

Abstract
Leptin is a 16-kDa hormone with an array of biologic actions. We, and others, have demonstrated that leptin is critical to the development of liver fibrogenesis both in vitro and in the lean littermates of ob/ob mice exposed to carbon tetrachloride (CCl4). Controversy exists as to whether leptin can act as a direct cytokine in the development of increased collagen expression, and whether ob/ob mice are resistant to potential injury from CCl4. Here, we provide evidence that strongly suggests that leptin acts to increase nascent production of mRNA for the α2(I) collagen gene based upon ribonuclease protection analysis (RPA). Actinomycin D, but not cyclohexamide, or the pan-neutralizing antibody to transforming growth factor beta one (TGFβ1), significantly diminished the effect of leptin on total α2(I) collagen mRNA levels. Further evidence that leptin acts directly on HSCs to alter gene expression in liver wounding is demonstrated by enhanced binding of phosphorylated signal transduction and activator of transcription factor 3 (pStat3) to a cis-inducible element (SIE) oligonucleotide by electrophoretic mobility shift assay (EMSA). This consensus sequence is responsible for production of a critical collagen transcription factor, AP-1. Finally, we have demonstrated from the ob/ob mouse model that these animals are at least as sensitive to CCl4 as their respective lean animals as assessed by serum alanine aminotransferase (ALT) measurements. Taken together, the current data provide a continued framework that leptin is a profibrogenic cytokine and plays a key role in liver fibrosis.
Keywords: leptin, collagen, fibrosis, stellate cells, fatty liver
Leptin is a 16-kDa hormone that has an array of biological affects as previously reported [Zhang et al., 1994; Campfield et al., 1995; Pelleymounter et al., 1995; Caro et al., 1996]. Recently, we, and others have shown that leptin plays a key role in the development of liver fibrosis, a dynamic process that ultimately leads to cirrhosis and liver failure, regardless of the etiology. Hepatic stellate cells (HSCs) are the principal collagen-producing cells in the liver but only upon their activation. This process results in HSC proliferation, enhanced gene transcription, the loss of retinoid stores, and a myofibroblasts-like phenotype characterized by an abundance of smooth muscle proteins including α-smooth muscle actin [Friedman, 2000].
In Western countries, cryptogenic liver disease has emerged as the third leading cause for cirrhosis and liver transplantation with chronic alcoholism and chronic hepatitis C virus remaining as the two major causes [Greeve et al., 1993]. Clinical data strongly suggests that obesity, type II diabetes mellitus, and dyslipidemia—clinical conditions characterized by both insulin and leptin resistance—may be epidemiological factors associating such metabolic derangements with cirrhosis for which no specific cause has been previously identified [Poonawala et al., 2000]. Furthermore, new clinical data suggests that hepatic steatosis, or non-alcoholic fatty liver, or steatohepatitis with necrosis, or non-alcoholic steatohepatitis, are also components of the metabolic syndrome X previously described [Diehl, 1999].
Previously, we have demonstrated that the activated, but not quiescent, HSCs contain leptin and such cells possess the long form of the leptin receptor (OB-RL or OB-Rb). Furthermore, we have subsequently demonstrated that in vitro leptin results in a significant increase in the mRNA for the α2(I) collagen gene as assessed by RNAse protection analysis. We have also reported that the lean littermates of ob/ob mice, but not the ob/ob mice themselves, treated with carbon tetrachloride (CCl4) developed fibrosis and had increased numbers of α-SMA positive cells [Saxena et al., 2002]. While other groups have shown similar findings both in vitro and in vivo, they postulate that leptin’s actions are mediated through endothelial cell or Kupffer cell signaling which act to release the potent mammalian profibrogenic cytokine, transforming growth factor beta one (TGFβ1) [Honda et al., 2002; Ikejima et al., 2002].
In the present study, we present data that support an independent role of leptin as a profibrogenic cytokine that acts, in vitro, to increase HSC collagen gene expression independent of TGFβ1. Further, we present follow-up data regarding the role of inflammation, in the previously reported in vivo study, which demonstrate that the presence of leptin, and not the presence of inflammation, is essential for the development of liver fibrosis.
MATERIALS AND METHODS
Isolation and Culture of Hepatic Stellate Cells
Activated rat HSCs were cultured as described previously [Friedman and Roll, 1987; Anania et al., 1995]. All rats received human care, and the Institutional Animal Care and Use Committee of the University of Maryland approved the HSC isolation protocol. In brief, after in situ perfusion of the liver with 200 mg/dl pronase E (Boehringer Mannheim, Indianapolis, IN) followed by perfusion with 80 mg/dl collagenase B (Crescent Chemical, Hauppauge, NY), dispersed cell suspensions were layered on a two-step discontinuous density gradient of 8.2 and 15.6% Nycodenz® (Sigma, St. Louis, MO). The resulting upper layer consisted of more than 95% stellate cells. Cells were placed in Dulbecco’s Modified Eagles Medium (DMEM Gibco-BRL, Grand Island, NY) containing 20% fetal bovine serum (FBS) [Flow Laboratories, Naperville, IL]. The purity of stellate cells was assessed by intrinsic auto fluorescence and detection of alpha smooth muscle actin (α-SMA). The viability of all cells was verified by phase-contrast microscopy as well as the ability to exclude propidium iodide. Cell viability of all cultures used for study was greater than 95%.
RNA Extraction and Ribonuclease Protection Analysis (RPA)
Total RNA was isolated from culture-activated HSCs as described previously [Chomczynski and Sacchi, 1987]. Two micrograms of total RNA from untreated HSCs was reverse-transcribed to cDNA using RETROscript First Strand Synthesis Kit for RT-PCR (Ambion, Austin, TX) as recommended by the manufacturer. A 533-base pair fragment of the α2(I) collagen gene with the T7 promoter was constructed by PCR from above RT reaction. Subsequently, a 533-nucleotide antisense riboprobe was transcribed with T7RNA polymerase (MAXI script kit, Ambion) by using [α32P] uridine triphosphate (specific activity, 800 Ci/mmol) [Amersham Pharmacia Biotech, Piscataway, NJ]. RPAs were performed using the RPAIII kit (Ambion). Gel-purified riboprobes (105 cpm for α2(I) collagen gene and 3 × 104 cpm for β-actin) were hybridized with 10 μg total cellular RNA extracted from primary cultured HSCs, or yeast transfer RNA at 42°C for 18 h followed by Ribonuclease A/T1 digestion at 37°C for 30 min. Protected fragments were heat denatured and separated on 3.5% denaturing urea/polyacrylamide gels. A rat β-actin antisense probe was used as a control. Radioactive signals were recorded and quantitated by the BioRad® Iamging System and Quantity One® quantitation software (Life Sciences Group, Hercules, CA). The experiments were performed with freshly prepared total RNA from HSC primary cultures after leptin treatment for various time intervals. Each α2(I) collagen signal was normalized to the β-actin signal from the same sample, and the normalized values were expressed relative to signals in the untreated primary cultured HSC (control) cells.
Treatment With Cyclohexamide or Actinomycin D
Primary HSCs were serum starved for 16 h and subsequently exposed to leptin alone as described previously, or a combination of leptin and cyclohexamide [10 μg/ml] or leptin and actinomycin D [10 μg/ml] for various time intervals before total RNA was harvested from HSC cultures and RPA was performed for both α2(I) collagen and β-actin.
Treatment With Pan-Neutralizing Antibody to TGFβ1
Primary HSCs were serum starved for 16 h and subsequently exposed to leptin alone or initially treated with a pan-neutralizing antibody to TGFβ1 [40 μg/plate] (R & D Systems, Minneapolis, MN) for 4 h prior to exposure to leptin. Total RNA was harvested as described above for specific time intervals and RPA was performed for both α2(I) collagen and β-actin.
TGFβ1 Enzyme-Linked Immunosorbent Assay (ELISA)
Primary HSCs were grown as described above. TGFβ1 secreted into the respective tissue culture media over the time course indicated was measured using a TGFβ1 ELISA assay (R & D Systems) according to the manufacturer’s instructions. A Becton-Dickinson ELISA plate reader was used to measure absorbance at 690 nm and concentrations were calculated using a standard curve developed simultaneously with study samples.
Isolation of Nuclear Proteins From Leptin-Treated HSCs
HSC cells were harvested after 16 h of incubation with media containing leptin [100 ng/ml] or 0.1% FBS. Nuclear protein extraction was performed as described elsewhere [Potter et al., 1991a,b]. Cells were washed with cold PBS, collected with a rubber policeman, and centrifuged at 1,100 × g. All steps were done on at ice at 4°C. The collected cells were placed in a solution containing 10 mM HEPES, pH 7.8, containing 2 mM MgCl2; 15 mM KCl; 0.1 mM EDTA; 1 mM dithiothreitol (DTT); and 1mM phenylmethylsulfonyl fluoride (PMSF). Cells were lysed in a Dounce Homogenizer and cell lysis was monitored by light microscopy. After isotonicity of the lysed cells was restored and centrifuged at 6,800 × g, the nuclear pellet was purified using a series of ammonium sulfate precipitations and was centrifuged at 100,000 × g for 1 h. The nuclear proteins were resuspended in 25 mM HEPES, pH 7.8, containing 50 mM KCl, 0.1 mM EDTA, 1 mM DTT, and 1 mM PMSF and 20% glycerol. The proteins were dialyzed in 100 volumes of this buffer for 4 to 6 h. Dialyzed proteins were aliquoted and stored at −70°C. Protein quantitation was performed by Lowry et al., 1951.
Electrophoretic Mobility Shift Assay (EMSA)
A single-stranded 20-base oligomer encoding the SIE, cis-inducible element, which contains a pStat3 consensus binding sequence [Robertson et al., 1995], and its complementary sequence was synthesized by the biopolymer facility at this institution. A double-stranded oligonucleotide was prepared using equimolar amounts of the complimentary oligonucleotides. The annealed oligo was diluted to a concentration of 2 μM. Blunt-end probes were end-labeled with [γ-32P] adenosine triphosphate [specific activity, 5,000 Ci/mmol, Amersham Biosciences, Piscataway, NJ]. DNA binding assays with isolated HSC nuclear protein extracts were performed with 20 μg as described [Anania et al., 1995]. Competition assays were performed using a 30-fold molar excess of unlabeled double-stranded DNA that was added to the reaction before the addition of labeled oligo. Super shift assays were performed with 2.5 μg rabbit polyclonal antibodies to phosphorylated Stat3 (Anti-pStat3) [Santa Cruz Biotechnologies, Santa Cruz, CA]. All completed reaction samples were electrophoresed on 5% non-denaturing polyacrylamide gels in a running buffer containing 1X TAE (40 mM Tris-acetate 1mM, EDTA; pH 8.0), dried, and subject to autoradiography.
CCl4 Treatment of ob/ob Mice and Their Lean Littermates
Male ob/ob mice and their lean littermates were purchased from Jackson Laboratories (Bar Harbor, ME). All animals received humane care, and the experimental protocol was approved by the Institutional Care and Use Committee of the University of Maryland. Mice were allowed free access to a laboratory chow diet and were housed for 1 week before CCl4 injections. Mice were weighed and given an intraperitoneal injection of 50% CCl4 (1 ml/kg) with olive oil twice weekly for 8 weeks. Control mice were also injected with 0.9% sterile saline and olive oil. All mice were killed for histologic analysis. Before sacrifice, blood was removed from the right heart. Serum ALT values were measured by Veterinary Resources using a serial multi-analyzer channel.
Statistical Analysis
All experiments were performed three times in triplicate. Data is expressed as means ±SEM and was considered significant if P < 0.05 as assessed by the Student’s t test.
RESULTS
Cyclohexamide Failed to Reduce α2(I) Collagen mRNA Expression Induced by Leptin as Assessed by RPA
To determine whether protein synthesis was critical to leptin-induced α2(I) collagen gene expression, 10 ng/ml cyclohexamide, a well-known inhibitor of translation, was used in combination with leptin and mRNA expression was compared to leptin-treated HSCs alone. As is apparent in Figure 1, the addition of low-dose cyclohexamide did not affect total steady-state levels of mRNA for the collagen gene induced by leptin.
Fig. 1.
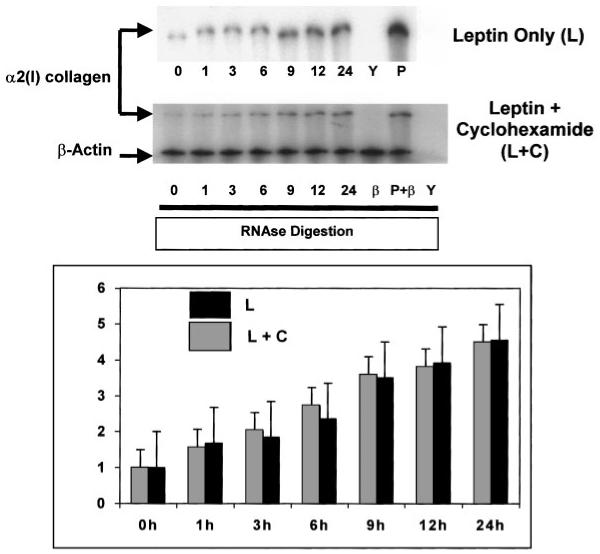
Cyclohexamide fails to significantly alter leptin-induced increases in total mRNA for α2(I) collagen in HSCs. Total RNA and experimental design is as described in the text, except that HSCs were either exposed to leptin (L) or leptin plus cyclohexamide (L + C). RPA for both α2(I) collagen and β-actin were performed and results, including densitometric analysis comparing the two treatments are shown. Results were quantitated by phosphoimaging analysis and presented as fold increase of total mRNA assayed from three independent experiments performed in triplicate. Increase mRNA was measured against quantitation of untreated HSCs only. As can be seen, no significant changes in total mRNA for α2(I) collagen are observed between either treatment indicating that protein synthesis is not required for the effect of leptin on the total mRNA for this gene. Y, yeast tRNA and riboprobe for α2(I) collagen with RNase reveals complete digestion of tRNA and α2(I) collagen riboprobe. P, mouse liver RNA and riboprobe for α2(I) collagen in the presence of RNase reveal protected fragments for the gene from mouse liver RNA. P + β, mouse liver RNA andriboprobe(s) for both α2(I) collagen and β-actin in the presence of Rnase reveal protected fragments for both genes from mouse liver RNA. β, β-actin mRNA was used for loading controls and as a housekeeping gene, and confirms a protected fragment from both stellate cell and mouse liver RNA. Time indicated in hours.
Actinomycin D Inhibits α2(I) Collagen mRNA Expression Induced by Leptin as Assessed by RPA
The RNA polymerase inhibitor, actinomycin D in combination with leptin, reduced by 50% the steady-state α2(I) collagen mRNA levels as compared to leptin-treated HSCs (Fig. 2).
Fig. 2.
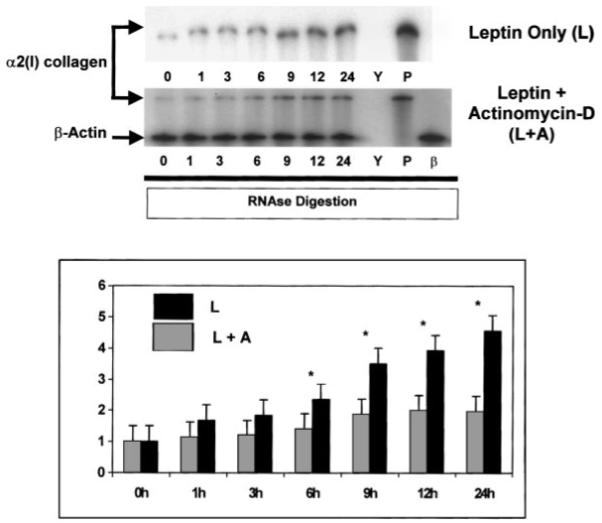
Actinomycin D significantly inhibits leptin-stimulated increases in α2(I) collagen mRNA. Total RNA was harvested from leptin (100 ng/ml)-treated [L] HSCs or leptin-treated HSCs also exposed to the transcription inhibitor Actinomycin D (10 μg/ml) [L + A] for times indicated in hours. RPA was performed for both α2(I) collagen and β-actin and results presented as in Figure 1. Y, yeast tRNA and riboprobe for α2(I) collagen with RNase reveals complete digestion of tRNA and α2(I) collagen riboprobe. P, mouse liver RNA and riboprobe for α2(I) collagen in the presence of RNase reveal protected fragments for the gene from mouse liver RNA. β, β-actin mRNA was used for loading controls and as a housekeeping gene, and confirms a protected fragment from stellate cells. Leptin alone resulted in a 4.5-fold increase over untreated HSCs, while the combination of leptin and actinomycin D resulted in less than a 2-fold increase in mRNA. *P < 0.05, Student’s t test. Time indicated in hours.
Pan-Neutralizing Antibody to TGFβ1 Failed to Alter α2(I) Collagen mRNA Levels as Assessed by RPA
Neutralizing antibodies were used to block TGFβ action from activated HSC production in culture. TGFβ1 autocrine feedback to activated HSCs is a well-described mechanism [Fibbi et al., 2001], which promotes hepatic fibrogenesis. As shown in Figure 3, a minor reduction in total mRNA for α2(I) collagen was observed in the presence of pan-neutralizing anti-TGFβ1, this effect was <10% of the 5-fold induced increase in collagen mRNA expression by leptin and was not found to be statistically significant.
Fig. 3.
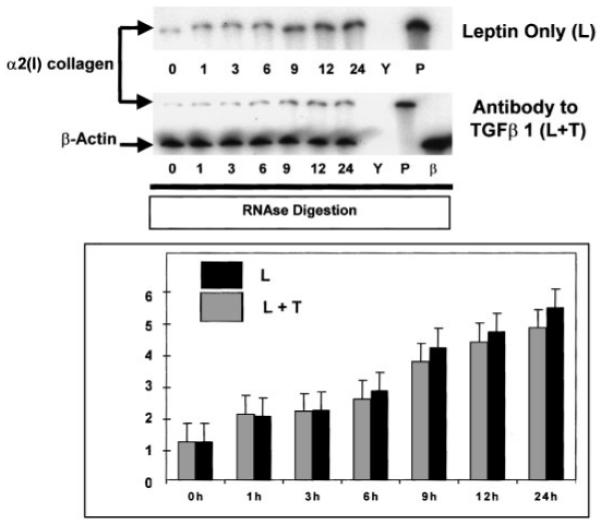
Pan-neutralizing antibody for TGFβ1 fails to significantly reduce leptin-induced total mRNA for α2(I) collagen in HSCs. RPA reported exactly as in Figures 1 and 2. The pan-neutralizing antibody [40 μg/plate] was added for 4 h prior to the addition of leptin. Following 24-h HSC exposure to leptin, while a decrease in total mRNA was demonstrated by the treatment combination (L + T), this effect was not significant, P=0.09. This figure is representative of three independent experiments performed in triplicate. RPA was performed for both α2(I) collagen and β-actin and results presented as in Figure 1. Y, yeast tRNA and riboprobe for α2(I) collagen with RNase reveals complete digestion of tRNA and α2(I) collagen riboprobe. P, mouse liver RNA and riboprobe for α2(I) collagen in the presence of RNase reveal a protected fragment for the gene from mouse liver RNA. β, β-actin mRNA was used for loading controls and as a housekeeping gene, and confirms a protected fragment from stellate cells. Time indicated in hours.
Leptin Does Not Significantly Alter TGFβ1 Levels Produced by HSCs in Culture Compared to Untreated HSCs
To determine whether leptin induces the production of a potent profibrogenic stimulus, total TGFβ1 levels (latent and active forms) were measured by ELISA over the time course as indicated in Figure 4. None of the TGFβ1 levels were significantly different in the untreated HSC cultures when compare to leptin-treated HSC cultures. Note that serum was used as the control group.
Fig. 4.
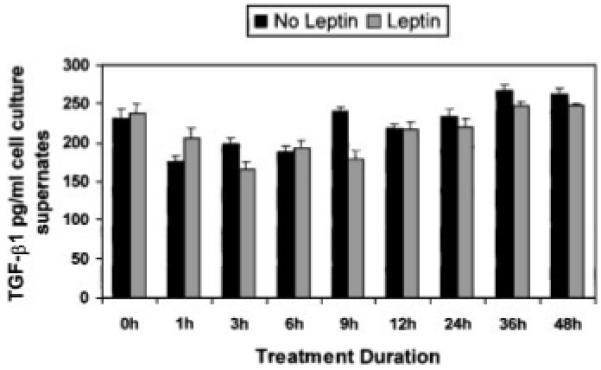
Leptin does not alter TGFβ1 production by HSCs in vitro when compared to cells grown in media with serum. HSCs were grown as described previously in either DMEM containing 15% FBS, or HSCs were grown in SF-DMEM with 100 ng/ml recombinant leptin. At times indicated media from cultures were collected and ELISA for TGFβ1 was performed. The experiments were performed twice in triplicate for all times indicated in hours. Although at 9 h, leptin may have resulted in a significant increase in TGFβ1 compared to HSCs in sera-containing media, this was not found at all other time points for which TGFβ1 cytokine activity was assayed.
EMSA Reveals That Leptin-Treated HSC Nuclear Extract Enhance pStat3 Binding to its Consensus Binding Sequence Along the c-fos Promoter
Nuclear extracts from untreated (serum-free) HSCs and extracts from leptin-treated cells were harvested and EMSA with super shift analysis were performed. Figure 5 is representative of results that reveal leptin-treated HSC nuclear extracts bind with higher affinity to the SIE consensus sequence. Unlabeled SIE oligo-nucleotide sequences abolish labeled binding indicating specificity for pStat3. Antibodies to the pStat3-oligonucleotide complex nearly retarded the entire binding complex (lane 7), confirming specificity for a large component of the nuclear protein-DNA binding complex. These data implicate leptin as an important mediator in c-fos promoter activation.
Fig. 5.
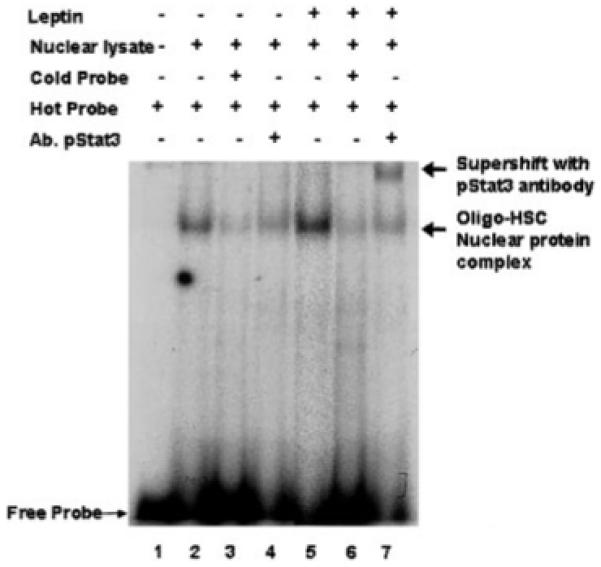
Electrophoretic mobility shift assay (EMSA) demonstrates that leptin enhances pStat3 binding activity from HSC-treated nuclear extracts to the c-fos promoter. Leptin-treated HSC nuclear extracts were harvested as described in Methods. Cultures were established as described previously, although the control, or untreated HSCs were exposed to DMEM with 0.1% FBS. Lane 1 is labeled free probe, an oligonucleotide known to bind AP-1. Lane 2, untreated HSC nuclear lysates binding activity to AP-1 oligo; lane 7, leptin-treated HSC nuclear lysates binding demonstrates significant activity over activity shown in lane 2. Lanes 3 and 6, demonstrate competition analysis with unlabeled oligonucleotide, which abrogates radioactive signal indicating specificity of the nuclear protein-oligonucleotide binding. Lanes 4 and 7 represent supershift analysis with a polyclonal antibody against AP-1 (Santa Cruz Biotechnologies, Santa Cruz, CA); lane 7, demonstrates significant supershift of leptin-treated HSC-AP-1 oligonucleotide complex, which is absent in lane 4 in the untreated HSC nuclear extracts.
Inflammation as Assessed by Serum Alanine Aminotransferase Measurements Reveals Higher Levels in ob/ob Mice Exposed to CCl4 but not in Their Treated Lean Littermates
Since NASH is defined as including necroinflammatory activity as a histologic hallmark [Diehl, 1999], ALT measurements were included in experiments which were completed and in which we previously published that the lean littermates, and not the ob/ob mice themselves, had significant fibrosis as assessed by pircosirius red staining [Janqueira et al., 1979], and had more activated α-smooth muscle positive cells (α-SMA) present by histologic analysis. Other groups using other hepatotoxins in animal studies have reported similar histologic data [Ikejima et al., 2002]. Figure 6 in this paper demonstrates that CCl4-treated ob/ob mice appear to be at least as sensitive to CCl4 treatment, and in fact have significantly increased ALT values than the lean littermates that developed significant histologic fibrosis and α-SMA positive cells.
Fig. 6.
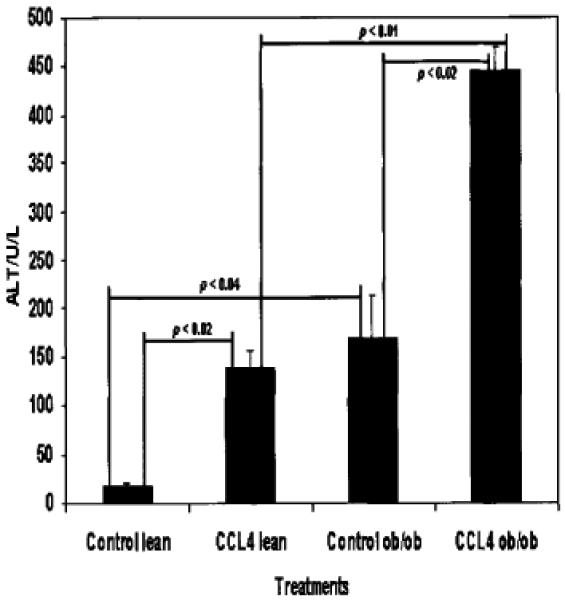
Serum ALT values from sera of wild-type ob/ob mice. Sera were collected from animals following CCl4 as described previously. Measurements were performed using a multi-analysis channel in the Veterinary Medicine Core. For each treatment condition, data shown are representative of three individual animals’ sera. The lean (wild-type) and ob/ob mice appear to respond significantly to CCl4 as assessed by ALT. Moreover, there is a significant difference between control ob/ob ALT values and CCl4-treated ob/ob mice. There is a significant difference between the CCl4-treated wild-type animals and either the CCl4-treated ob/ob mice. Data analysis: paired Student’s t test.
DISCUSSION
The results of these experiments support the hypothesis that leptin is a profibrogenic substance that results in a net increase of α2(I) collagen mRNA. This effect does not appear to be dependent on translation but rather transcription given the fact that actinomycin D significantly reduced 2(I) collagen mRNA levels compared to HSCs treated with leptin alone. These results warrant more rigorous molecular analyses, including nuclear run-on experiments, to determine if in fact leptin results in a nascent increase in α2(I) collagen mRNA or whether leptin acts to stabilize the half-life of the mRNA. mRNA stabilization for the type I collagen genes is known to occur by 3′ untranslated region binding to CAP proteins as suggested recently [Maata et el., 1994; Stefanovic et al., 1997; Lindquist et al., 2000]. Several groups now agree that leptin is a profibrogenic substance in the development of liver fibrosis but the mechanism and cell types targeted are subject to dispute. One hypothesis suggests that Kupffer cells or sinusoidal endothelial cells possess leptin receptors capable of signaling and thus in turn release transforming growth factor beta to mediate the effect of leptin on activated HSCs. We support the hypothesis that leptin acts directly on HSCs and target specific signal transduction systems to alter HSC gene expression.
If in fact leptin can only act indirectly on HSCs, then the pan-neutralizing antibody study should have significantly abrogated the net increase in α2(I) collagen mRNA in Figure 3 since it is widely published that culture-activated primary HSCs produce TGFβ1 in vitro [Bissell et al., 1995] and in vivo [Kanzler et al., 1999]. Figure 4 demonstrates that leptin-treated HSCs do not appear to result in significantly increased TGFβ1 production. Data published recently [Tang et al., 2002] also indicate that leptin does not result in enhanced TGFβ1 production, but rather increased receptor type II. An increase in TGFβ1 receptor isoforms may be also increased by HSC leptin exposure as has been reported occurs in renal mesangial cells [Wolf et al., 2002]. Hence, an emerging consensus suggests that leptin may enhance HSC collagen gene expression directly, and via heightened HSC sensitization to TGF β1. Additional studies will need to be performed related to the independent role of leptin as a profibrogenic cytokine for the following reasons. The major portion of TGF-beta is secreted as part of an inactive complex and the details of the activation process in liver have not yet been elucidated. The initially striking simplicity of the core TGF-beta/Smad signaling pathways is rapidly giving way to a much more complex view of intracellular signal transduction mechanisms and recent work has demonstrated the importance of cross-talk among different signaling pathways to either specify, enhance, or inhibit TGF-beta responses [Gressner et al., 2002].
In previous work, we have shown that Stat3 is phosphorylated in HSCs when exposed to leptin. Here we show by EMSA, that leptin specifically enhances pStat3-binding activity from leptin-treated HSC nuclear extracts to the SIE, which results in enhanced AP-1 production. While controversy remains as to the role of potential signal transduction in HSCs, this is the first time leptin has been shown to alter nuclear protein binding activity in HSCs. Other groups report leptin’s action on HSCs but have only detected the short form of the leptin receptor (Ob-RS or Ob-Ra) [Ikejima et al., 2002; Tang et al., 2002]. To date, data making a case for Ob-Ra receptor signaling is limited [Wolf et al., 2002].
The constituents of each AP-1 complex result from the dimerization of members of the Jun and fos families of immediate early genes [Gonzales and Bowden, 2002]. AP-1 is widely known to be critical to collagen gene regulation [Armendariz-Borunda et al., 1994; Chung et al., 1996; Vergeer et al., 2000]. The data presented here implicate that leptin does directly affect collagen gene expression and signals via at least Stat3 to activate a critical component of the AP-1 transcription factor. It is possible that leptin may act to alter gene expression or extracellular matrix remodeling during chronic liver injury by acting both directly on HSCs and on nearby Kupffer cells and possibly sinusoidal endothelial cells since other recently published reports indicate that endothelial cells play a key role in leptin-initiated fibrosis in other mammalian organ systems [Sierra-Honigmann et al., 1998; Wolf et al., 2002].
Finally, we examined, in our ongoing in vivo work, whether ob/ob mice are resistant to inflammation by a well-studied hepatotoxin, CCl4. We have previously shown that despite the presence of significant steatosis, but in the absence of leptin, ob/ob mice did not appear to develop significant fibrosis or activation of HSCs as assessed by α-SMA positive cell staining [Saxena et al., 2002]. In fact, by serum ALT measurements in these animals, we find that to the contrary of other published reports suggesting ob/ob animals are resistant to inflammation [Leclercq et al., 2000] and may, therefore, be resistant to inflammation at baseline, actually have a more robust degree of inflammation based on the ALT values before exposure to CCl4. Following CCl4, administration the ALT values in the ob/ob mice were significantly greater than in their lean littermates. These findings did not translate into either significant collagen deposition or localization of α-SMA positive cells and there was similar data related to aspartate aminotransferase (AST) values.
In alcoholic liver disease, it is well established that necro-inflammatory activity is not required for fibrogenesis to occur [French, 1989; Reeves et al., 1996] and thus the relationship between inflammation in NASH and liver fibrogenesis to leptin will need further clarification. Since obesity involves a state of leptin resistance and not a state of deficiency, models of resistance, e.g., impaired leptin signaling models, must also be studied. It is also not known whether leptin production is a local phenomenon and is not related to systemic leptin resistance in patients with the metabolic syndrome X—obesity, type II diabetes mellitus, hyperlipidemia, etc. Alternatively, work is ongoing to determine whether steatosis, or steatohepatitis is a marker for fibrosis or whether the presence of fat-laden hepatocytes results in the generation of reactive oxidant species—or a ‘second hit’ [Chitturi and Farrell, 2001; Reid, 2001] that may lead to HSC activation and development of fibrosis.
Acknowledgments
Grant sponsor: NIAAA; Grant number: 12933; Grant sponsor: NIDDK; Grant number: 062092; Grant sponsor: Department of Medicine, University of Maryland.
REFERENCES
- Anania FA, Potter JJ, Rennie-Tankersley L, Mezey E. Effects of acetaldehyde on nuclear protein binding to the nuclear factor I consensus sequence in the α2(I) collagen promoter. Hepatology. 1995;21:1640–1648. [PubMed] [Google Scholar]
- Armendariz-Borunda J, Simkevich CP, Roy N, Raghow R, Kang AH, Seyer JM. Activation of Ito cells involves regulation of AP-1 binding proteins and induction of type I collagen gene expression. Biochem J. 1994;304:817–824. doi: 10.1042/bj3040817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell DM, Wang SS, Jarnagin WR, Roll FJ. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest. 1995;96:447–455. doi: 10.1172/JCI118055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: Evidence for a peripheral signal linkijng adiposity and central neural networks. Science. 1995;269:546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- Caro JF, Sinha MK, Koaczynski JW, Zhang PL, Considine RV. Leptin: The tale of an obesity gene. Diabetes. 1996;45:1455–1462. doi: 10.2337/diab.45.11.1455. [DOI] [PubMed] [Google Scholar]
- Chitturi S, Farrell GC. Etiopathogenesis of non-alcoholic steatohepatitis. Semin Liver Dis. 2001;21:27–41. doi: 10.1055/s-2001-12927. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Chung KY, Agarwal A, Uitto J, Mauviel A. An AP-1 binding sequence is essential for regulation of the human alpha2(I) collagen (COL1A2) promoter activity by transforming growth factor-beta. J Biol Chem. 1996;271:3272–3278. doi: 10.1074/jbc.271.6.3272. [DOI] [PubMed] [Google Scholar]
- Diehl AM. Non-alcoholic steatohepatitis. Semin Liver Dis. 1999;19:221–229. doi: 10.1055/s-2007-1007111. [DOI] [PubMed] [Google Scholar]
- Fibbi G, Pucci M, D’Alessio S, Grappone C, Pellegrini G, Salzano R, Casini A, Milani S, Del Rosso M. Transforming growth factor beta-1 stimulates invasivity of hepatic stellate cells by engagement of the cell-associated fibrinolytic system. Growth Factors. 2001;19:87–100. doi: 10.3109/08977190109001078. [DOI] [PubMed] [Google Scholar]
- French SW. Biochemical basis for alcohol-induced liver injury. Clin Biochem. 1989;22:41–49. doi: 10.1016/s0009-9120(89)80067-0. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;75:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- Friedman SL, Roll FJ. Isolation and culture of hepatic lipocytes, Kupffer cells, and sinusoidal endothelial cells by density gradient centrifugation with stractan. Anal Biochem. 1987;161:207–218. doi: 10.1016/0003-2697(87)90673-7. [DOI] [PubMed] [Google Scholar]
- Gonzales M, Bowden GT. Ultraviolet B (UVB) induction of the c-fos promoter is mediated by phospho-cAMP response element binding protein (CREB) binding to the CRE and c-fos activator protein 1 site (FAP1) cis elements. Gene. 2002;293:169–179. doi: 10.1016/s0378-1119(02)00723-0. [DOI] [PubMed] [Google Scholar]
- Greeve M, Ferrell L, Kim M, Combs C, Roberts J, Ascher N, Wright TL. Cirrhosis of undefined pathogenesis: Absence of evidence for unknown viruses or autoimmune processes. Hepatology. 1993;17:593–598. doi: 10.1002/hep.1840170411. [DOI] [PubMed] [Google Scholar]
- Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:d793–d807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- Honda H, Ikejima K, Hirose M, Yoshikawa M, Lang T, Enomoto N, Kitamura T, Takei Y, Sato N. Leptin is required for fibrogenic responses induced by thioacetamide in the murine liver. Hepatology. 2002;36:12–21. doi: 10.1053/jhep.2002.33684. [DOI] [PubMed] [Google Scholar]
- Ikejima K, Takei Y, Honda H, Hirose M, Yoshikawa M, Zhang YJ, Lang T, Fukuda T, Yamashina S, Kitamura T, Sato N. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122:1399–1410. doi: 10.1053/gast.2002.32995. [DOI] [PubMed] [Google Scholar]
- Janqueira L, Bignolas G, Brentani R. Picrosirius staining plus polarization microscopy: A specific method for collagen detection in tissue sections. Histochem J. 1979;11:447–455. doi: 10.1007/BF01002772. [DOI] [PubMed] [Google Scholar]
- Kanzler S, Lohse AW, Keil A, Henninger J, Dienes HP, Schirmacher P, Rose-John S, zum Buschenfelde KH, Blessing M. TGF-beta1 in liver fibrosis: An inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol. 1999;276:G1059–G1068. doi: 10.1152/ajpgi.1999.276.4.G1059. [DOI] [PubMed] [Google Scholar]
- Leclercq IA, Field J, Enriquez A, Farrell GC, Robertson GR. Constitutive and inducible expression of hepatic CYP2E1 in leptin-deficient ob/ob mice. Biochem Biophys Res Commun. 2000;268:337–344. doi: 10.1006/bbrc.2000.2125. [DOI] [PubMed] [Google Scholar]
- Lindquist JN, Kauschke SG, Stefanovic B, Burchardt ER, Brenner DA. Characterization of the interaction between alphaCP(2) and the 3′-untranslated region of collagen alpha1(I) mRNA. Nucleic Acids Res. 2000;28:4306–4316. doi: 10.1093/nar/28.21.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Maatta A, Penttinen RP. Nuclear and cytoplasmic alpha 1 (I) collagen mRNA-binding proteins. FEBS Lett. 1994;340:71–77. doi: 10.1016/0014-5793(94)80175-4. [DOI] [PubMed] [Google Scholar]
- Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- Poonawala A, Nair SP, Thuluvath PJ. Prevalence of obesity and diabetes in patients with cryptogenic cirrhosis: A case-control study. Hepatology. 2000;32:689–692. doi: 10.1053/jhep.2000.17894. [DOI] [PubMed] [Google Scholar]
- Potter JJ, Cheneval D, Dang CV, Resar LMS, Mezey E, Yang VW. The upstream stimulatory factor binds to and activate the promoter of the rat class I alcohol dehydrogenase gene. J Biol Chem. 1991a;266:15457–15463. [PubMed] [Google Scholar]
- Potter JJ, Mezey E, Christy RJ, Crabb DW, Stein PH, Yang VW. CCAAT/enhancer binding protein binds and activates the promoter of the class I alcohol dehydrogenase gene. Arch Biochem Biophys. 1991b;285:246–251. doi: 10.1016/0003-9861(91)90356-n. [DOI] [PubMed] [Google Scholar]
- Reeves HL, Burt AD, Wood S, Day CP. Hepatic stellate cell activation occurs in the absence of hepatitis in alcoholic liver disease and correlates with the severity of steatosis. J Hepatol. 1996;25:677–683. doi: 10.1016/s0168-8278(96)80238-8. [DOI] [PubMed] [Google Scholar]
- Reid AE. Non-alcoholic steatohepatitis. Gastroenterology. 2001;121:710–723. doi: 10.1053/gast.2001.27126. [DOI] [PubMed] [Google Scholar]
- Robertson LM, Kerppola TK, Vendrell M, Luk D, Smeyne RJ, Bocchiaro C, Morgan JI, Curran T. Regulation of c-fos expression in transgenic mice requires multiple interdependent transcription control elements. Neuron. 1995;14:241–252. doi: 10.1016/0896-6273(95)90282-1. [DOI] [PubMed] [Google Scholar]
- Saxena NK, Ikeda K, Rockey DC, Friedman SL, Anania FA. Leptin in hepatic fibrosis: Evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35:762–771. doi: 10.1053/jhep.2002.32029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra-Honigmann MR, Nath AK, Murakami C, Garcia-Cardena G, Papapetropoulos A, Sessa WC, Madge LA, Schechner JS, Schwabb MB, Polverini PJ, Flores-Riveros JR. Biological action of leptin as an angiogenic factor. Science. 1998;281:1683–1686. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- Stefanovic B, Hellerbrand C, Holcik M, Briendl M, Aliebhaber S, Brenner DA. Posttranscriptional regulation of collagen alpha1(I) mRNA in hepatic stellate cells. Mol Cell Biol. 1997;17:5201–5209. doi: 10.1128/mcb.17.9.5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Potter J, Mezey E. Leptin enhances the effect of transforming growth factor beta in increasing type I collagen formation. Biochem Biophys Res Commun. 2002;297:906. doi: 10.1016/s0006-291x(02)02300-8. [DOI] [PubMed] [Google Scholar]
- Vergeer WP, Sogo JM, Pretorius PJ, de Vries WN. Interaction of Ap1, Ap2, and Sp1 with the regulatory regions of the human pro-alpha1(I) collagen gene. Arch Biochem Biophys. 2000;377:69–79. doi: 10.1006/abbi.2000.1760. [DOI] [PubMed] [Google Scholar]
- Wolf G, Chen S, Han DC, Ziyadeh FN. Leptin and renal disease. Am J Kidney Dis. 2002;39:1–11. doi: 10.1053/ajkd.2002.29865. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]