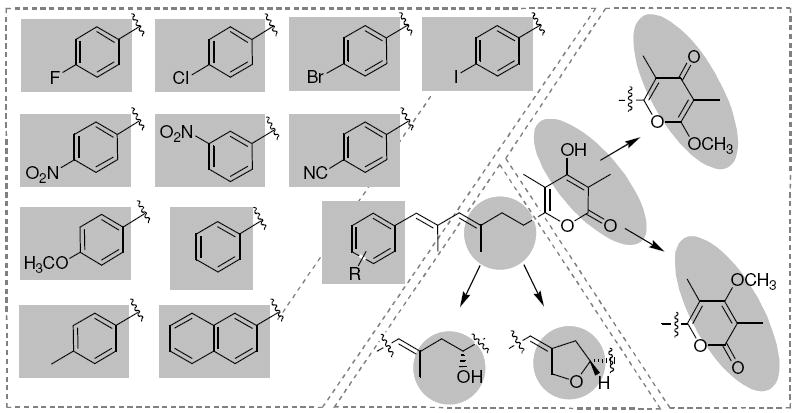
Survey of conceivable aureothin derivatives.
Acknowledgments
This work has been financially supported by the DFG within the framework of SPP1152 and the NIH (AI47818). We thank C. Heiden and M. Steinacker for performing fermentation and downstream processings, U. Wohlfeld and M.G. Schwinger for antimicrobial assays, E.-M. Neumann for antiproliferative and cytotoxic assays, and A. Perner and F.A. Gollmick are acknowledged for MS and NMR measurements, respectively.
Footnotes
SUPPORTING INFORMATION AVAILABLE: Experimental details, physicochemical data, biological assays, and NMR spectra. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 2.Cragg GM, Grothaus PG, Newman DJ. Chem Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 3.Osada H, Hertweck C. Curr Opin Chem Biol. 2009;13:133–134. doi: 10.1016/j.cbpa.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 4.John JE. Drug Discov Today. 2010 doi: 10.1016/j.drudis.2010.02.008. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 5.Newman DJ, Cragg GM, Snader KM. Nat Prod Rep. 2000;17:215–234. doi: 10.1039/a902202c. [DOI] [PubMed] [Google Scholar]
- 6.von Nussbaum F, Brands M, Hinzen B, Weigand S, Häbich D. Angew Chem Int Ed. 2006;45:5072–5129. doi: 10.1002/anie.200600350. [DOI] [PubMed] [Google Scholar]
- 7.Weist S, Süssmuth RD. Appl Microbiol Biotechnol. 2005;68:141–150. doi: 10.1007/s00253-005-1891-8. [DOI] [PubMed] [Google Scholar]
- 8.Recent examples include enterocin: Kalaitzis JA, Izumikawa M, Xiang L, Hertweck C, Moore BS. J Am Chem Soc. 2003;125:9290–9291. doi: 10.1021/ja035973o.; rapamycin: Gregory MA, Petkovic H, Lill RE, Moss SJ, Wilkinson B, Gaisser S, Leadlay PF, Sheridan RM. Angew Chem Int Ed. 2005;44:4757–4760. doi: 10.1002/anie.200462784.; borrelidin: Moss SJ, Carletti I, Olano C, Sheridan RM, Ward M, Math V, Nur EAM, Brana AF, Zhang MQ, Leadlay PF, Mendez C, Salas JA, Wilkinson B. Chem Commun. 2006:2341–2343. doi: 10.1039/b602931k.; ansamitocin: Taft F, Brünjes M, Floss HG, Czempinski N, Grond S, Sasse F, Kirschning A. ChemBioChem. 2008;9:1057–1060. doi: 10.1002/cbic.200700742.; geldanamycin: Kim W, Lee JS, Lee D, Cai XF, Shin JC, Lee K, Lee CH, Ryu S, Paik SG, Lee JJ, Hong YS. ChemBioChem. 2007;8:1491–1494. doi: 10.1002/cbic.200700196.; Eichner S, Floss HG, Sasse F, Kirschning A. ChemBioChem. 2009;10:1801–1805. doi: 10.1002/cbic.200900246.; myxalamid: Bode HB, Meiser P, Klefisch T, Cortina NS, Krug D, Göhring A, Schwär G, Mahmud T, Elnakady YA, Müller R. ChemBioChem. 2007;8:2139–2144. doi: 10.1002/cbic.200700401.; pikromycin: Gupta S, Lakshmanan V, Kim BS, Fecik R, Reynolds KA. ChemBioChem. 2008;9:1609–1616. doi: 10.1002/cbic.200700635.; spinosyn: Sheehan LS, Lill RE, Wilkinson B, Sheridan RM, Vousden WA, Kaja AL, Crouse GD, Gifford J, Graupner PR, Karr L, Lewer P, Sparks TC, Leadlay PF, Waldron C, Martin CJ. J Nat Prod. 2006;69:1702–1710. doi: 10.1021/np0602517., and salinosporamide: Eustáquio AS, Moore BS. Angew Chem Int Ed. 2008;47:3936–3938. doi: 10.1002/anie.200800177.; Nett M, Gulder TA, Kale AJ, Hughes CC, Moore BS. J Med Chem. 2009;52:6163–6167. doi: 10.1021/jm901098m.; Liu Y, Hazzard C, Eustáquio AS, Reynolds KA, Moore BS. J Am Chem Soc. 2009;131:10376–10377. doi: 10.1021/ja9042824..
- 9.Müller M. Curr Opin Biotechnol. 2004;15:591–598. doi: 10.1016/j.copbio.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Floss HG. J Biotechnol. 2006;124:242–257. doi: 10.1016/j.jbiotec.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menzella HG, Reeves CD. Curr Opin Microbiol. 2007;10:238–245. doi: 10.1016/j.mib.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Zhang W, Tang Y. J Med Chem. 2008;51:2629–2633. doi: 10.1021/jm701269v. [DOI] [PubMed] [Google Scholar]
- 13.Hertweck C. Angew Chem Int Ed. 2009;48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- 14.Khosla C, Keasling JD. Nat Rev Drug Discov. 2003;2:1019–1025. doi: 10.1038/nrd1256. [DOI] [PubMed] [Google Scholar]
- 15.Wilkinson B, Moss SJ. Curr Opin Drug Discov Devel. 2005;8:748–756. [PubMed] [Google Scholar]
- 16.Wilkinson B, Micklefield J. Nat Chem Biol. 2007;3:379–386. doi: 10.1038/nchembio.2007.7. [DOI] [PubMed] [Google Scholar]
- 17.Kirschning A, Taft F, Knobloch T. Org Biomol Chem. 2007;5:3245–3259. doi: 10.1039/b709549j. [DOI] [PubMed] [Google Scholar]
- 18.Zhou H, Xie X, Tang Y. Curr Opin Biotechnol. 2008;19:590–596. doi: 10.1016/j.copbio.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 19.McGlacken GP, Fairlamb IJ. Nat Prod Rep. 2005;22:369–385. doi: 10.1039/b416651p. [DOI] [PubMed] [Google Scholar]
- 20.Wilk W, Waldmann H, Kaiser M. Bioorg Med Chem. 2008;17:2304–2309. doi: 10.1016/j.bmc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Busch B, Hertweck C. Phytochemistry. 2009;70:1833–1840. doi: 10.1016/j.phytochem.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 22.Hirata Y, Nakata H, Yamada K, Okuhara K, Naito T. Tetrahedron. 1961;14:252–274. [Google Scholar]
- 23.Taginuchi M, Watanabe M, Nagai K, Suzumura KI, Suzuki KI, Tanaka A. J Antibiot. 2000;53:844–847. doi: 10.7164/antibiotics.53.844. [DOI] [PubMed] [Google Scholar]
- 24.Cassinelli G, Grein A, Orezzi P, Pennella P, Sanfilippo A. Arch Microbiol. 1967;55:358. doi: 10.1007/BF00406442. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi K, Tsuda E, Kurosawa K. J Antibiot. 2001;54:548–553. doi: 10.7164/antibiotics.54.548. [DOI] [PubMed] [Google Scholar]
- 26.Müller M, Kusebauch B, Liang G, Beaudry CM, Trauner D, Hertweck C. Angew Chem Int Ed. 2006;45:7835–7838. doi: 10.1002/anie.200602840. [DOI] [PubMed] [Google Scholar]
- 27.He J, Hertweck C. Chem Biol. 2003;10:1225–1232. doi: 10.1016/j.chembiol.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 28.Traitcheva N, Jenke-Kodama H, He J, Dittmann E, Hertweck C. ChemBioChem. 2007;8:1841–1849. doi: 10.1002/cbic.200700309. [DOI] [PubMed] [Google Scholar]
- 29.He J, Hertweck C. J Am Chem Soc. 2004;126:3694–3695. doi: 10.1021/ja039328t. [DOI] [PubMed] [Google Scholar]
- 30.Winkler R, Hertweck C. Angew Chem. 2005;117:4152–4155. doi: 10.1002/anie.200500365. [DOI] [PubMed] [Google Scholar]
- 31.Winkler R, Richter M, Knüpfer U, Merten D, Hertweck C. Angew Chem Int Ed. 2006;45:8016–8018. doi: 10.1002/anie.200603060. [DOI] [PubMed] [Google Scholar]
- 32.He J, Hertweck C. ChemBioChem. 2005;6:908–912. doi: 10.1002/cbic.200400333. [DOI] [PubMed] [Google Scholar]
- 33.Müller M, He J, Hertweck C. ChemBioChem. 2006;7:37–39. doi: 10.1002/cbic.200500161. [DOI] [PubMed] [Google Scholar]
- 34.He J, Müller M, Hertweck C. J Am Chem Soc. 2004;126:16742–16743. doi: 10.1021/ja046104h. [DOI] [PubMed] [Google Scholar]
- 35.Richter MEA, Traitcheva N, Knüpfer U, Hertweck C. Angew Chem Int Ed. 2008;47:8872–8875. doi: 10.1002/anie.200803714. [DOI] [PubMed] [Google Scholar]
- 36.Moore BS, Hertweck C. Nat Prod Rep. 2002;19:70–99. doi: 10.1039/b003939j. [DOI] [PubMed] [Google Scholar]
- 37.Ziehl M, He J, Dahse H-M, Hertweck C. Angew Chem Int Ed. 2005;44:1202–1205. doi: 10.1002/anie.200461990. [DOI] [PubMed] [Google Scholar]
- 38.Piel J, Hertweck C, Shipley P, Hunt DS, Newman MS, Moore BS. Chem Biol. 2000;7:943–955. doi: 10.1016/s1074-5521(00)00044-2. [DOI] [PubMed] [Google Scholar]
- 39.Cheng Q, Xiang L, Izumikawa M, Meluzzi D, Moore BS. Nat Chem Biol. 2007;3:557–558. doi: 10.1038/nchembio.2007.22. [DOI] [PubMed] [Google Scholar]
- 40.Werneburg M, Hertweck C. Chembiochem. 2008;9:2064–2066. doi: 10.1002/cbic.200800301. [DOI] [PubMed] [Google Scholar]
- 41.Lima LMA, Barreiro EJ. Curr Med Chem. 2005;12:23–49. doi: 10.2174/0929867053363540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.