

Abstract
S-nitrosothiols have a number of potential clinical applications, among which their use as antithrombotic agents has been emphasized. This is largely because of their well-documented platelet inhibitory effects, which show a degree of platelet selectivity, although the mechanism of this remains undefined. Recent progress in understanding how nitric oxide (NO)-related signalling is delivered into cells from stable S-nitrosothiol compounds has revealed a variety of pathways, in particular denitrosation by enzymes located at the cell surface, and transport of intact S-nitrosocysteine via the amino acid transporter system-L (L-AT). Differences in the role of these pathways in platelets and vascular cells may in part explain the reported platelet-selective action. In addition, emerging evidence that S-nitrosothiols regulate key targets on the exofacial surfaces of cells involved in the thrombotic process (for example, protein disulphide isomerase, integrins and tissue factor) suggests novel antithrombotic actions, which may not even require transmembrane delivery of NO.
Keywords: S-nitrosothiols, nitric oxide, platelets, coagulation, protein disulphide isomerase, L-AT, nitrosation
Introduction
Cardiovascular disease is the most common cause of death in developed countries and arterial thrombosis, following rupture of an atherosclerotic plaque, underlies most cases of myocardial infarction and stroke. Thrombus formation involves the rapid accumulation of blood platelets and fibrin into an occlusive mass within the blood vessel. Platelets adhere to collagen and von Willebrand factor exposed in the ruptured plaque, and their activation is up-regulated by locally generated thrombin, thromboxane and ADP, with subsequent surface display of integrin adhesion molecules (principally αIIbβ3) in their active conformation, allowing fibrinogen binding and platelet aggregation (Mackman, 2008). Thrombin-mediated fibrin formation follows triggering of the blood coagulation cascade by exposure (Steffel et al., 2006) or de-encryption (Bach, 2006) of tissue factor, either located within the plaque itself or arriving with the cells accumulated by the thrombus. Following these initiating events, thrombin generation is amplified and propagated by assembly of coagulation enzyme complexes on the surface of activated platelets and other cells (Hoffman and Monroe, 2001) with resulting fibrin deposition.
Endothelial dysfunction and loss of nitric oxide (NO) bioactivity
Intravascular platelet activation is evident in ischaemic syndromes affecting both the coronary (Gurbel et al., 2004) and cerebral circulations (Badimon and Vilahur, 2007), and interactions between activated platelets and the vessel wall are thought to contribute not only to the final thrombotic events in atherosclerotic disease, but also in the initiation and progression of atheroma (Langer and Gawaz, 2008). Failure to control platelets in such circumstances is because of endothelial dysfunction, in particular the loss of NO activity (Vanhoutte et al., 2009). Common conditions such as diabetes mellitus, hypertension and renal failure are characterized by chronic oxidative stress, which increases cardiovascular risk, in part by diminishing the availability of bioactive NO and thus permitting platelets to contribute to thrombosis (Freedman and Loscalzo, 2003). These considerations provide a rationale for NO supplementation using donor agents to limit thrombotic risk and a variety of compounds are available for this purpose (Miller and Megson, 2007). This review will focus on the antithrombotic potential of S-nitrosothiols (RSNOs), and particular attention will be paid to the mechanism by which these compounds deliver NO to platelets and other cells of the vascular compartment, since there has been progress in this area that may explain reports of selective anti-platelet action from certain RSNO molecules.
S-nitrosothiols
S-nitrosothiols are a class of compounds produced by the S-nitrosation of sulphydryl groups (usually cysteine thiols), and with the general formula R-SNO. They are sometimes referred to as thionitrites in the older chemical literature and their general chemistry and biological properties are documented in several review articles (Al-Sa'doni and Ferro, 2000; Hogg, 2000), as are prospects for their therapeutic use (Richardson and Benjamin, 2002). Their attractiveness as therapeutic agents is increased by the fact that they occur naturally in blood and tissues as endogenous metabolites of NO, suggesting that toxicity associated with their use might be low.
Following the identification of NO as the active component of nitrovasodilator drugs (Murad, 1999) and the molecule responsible for the endogenous activity known as endothelium derived relaxing factor (Palmer et al., 1987), it was suggested that physiological RSNO formation might provide a means of stabilizing and extending the activity of NO (Myers et al., 1990; Stamler et al., 1992a). Despite the fact that NO itself is a poor nitrosating agent, plausible mechanisms exist for RSNO formation within the biological environment (Zhang and Hogg, 2005) and these compounds, principally in the form of S-nitrosoalbumin and S-nitrosohaemoglobin, but also as low molecular weight forms (S-nitrosocysteine, s-nitrosocysteinylglycine, S-nitrosoglutathione), are found to occur naturally in blood and tissues (Giustarini et al., 2003). Circulating RSNO concentrations are a matter of dispute, largely through differences in methodology (Stamler, 2004); however, most reports put them in the low nanomolar range.
RSNOs are simple and cheap to synthesize in the laboratory, an advantage if they were to be employed as drugs. Among the suggested clinical uses of RSNOs, their potential as antithrombotic agents has often been highlighted. There is a substantial literature recording the platelet-inhibitory action of RSNOs, and a number of mechanisms have been identified. In addition, a smaller number of reports have suggested that other components of the haemostatic system (for example coagulation and fibrinolysis) may be influenced by these agents.
Antiplatelet actions of RSNOs
Very soon after the identification of NO as an endogenous mediator, it was shown that NO inhibited platelet function and that this inhibition coincided with stimulation of soluble guanylyl cyclase (sGC) and intra-platelet cyclic GMP accumulation (Radomski et al., 1987). Nitrovasodilator drugs, including various RSNO compounds, had already been shown to suppress platelet aggregation via sGC stimulation (Mellion et al., 1983), and further reports documented cyclic GMP-mediated inhibition of platelet adhesion, aggregation, granule secretion and fibrinogen binding by S-nitrosocysteine (cysNO) and S-nitrosoglutathione (GSNO) (Mendelsohn et al., 1990; Lieberman et al., 1991; Radomski et al., 1992). Accumulation of the second messenger cyclic GMP influences platelet function by activation of protein kinase G, with consequent phosphorylation of numerous intracellular targets and inhibition of processes including calcium mobilization, integrin αIIbβ3 activation, cytoskeleton re-arrangement, granule secretion (Schwarz et al., 2001), activity of thromboxane receptors (Wang et al., 1998) and phosphoinositide 3-kinase (Pigazzi et al., 1999). It should be noted that cyclic GMP-induced platelet inhibition can under some circumstances occur independently of NO (Riba et al., 2008); however, the physiological importance of this mechanism, relative to endothelial NO release, is not yet clear.
Although sGC stimulation probably represents the primary mode of action of NO donor agents, including RSNOs, it has nevertheless become evident that platelet control is also exerted by cyclic GMP-independent mechanisms (Gordge et al., 1998b; Pawloski et al., 1998; Sogo et al., 2000). A variety of molecular alterations have been proposed to mediate this process, including prevention of thromboxane synthesis (Tsikas et al., 1999), nitration of α-actinin (Marcondes et al., 2006), inhibition of the platelet P2Y12 ADP receptor (or, more precisely its cellular signalling partners) (Kokkola et al., 2005) and either S-nitrosylation (Walsh et al., 2007) or altered phosphorylation (Oberprieler et al., 2007) of the important platelet integrin αIIbβ3. There appears to be a requirement for extracellular generation of NO to occur before cyclic GMP-independent inhibition of calcium signalling and platelet aggregation can be brought about by NO donor compounds, including RSNOs (Crane et al., 2005).
Other effects of RSNOs on the haemostasis process
Haemostasis involves the interaction of a variety of components, including platelets, vascular cells and proteins of the coagulation and fibrinolytic systems. However, relatively little work has been published on the direct influence of NO on coagulation and fibrinolysis. Clot formation via thrombin-induced polymerization of fibrin and its subsequent crosslinking by factor XIII is accelerated by nitrating, but not non-nitrating oxidants, suggesting that nitrosative stress may bring about a pro-thrombotic state (Vadseth et al., 2004), although paradoxically others have reported inhibition of fibrin polymerization by peroxynitrite (a well-known nitrating agent) (Lupidi et al., 1999). Conversely, exposure of fibrinogen to the RSNO compound GSNO suppresses fibrin polymerization (i.e. an antithrombotic effect) through what appears to be an allosteric interaction separate from covalent modification of the fibrinogen molecule (Akhter et al., 2002; Geer et al., 2008). RSNOs may further oppose thrombosis by inhibiting the action of transglutaminase enzymes, including coagulation factor XIII (Catani et al., 1998; Lai et al., 2001). The major vascular initiator of fibrinolysis is tissue plasminogen activator (tPA), and S-nitrosylation of tPA, as might be brought about via transnitrosation from RSNO molecules, confers antiplatelet properties on tPA without altering its fibrinolytic action (Stamler et al., 1992b). A further intriguing, and possibly crucial aspect of coagulation control by RSNOs involves their involvement in switching tissue factor (the main physiological trigger for coagulation) into a coagulation inactive form (Ahamed et al., 2006). This fits into an emerging paradigm in which tissue factor activity is regulated by a variety of post-translational modifications (Egorina et al., 2008), in particular redox modification of an allosteric disulphide bond (Chen et al., 2006). It should be noted, however, that the concept of redox regulation of tissue factor is contested (Pendurthi et al., 2007).
Thus, via a variety of pathways beyond simple inhibition of platelet function, RSNO compounds show potential for antithrombotic action.
Antithrombotic action of RSNOs –in vivo studies
Both low molecular weight and protein forms of RSNO suppress platelet activation in animal models (Radomski et al., 1992; Keaney et al., 1993), and the anti-platelet action of GSNO coincided with improved tissue survival in a rat model of ischaemia/reperfusion injury (Kuo et al., 2004). Novel antithrombotic RSNO molecules with chemical modifications conferring selectivity for areas of vascular injury (Miller et al., 2003) or for platelets (Vilahur et al., 2004) have been developed, and shown to be effective in rabbit and porcine models.
In human patients, GSNO limits platelet activation in severe pre-eclampsia (Lees et al., 1996). In addition, a number of small clinical trials have documented a significant antithrombotic and/or anti-embolic effect of GSNO administration, following surgical procedures such as coronary artery bypass grafting (Salas et al., 1998), carotid enderartectomy (Molloy et al., 1998) and carotid angioplasty (Kaposzta et al., 2002). An interesting property of GSNO identified during human in vivo studies is that it shows a degree of platelet selectivity, in that platelet inhibition could be demonstrated with doses of GSNO that failed to produce significant vasodilatation (De Belder et al., 1994). The mechanism of this platelet selective behaviour was not fully defined, but the authors speculated that it might relate to different abilities of platelets and other vascular cell types to mediate enzymatic release of NO from GSNO.
Delivery of NO signalling by RSNO compounds
The tissue effects of RSNOs (cyclic GMP generation, vasodilatation, platelet aggregation inhibition) are not shown by the non-nitrosated parent thiol compounds (Mathews and Kerr, 1993), implying that RSNOs must act via transmission of NO-related signals. Nevertheless, in the early 1990s it was recognized that the rate of spontaneous NO release from different RSNOs failed to correlate with their corresponding potencies in bioassay systems therefore spontaneous NO liberation could not explain the biological actions of RSNOs (Kowaluk and Fung, 1990; Mathews and Kerr, 1993). This lack of correlation probably reflects the fact that NO release from RSNOs into solution almost always results from catalysis by copper (I) ions, whereas in vivo other pathways are involved. The mode of intracellular delivery of NO from RSNOs is more complex and RSNOs cannot be viewed as simple ‘NO donors’.
Cellular metabolism of RSNOs
Cellular metabolism is one possible means of NO delivery and a number of studies have documented NO release from RSNOs mediated by intact platelets and other cell types (Simon et al., 1993; Gordge et al., 1998a; Liu et al., 2001; Zeng et al., 2001; Cornwell et al., 2003; Shah et al., 2003). In addition, RSNOs are substrates for a variety of enzymes including glutathione peroxidase (Freedman et al., 1995), a copper (I)-dependent enzyme (Gordge et al., 1996), γ-glutamyl transferase (Hogg et al., 1997), thioredoxin reductase (Nikitovic and Holmgren, 1996), superoxide dismutase (Jourd'heuil et al., 1999), protein disulphide isomerase (Sliskovic et al., 2005), cytoplasmic metalloprotein (Mani et al., 2006) and GSNO reductase (glutathione-dependent formaldehyde reductase, or alcohol dehydrogenase 3) (Liu et al., 2001; 2004;). This latter enzyme appears to play a crucial role in regulating nitrosative stress via adjustment of intracellular levels of S-nitrosylated proteins (Foster et al., 2009; Staab et al., 2009); however, there is no direct evidence yet for a role in the transfer of NO signalling from extracellular RSNOs.
Cell surface protein disulphide isomerase promotes NO delivery across the plasma membrane
Of the enzymes mentioned earlier, protein disulphide isomerase (PDI) has perhaps the best credentials as a mediator of RSNO signalling. PDI was originally characterized as a resident of the endoplasmic reticulum, assisting in the correct folding of nascent proteins (Gruber et al., 2006). In recent years PDI has been documented at locations outside the ER, including the cell surface, cytosol and nucleus (Turano et al., 2002). Cell surface isomerases, including PDI (csPDI), have attracted particular research interest through their involvement in infectious disease (Conant and Stephens, 2007), HIV entry into CD4 positive lymphocytes (Barbouche et al., 2003), platelet aggregation (Burgess et al., 2000; Lahav et al., 2003; Jordan et al., 2005; Robinson et al., 2006; Manickam et al., 2008), and control of tissue factor activity (Chen et al., 2006; Versteeg and Ruf, 2007). Studies in mice have confirmed an in vivo role for csPDI in both fibrin generation and platelet thrombus formation (Cho et al., 2008), thus csPDI has a direct bearing on haemostatic regulation.
A further line of research has shown that csPDI plays an important role in NO signalling, specifically the transfer of NO from extracellular membrane-impermeant RSNOs across the plasma membrane of target cells (Zai et al., 1999; Ramachandran et al., 2001; Bell et al., 2007). The best-developed model described so far postulates that csPDI denitrosates RSNO molecules in the vicinity of the plasma membrane, releasing NO that then enters the membrane by virtue of its lipophilicity and combines there with oxygen to produce the nitrosating agent N2O3. When this, in turn, nitrosates target molecules on the cytoplasmic side of the plasma membrane the goal of NO internalization is achieved (Ramachandran et al., 2001). Our own experimental studies have confirmed the role of csPDI in delivery into platelets of NO-related signalling from RSNOs. However, we also found that active csPDI was necessary for signal delivery from donors of nitroxyl (NO-) and of NO (Bell et al., 2007). csPDI-mediated denitrosation should not be required for entry of NO, and further work is therefore needed to reconcile these results. Redox mechanisms involving the vicinal thiols of the csPDI active site underlie the process of RSNO denitrosation (Sliskovic et al., 2005). The published scheme shows plausibly how a single enzyme turnover brings about NO release, but the mechanism of active site thiol regeneration, required to continue RSNO signalling, is not yet defined. Several studies have documented thiol oxidation within csPDI and loss of enzyme activity as a result of the interaction with RSNO (Zai et al., 1999; Root et al., 2004; Shah et al., 2007). Redox regeneration of csPDI may derive from both internal sources, via trans-membrane oxidoreductases such as NAD(P)H oxidase, and/or from reducing equivalents present in blood plasma. The relative importance of these various systems, and the effects of oxidative/nitrosative stress on csPDI-mediated processes, need to be known for a full understanding of csPDI pathophysiology.
Some RSNO molecules are delivered intact via membrane transporters
An alternative means of RSNO-mediated signalling is by cellular uptake of an intact RSNO molecule via a membrane transporter. Evidence has emerged from a number of different laboratories showing that the low molecular weight RSNO compounds cysNO and S-nitrosohomocysteine act as substrates for the widely-distributed amino acid transporter system-L (L-AT). This mechanism for transmembrane transport of cysNO could explain why stereoselective haemodynamic effects are seen following administration of l-cysNO and d-cysNO to rats (Davisson et al., 1996), and experimental studies carried out in vitro have confirmed its presence in a number of cell types including erythrocytes (Sandmann et al., 2005), endothelial cells (Broniowska et al., 2006), vascular smooth muscle cells (Li and Whorton, 2007; Riego et al., 2009), epithelial cells (Granillo et al., 2008), and various transformed cell lines (Zhang and Hogg, 2004; Li and Whorton, 2005), although to date, there has been no direct demonstration of cysNO uptake via L-AT in platelets. In these published studies, other forms of RSNOs, including GSNO, S-nitroso-cysteinyl-glycine, S-nitroso-N-acetyl-penicillamine and S-nitrosoalbumin, failed to be transported via L-AT, nor could they mediate NO-related signalling in target cells unless extracellular cysteine was supplied. In the presence of extracellular cysteine, cysNO is formed from the inert RSNO by a process of transnitrosation, with subsequent uptake on the L-AT system and intracellular signal transmission. If only cystine is available, a cystine–cysteine shuttle mediated by the Xc- aminoacid transport system can import cystine and subsequently release cysteine into the surrounding medium following intracellular reduction, thus providing substrate for transnitrosation and L-AT mediated uptake of cysNO (Zhang and Hogg, 2004; Li and Whorton, 2005). This mechanism has been shown to be relevant for a wide range of signalling events and also for the accumulation of intracellular RSNOs. Experiments have generally been performed using relatively high RSNO concentrations (20 µM upwards) and endpoints measured after RSNO exposure for at least 15 min. An interesting feature to emerge from these studies is that in general, cellular effects mediated by cysNO/L-AT are insensitive to the presence in the extracellular medium of NO scavengers, such as oxyhaemoglobin, thus excluding NO release from the mechanism (Zhang and Hogg, 2004; Zhu et al., 2008). An exception is when cysNO-mediated stimulation of sGC is considered – this process is inhibited by oxyhaemoglobin, but only because intracellular reduction of cysNO to NO is required before a cyclic GMP response can occur (Riego et al., 2009).
Different modes of RSNO delivery may explain their selective antithrombotic action
RSNOs are potent platelet inhibitors (see above) but it is not yet clear that their antiplatelet actions require prior conversion of RSNO to cysNO and transport into the platelet via the L-AT system. In vitro aggregation of washed platelet suspensions is inhibited by a range of RSNO molecules without the need for addition to the surrounding medium of cysteine or cystine (Radomski et al., 1992; Mathews and Kerr, 1993; Simon et al., 1993; Gordge et al., 1998b), and unlike L-AT-mediated actions, platelet inhibition (both cyclic GMP-dependent and -independent) is inhibited by haemoglobin (Radomski et al., 1992; Megson et al., 2000; Crane et al., 2005), implying that release of free NO must occur as part of the process. For protein RSNOs, such as S-nitrosoalbumin, to inhibit platelet aggregation, there does appear to be a requirement for prior transnitrosation to a low molecular weight thiol for the anti-platelet action to be realized; however, this function can be fulfilled as efficiently by glutathione or cysteinyl-glycine as by cysteine (Crane et al., 2002). Studies using washed platelets have not included measurement of cysteine concentrations in the surrounding medium, and it is therefore possible that cysteine released by the platelets themselves mediates NO signal transfer via L-AT. However, this seems unlikely as the antiplatelet action of GSNO, for example, is evident at lower concentrations (10 µM or less) and within a more rapid timeframe (<2 min) than effects reported for L-AT-mediated signalling. Furthermore, if the inhibitory action of GSNO depended upon cysNO/L-AT then it might be expected to be more potent in platelet-rich plasma (where plasma cysteine/cystine is available) than in washed platelet suspensions, whereas in fact the reverse is true (Radomski et al., 1992). In a recent publication that addressed the possible modes of intra-platelet transport of NO, neither cyclic GMP accumulation nor DAF-FM fluorescence in response to GSNO was significantly inhibited by the L-AT inhibitors BCH or l-leucine (Bell et al., 2007). Therefore, despite a wealth of evidence for the importance of the cysNO/L-AT system in endothelial and smooth muscle cells of the vascular wall, platelets appear to respond to RSNOs in a different way (Figure 1). If this anomalous behaviour of platelets, compared with other vascular cells, can be confirmed then a possible explanation for the reported platelet-selectivity of GSNO is suggested (De Belder et al., 1994), as, at low concentrations, this molecule may have access to a direct antithrombotic action on platelets that is not available to endothelial or smooth muscle cells.
Figure 1.
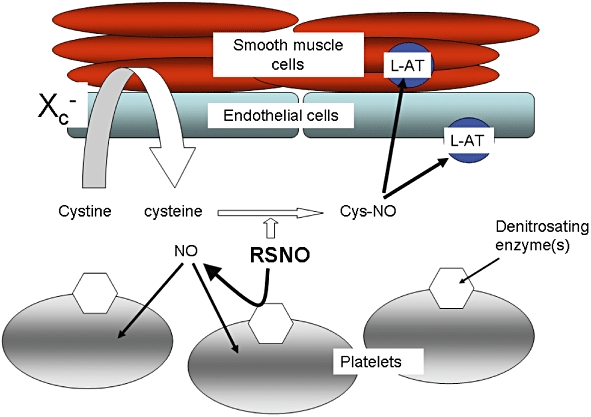
Different modes of nitric oxide (NO) delivery from S-nitrosothiols (RSNOs). Cells of the vascular wall import NO principally via uptake of S-nitrosocysteine (cysNO) on the amino acid transporter system L (L-AT), following an extracellular process of transnitrosation from RSNO to cysteine. A cystine–cysteine shuttle mediated by the Xc- transporter may act as a supply of extracellular reduced cysteine. In contrast NO delivery into platelets relies on the activity of cell surface denitrosating enzymes, such as cell surface isomerases (csPDI). This scheme indicates the main routes of NO uptake, but does not exclude the possibility that alternative or additional routes are available for each cell type.
The susceptibility of GSNO's antiplatelet action to NO scavenging by haemoglobin suggests instead that it undergoes platelet-mediated metabolism, either by a (so far uncharacterized) copper-dependent surface enzyme (Gordge et al., 1995; 1996;), or by csPDI, which is known to be present on platelets (Essex et al., 1995) and capable of releasing NO (Root et al., 2004). If differences exist in csPDI expression between platelets and vascular cells, then it might be possible to exploit this to provide selective antithrombotic action. The inhibition of platelet csPDI that results from interaction with RSNO molecules (Shah et al., 2007) is potentially a major antithrombotic mechanism, as there is abundant evidence that active csPDI is required for platelets to function efficiently during haemostasis (Essex, 2004).
Another possible reason why RSNOs might mediate selective antithrombotic effects is that a number of functionally important nitrosation targets exist on the external surface of platelets. These include csPDI itself but also the adhesion molecules glycoprotein 1b (Burgess et al., 2000) and integrin αIIbβ3 (Yan and Smith, 2000; Walsh et al., 2007). Allosteric disulphides on tissue factor and other haemostatically active extracellular proteins (Chen and Hogg, 2006) present further possible targets for alteration by plasma RSNOs. Thus although RSNO-mediated signalling to the vessel wall via intracellular thiol modification requires processing via the cysNO/L-AT system, the ability of GSNO (and other RSNOs) to modify important exofacial targets on platelets and other cells may confer selective antithrombotic action (Figure 2).
Figure 2.
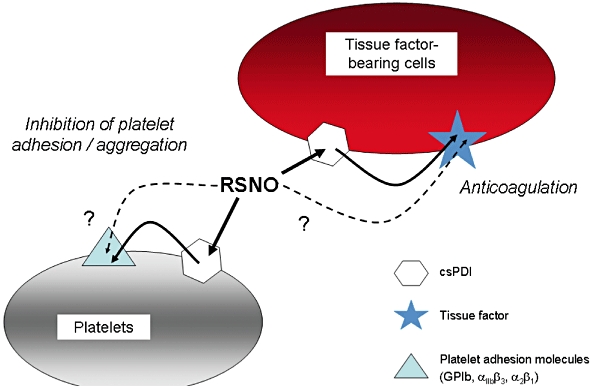
Cell surface targets for the antithrombotic action of S-nitrosothiols (RSNOs). Inhibition of the thrombotic process may be mediated without the need for intracellular nitric oxide (NO) entry, by inactivation of platelet surface adhesion molecules and/or of tissue factor exposed on the surface of the damaged vascular wall, on activated monocytes or circulating microparticles. These modifications occur indirectly via RSNO-induced inhibition of cell surface protein disulphide isomerase (csPDI), and also possibly via direct transnitrosation of the target molecule.
Conclusion
Possible mechanisms for selective antithrombotic action of RSNOs may be summarized as:
Differences between platelets and cells of the vascular wall in expression of RSNO metabolizing enzymes, such as csPDI, and/or dependence on csPDI for signal transmission.
Differences between platelets and cells of the vascular wall in expression of the L-AT system, or dependence on cysNO/L-AT as a mode of NO delivery.
A heightened role in platelets, compared with cells of the vascular wall, for modification of target proteins on the external surface of the plasma membrane, thus allowing at least partial inhibitory effects to be achieved without the need for intracellular delivery of NO.
RSNO-mediated regulation of blood coagulation, in particular tissue factor exposed at sites of vessel injury or on circulating monocytes or platelet microparticles. This might be either a direct effect or secondary to csPDI modification.
There are few experimental data directly comparing these different mechanisms between platelets and cells of the vascular wall, so it is difficult to grade the mechanisms in order of importance. Nevertheless, the evidence currently available suggests a scenario in which denitrosating enzymes on platelets permit low concentrations of RSNO to mediate antithrombotic NO signalling, whereas higher concentrations of RSNO are required for vasodilatory signalling via the cysNO/L-AT mechanism. A further impeding factor may be the need for RSNO to cross the endothelial monolayer to gain entry into vascular smooth muscle, both of which steps involve cysNO/L-AT. At present these ideas remain speculative, however, they do suggest lines of enquiry that might help define and realize the antithrombotic potential of RSNO compounds.
Acknowledgments
We acknowledge the support of the British Heart Foundation for work performed in this department.
Glossary
Abbreviations:
- csPDI
cell surface protein disulphide isomerase
- cyclic GMP
guanosine 3′:5′-cyclic monophosphate
- cysNO
S-nitrosocysteine
- DAF-FM
4-amino-5-methylamino-2′7′-difluorofluorescein
- GSNO
S-nitrosoglutathione
- L-AT
amino acid transporter system-L
- PDI
protein disulphide isomerase
- RSNO
S-nitrosothiol
- sGC
soluble guanylate cyclase
- tPA
tissue plasminogen activator
Statement of conflicts of interest
None.
References
- Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ, et al. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci USA. 2006;103:13932–13937. doi: 10.1073/pnas.0606411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhter S, Vignini A, Wen Z, English A, Wang PG, Mutus B. Evidence for S-nitrosothiol-dependent changes in fibrinogen that do not involve transnitrosation or thiolation. Proc Natl Acad Sci USA. 2002;99:9172–9177. doi: 10.1073/pnas.142136499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sa'doni H, Ferro A. S-Nitrosothiols: a class of nitric oxide-donor drugs. Clin Sci (Lond) 2000;98:507–520. [PubMed] [Google Scholar]
- Bach RR. Tissue factor encryption. Arterioscler Thromb Vasc Biol. 2006;26:456–461. doi: 10.1161/01.ATV.0000202656.53964.04. [DOI] [PubMed] [Google Scholar]
- Badimon L, Vilahur G. Platelets, arterial thrombosis and cerebral ischemia. Cerebrovasc Dis. 2007;24(Suppl. 1):30–39. doi: 10.1159/000107377. [DOI] [PubMed] [Google Scholar]
- Barbouche R, Miquelis R, Jones IM, Fenouillet E. Protein-disulfide isomerase-mediated reduction of two disulfide bonds of HIV envelope glycoprotein 120 occurs post-CXCR4 binding and is required for fusion. J Biol Chem. 2003;278:3131–3136. doi: 10.1074/jbc.M205467200. [DOI] [PubMed] [Google Scholar]
- De Belder AJ, MacAllister R, Radomski MW, Moncada S, Vallance PJ. Effects of S-nitroso-glutathione in the human forearm circulation: evidence for selective inhibition of platelet activation. Cardiovasc Res. 1994;28:691–694. doi: 10.1093/cvr/28.5.691. [DOI] [PubMed] [Google Scholar]
- Bell SE, Shah CM, Gordge MP. Protein disulfide-isomerase mediates delivery of nitric oxide redox derivatives into platelets. Biochem J. 2007;403:283–288. doi: 10.1042/BJ20061146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broniowska KA, Zhang Y, Hogg N. Requirement of transmembrane transport for S-nitrosocysteine-dependent modification of intracellular thiols. J Biol Chem. 2006;281:33835–33841. doi: 10.1074/jbc.M603248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess JK, Hotchkiss KA, Suter C, Dudman NP, Szollosi J, Chesterman CN, et al. Physical proximity and functional association of glycoprotein 1balpha and protein-disulfide isomerase on the platelet plasma membrane. J Biol Chem. 2000;275:9758–9766. doi: 10.1074/jbc.275.13.9758. [DOI] [PubMed] [Google Scholar]
- Catani MV, Bernassola F, Rossi A, Melino G. Inhibition of clotting factor XIII activity by nitric oxide. Biochem Biophys Res Commun. 1998;249:275–278. doi: 10.1006/bbrc.1998.9130. [DOI] [PubMed] [Google Scholar]
- Chen VM, Hogg PJ. Allosteric disulfide bonds in thrombosis and thrombolysis. J Thromb Haemost. 2006;4:2533–2541. doi: 10.1111/j.1538-7836.2006.02236.x. [DOI] [PubMed] [Google Scholar]
- Chen VM, Ahamed J, Versteeg HH, Berndt MC, Ruf W, Hogg PJ. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry. 2006;45:12020–12028. doi: 10.1021/bi061271a. [DOI] [PubMed] [Google Scholar]
- Cho J, Furie BC, Coughlin SR, Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest. 2008;118:1123–1131. doi: 10.1172/JCI34134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant CG, Stephens RS. Chlamydia attachment to mammalian cells requires protein disulfide isomerase. Cell Microbiol. 2007;9:222–232. doi: 10.1111/j.1462-5822.2006.00783.x. [DOI] [PubMed] [Google Scholar]
- Cornwell TL, Ceaser EK, Li J, Marrs KL, Darley-Usmar VM, Patel RP. S-nitrosothiols inhibit uterine smooth muscle cell proliferation independent of metabolism to NO and cyclic GMP formation. Am J Physiol Cell Physiol. 2003;284:C1516–C1524. doi: 10.1152/ajpcell.00268.2002. [DOI] [PubMed] [Google Scholar]
- Crane MS, Ollosson R, Moore KP, Rossi AG, Megson IL. Novel role for low molecular weight plasma thiols in nitric oxide-mediated control of platelet function. J Biol Chem. 2002;277:46858–46863. doi: 10.1074/jbc.M208608200. [DOI] [PubMed] [Google Scholar]
- Crane MS, Rossi AG, Megson IL. A potential role for extracellular nitric oxide generation in cyclic GMP-independent inhibition of human platelet aggregation: biochemical and pharmacological considerations. Br J Pharmacol. 2005;144:849–859. doi: 10.1038/sj.bjp.0706110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davisson RL, Travis MD, Bates JN, Lewis SJ. Hemodynamic effects of L- and D-S-nitrosocysteine in the rat. Stereoselective S-nitrosothiol recognition sites. Circ Res. 1996;79:256–262. doi: 10.1161/01.res.79.2.256. [DOI] [PubMed] [Google Scholar]
- Egorina EM, Sovershaev MA, Osterud B. Regulation of tissue factor procoagulant activity by post-translational modifications. Thromb Res. 2008;122:831–837. doi: 10.1016/j.thromres.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Essex DW. The role of thiols and disulfides in platelet function. Antioxid Redox Signal. 2004;6:736–746. doi: 10.1089/1523086041361622. [DOI] [PubMed] [Google Scholar]
- Essex DW, Chen K, Swiatkowska M. Localization of protein disulfide isomerase to the external surface of the platelet plasma membrane. Blood. 1995;86:2168–2173. [PubMed] [Google Scholar]
- Foster MW, Liu L, Zeng M, Hess DT, Stamler JS. A genetic analysis of nitrosative stress. Biochemistry. 2009;48:792–799. doi: 10.1021/bi801813n. [DOI] [PubMed] [Google Scholar]
- Freedman JE, Loscalzo J. Nitric oxide and its relationship to thrombotic disorders. J Thromb Haemost. 2003;1:1183–1188. doi: 10.1046/j.1538-7836.2003.00180.x. [DOI] [PubMed] [Google Scholar]
- Freedman JE, Frei B, Welch GN, Loscalzo J. Glutathione peroxidase potentiates the inhibition of platelet function by S-nitrosothiols. J Clin Invest. 1995;96:394–400. doi: 10.1172/JCI118047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer CB, Stasko NA, Rus IA, Lord ST, Schoenfisch MH. Influence of glutathione and its derivatives on fibrin polymerization. Biomacromolecules. 2008;9:1876–1882. doi: 10.1021/bm800146j. [DOI] [PubMed] [Google Scholar]
- Giustarini D, Milzani A, Colombo R, Dalle-Donne I, Rossi R. Nitric oxide and S-nitrosothiols in human blood. Clin Chim Acta. 2003;330:85–98. doi: 10.1016/s0009-8981(03)00046-9. [DOI] [PubMed] [Google Scholar]
- Gordge MP, Meyer DJ, Hothersall J, Neild GH, Payne NN, Noronha-Dutra A. Copper chelation-induced reduction of the biological activity of S-nitrosothiols. Br J Pharmacol. 1995;114:1083–1089. doi: 10.1111/j.1476-5381.1995.tb13317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordge MP, Hothersall JS, Neild GH, Dutra AA. Role of a copper (I)-dependent enzyme in the anti-platelet action of S-nitrosoglutathione. Br J Pharmacol. 1996;119:533–538. doi: 10.1111/j.1476-5381.1996.tb15704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordge MP, Addis P, Noronha-Dutra AA, Hothersall JS. Cell-mediated biotransformation of S-nitrosoglutathione. Biochem Pharmacol. 1998a;55:657–665. doi: 10.1016/s0006-2952(97)00498-x. [DOI] [PubMed] [Google Scholar]
- Gordge MP, Hothersall JS, Noronha-Dutra AA. Evidence for a cyclic GMP-independent mechanism in the anti-platelet action of S-nitrosoglutathione. Br J Pharmacol. 1998b;124:141–148. doi: 10.1038/sj.bjp.0701821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granillo OM, Brahmajothi MV, Li S, Whorton AR, Mason SN, McMahon TJ, et al. Pulmonary alveolar epithelial uptake of S-nitrosothiols is regulated by l-type amino acid transporter. Am J Physiol Lung Cell Mol Physiol. 2008;295:L38–L43. doi: 10.1152/ajplung.00280.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber CW, Cemazar M, Heras B, Martin JL, Craik DJ. Protein disulfide isomerase: the structure of oxidative folding. Trends Biochem Sci. 2006;31:455–464. doi: 10.1016/j.tibs.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Gurbel PA, Bliden KP, Hayes KM, Tantry U. Platelet activation in myocardial ischemic syndromes. Expert Rev Cardiovasc Ther. 2004;2:535–545. doi: 10.1586/14779072.2.4.535. [DOI] [PubMed] [Google Scholar]
- Hoffman M, Monroe DM., 3rd A cell-based model of hemostasis. Thromb Haemost. 2001;85:958–965. [PubMed] [Google Scholar]
- Hogg N. Biological chemistry and clinical potential of S-nitrosothiols. Free Radic Biol Med. 2000;28:1478–1486. doi: 10.1016/s0891-5849(00)00248-3. [DOI] [PubMed] [Google Scholar]
- Hogg N, Singh RJ, Konorev E, Joseph J, Kalyanaraman B. S-Nitrosoglutathione as a substrate for gamma-glutamyl transpeptidase. Biochem J. 1997;323:477–481. doi: 10.1042/bj3230477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan PA, Stevens JM, Hubbard GP, Barrett NE, Sage T, Authi KS, et al. A role for the thiol isomerase protein ERP5 in platelet function. Blood. 2005;105:1500–1507. doi: 10.1182/blood-2004-02-0608. [DOI] [PubMed] [Google Scholar]
- Jourd'heuil D, Laroux FS, Miles AM, Wink DA, Grisham MB. Effect of superoxide dismutase on the stability of S-nitrosothiols. Arch Biochem Biophys. 1999;361:323–330. doi: 10.1006/abbi.1998.1010. [DOI] [PubMed] [Google Scholar]
- Kaposzta Z, Clifton A, Molloy J, Martin JF, Markus HS. S-nitrosoglutathione reduces asymptomatic embolization after carotid angioplasty. Circulation. 2002;106:3057–3062. doi: 10.1161/01.cir.0000041251.07332.28. [DOI] [PubMed] [Google Scholar]
- Keaney JF, Jr, Simon DI, Stamler JS, Jaraki O, Scharfstein J, Vita JA, et al. NO forms an adduct with serum albumin that has endothelium-derived relaxing factor-like properties. J Clin Invest. 1993;91:1582–1589. doi: 10.1172/JCI116364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkola T, Savinainen JR, Monkkonen KS, Retamal MD, Laitinen JT. S-nitrosothiols modulate G protein-coupled receptor signaling in a reversible and highly receptor-specific manner. BMC Cell Biol. 2005;6:21. doi: 10.1186/1471-2121-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaluk EA, Fung HL. Spontaneous liberation of nitric oxide cannot account for in vitro vascular relaxation by S-nitrosothiols. J Pharmacol Exp Ther. 1990;255:1256–1264. [PubMed] [Google Scholar]
- Kuo YR, Wang FS, Jeng SF, Huang HC, Wei FC, Yang KD. Nitrosoglutathione modulation of platelet activation and nitric oxide synthase expression in promotion of flap survival after ischemia/reperfusion injury. J Surg Res. 2004;119:92–99. doi: 10.1016/j.jss.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Lahav J, Wijnen EM, Hess O, Hamaia SW, Griffiths D, Makris M, et al. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin alpha2beta1. Blood. 2003;102:2085–2092. doi: 10.1182/blood-2002-06-1646. [DOI] [PubMed] [Google Scholar]
- Lai TS, Hausladen A, Slaughter TF, Eu JP, Stamler JS, Greenberg CS. Calcium regulates S-nitrosylation, denitrosylation, and activity of tissue transglutaminase. Biochemistry. 2001;40:4904–4910. doi: 10.1021/bi002321t. [DOI] [PubMed] [Google Scholar]
- Langer HF, Gawaz M. Platelet-vessel wall interactions in atherosclerotic disease. Thromb Haemost. 2008;99:480–486. doi: 10.1160/TH07-11-0685. [DOI] [PubMed] [Google Scholar]
- Lees C, Langford E, Brown AS, de Belder A, Pickles A, Martin JF, et al. The effects of S-nitrosoglutathione on platelet activation, hypertension, and uterine and fetal Doppler in severe preeclampsia. Obstet Gynecol. 1996;88:14–19. doi: 10.1016/0029-7844(96)00070-1. [DOI] [PubMed] [Google Scholar]
- Li S, Whorton AR. Identification of stereoselective transporters for S-nitroso-L-cysteine: role of LAT1 and LAT2 in biological activity of S-nitrosothiols. J Biol Chem. 2005;280:20102–20110. doi: 10.1074/jbc.M413164200. [DOI] [PubMed] [Google Scholar]
- Li S, Whorton AR. Functional characterization of two S-nitroso-L-cysteine transporters, which mediate movement of NO equivalents into vascular cells. Am J Physiol Cell Physiol. 2007;292:C1263–C1271. doi: 10.1152/ajpcell.00382.2006. [DOI] [PubMed] [Google Scholar]
- Lieberman EH, O'Neill S, Mendelsohn ME. S-nitrosocysteine inhibition of human platelet secretion is correlated with increases in platelet cyclic GMP levels. Circ Res. 1991;68:1722–1728. doi: 10.1161/01.res.68.6.1722. [DOI] [PubMed] [Google Scholar]
- Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- Lupidi G, Angeletti M, Eleuteri AM, Tacconi L, Coletta M, Fioretti E. Peroxynitrite-mediated oxidation of fibrinogen inhibits clot formation. FEBS Lett. 1999;462:236–240. doi: 10.1016/s0014-5793(99)01500-8. [DOI] [PubMed] [Google Scholar]
- Mackman N. Triggers, targets and treatments for thrombosis. Nature. 2008;451:914–918. doi: 10.1038/nature06797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani AR, Ebrahimkhani MR, Ippolito S, Ollosson R, Moore KP. Metalloprotein-dependent decomposition of S-nitrosothiols: studies on the stabilization and measurement of S-nitrosothiols in tissues. Free Radic Biol Med. 2006;40:1654–1663. doi: 10.1016/j.freeradbiomed.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Manickam N, Sun X, Li M, Gazitt Y, Essex DW. Protein disulphide isomerase in platelet function. Br J Haematol. 2008;140:223–229. doi: 10.1111/j.1365-2141.2007.06898.x. [DOI] [PubMed] [Google Scholar]
- Marcondes S, Cardoso MH, Morganti RP, Thomazzi SM, Lilla S, Murad F, et al. Cyclic GMP-independent mechanisms contribute to the inhibition of platelet adhesion by nitric oxide donor: a role for alpha-actinin nitration. Proc Natl Acad Sci USA. 2006;103:3434–3439. doi: 10.1073/pnas.0509397103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews WR, Kerr SW. Biological activity of S-nitrosothiols: the role of nitric oxide. J Pharmacol Exp Ther. 1993;267:1529–1537. [PubMed] [Google Scholar]
- Megson IL, Sogo N, Mazzei FA, Butler AR, Walton JC, Webb DJ. Inhibition of human platelet aggregation by a novel S-nitrosothiol is abolished by haemoglobin and red blood cells in vitro: implications for antithrombotic therapy. Br J Pharmacol. 2000;131:1391–1398. doi: 10.1038/sj.bjp.0703731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellion BT, Ignarro LJ, Myers CB, Ohlstein EH, Ballot BA, Hyman AL, et al. Inhibition of human platelet aggregation by S-nitrosothiols. Heme-dependent activation of soluble guanylate cyclase and stimulation of cyclic GMP accumulation. Mol Pharmacol. 1983;23:653–664. [PubMed] [Google Scholar]
- Mendelsohn ME, O'Neill S, George D, Loscalzo J. Inhibition of fibrinogen binding to human platelets by S-nitroso-N-acetylcysteine. J Biol Chem. 1990;265:19028–19034. [PubMed] [Google Scholar]
- Miller MR, Megson IL. Recent developments in nitric oxide donor drugs. Br J Pharmacol. 2007;151:305–321. doi: 10.1038/sj.bjp.0707224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MR, Hanspal IS, Hadoke PW, Newby DE, Rossi AG, Webb DJ, et al. A novel S-nitrosothiol causes prolonged and selective inhibition of platelet adhesion at sites of vascular injury. Cardiovasc Res. 2003;57:853–860. doi: 10.1016/s0008-6363(02)00779-4. [DOI] [PubMed] [Google Scholar]
- Molloy J, Martin JF, Baskerville PA, Fraser SC, Markus HS. S-nitrosoglutathione reduces the rate of embolization in humans. Circulation. 1998;98:1372–1375. doi: 10.1161/01.cir.98.14.1372. [DOI] [PubMed] [Google Scholar]
- Murad F. Discovery of some of the effects of nitric oxide and its role in cell signalling. Biosci Rep. 1999;19:133–154. doi: 10.1023/a:1020265417394. [DOI] [PubMed] [Google Scholar]
- Myers PR, Minor RL, Jr, Guerra R, Jr, Bates JN, Harrison DG. Vasorelaxant properties of the endothelium-derived relaxing factor more closely resemble S-nitrosocysteine than nitric oxide. Nature. 1990;345:161–163. doi: 10.1038/345161a0. [DOI] [PubMed] [Google Scholar]
- Nikitovic D, Holmgren A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J Biol Chem. 1996;271:19180–19185. doi: 10.1074/jbc.271.32.19180. [DOI] [PubMed] [Google Scholar]
- Oberprieler NG, Roberts W, Riba R, Graham AM, Homer-Vanniasinkam S, Naseem KM. cyclic GMP-independent inhibition of integrin alphaIIbbeta3-mediated platelet adhesion and outside-in signalling by nitric oxide. FEBS Lett. 2007;581:1529–1534. doi: 10.1016/j.febslet.2007.02.072. [DOI] [PubMed] [Google Scholar]
- Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- Pawloski JR, Swaminathan RV, Stamler JS. Cell-free and erythrocytic S-nitrosohemoglobin inhibits human platelet aggregation. Circulation. 1998;97:263–267. doi: 10.1161/01.cir.97.3.263. [DOI] [PubMed] [Google Scholar]
- Pendurthi UR, Ghosh S, Mandal SK, Rao LV. Tissue factor activation: is disulfide bond switching a regulatory mechanism? Blood. 2007;110:3900–3908. doi: 10.1182/blood-2007-07-101469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigazzi A, Heydrick S, Folli F, Benoit S, Michelson A, Loscalzo J. Nitric oxide inhibits thrombin receptor-activating peptide-induced phosphoinositide 3-kinase activity in human platelets. J Biol Chem. 1999;274:14368–14375. doi: 10.1074/jbc.274.20.14368. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Palmer RM, Moncada S. The role of nitric oxide and cyclic GMP in platelet adhesion to vascular endothelium. Biochem Biophys Res Commun. 1987;148:1482–1489. doi: 10.1016/s0006-291x(87)80299-1. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Rees DD, Dutra A, Moncada S. S-nitroso-glutathione inhibits platelet activation in vitro and in vivo. Br J Pharmacol. 1992;107:745–749. doi: 10.1111/j.1476-5381.1992.tb14517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran N, Root P, Jiang XM, Hogg PJ, Mutus B. Mechanism of transfer of NO from extracellular S-nitrosothiols into the cytosol by cell-surface protein disulfide isomerase. Proc Natl Acad Sci USA. 2001;98:9539–9544. doi: 10.1073/pnas.171180998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riba R, Patel B, Aburima A, Naseem KM. Globular adiponectin increases cyclic GMP formation in blood platelets independently of nitric oxide. J Thromb Haemost. 2008;6:2121–2131. doi: 10.1111/j.1538-7836.2008.03179.x. [DOI] [PubMed] [Google Scholar]
- Richardson G, Benjamin N. Potential therapeutic uses for S-nitrosothiols. Clin Sci (Lond) 2002;102:99–105. [PubMed] [Google Scholar]
- Riego JA, Broniowska KA, Kettenhofen NJ, Hogg N. Activation and inhibition of soluble guanylyl cyclase by S-nitrosocysteine: involvement of amino acid transport system L. Free Radic Biol Med. 2009;47:269–274. doi: 10.1016/j.freeradbiomed.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson A, O'Neill S, Kiernan A, O'Donoghue N, Moran N. Bacitracin reveals a role for multiple thiol isomerases in platelet function. Br J Haematol. 2006;132:339–348. doi: 10.1111/j.1365-2141.2005.05878.x. [DOI] [PubMed] [Google Scholar]
- Root P, Sliskovic I, Mutus B. Platelet cell surface protein disulfide isomerase mediated S-nitrosoglutathione consumption. Biochem J. 2004;382:575–580. doi: 10.1042/BJ20040759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas E, Langford EJ, Marrinan MT, Martin JF, Moncada S, de Belder AJ. S-nitrosoglutathione inhibits platelet activation and deposition in coronary artery saphenous vein grafts in vitro and in vivo. Heart. 1998;80:146–150. doi: 10.1136/hrt.80.2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmann J, Schwedhelm KS, Tsikas D. Specific transport of S-nitrosocysteine in human red blood cells: implications for formation of S-nitrosothiols and transport of NO bioactivity within the vasculature. FEBS Lett. 2005;579:4119–4124. doi: 10.1016/j.febslet.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Schwarz UR, Walter U, Eigenthaler M. Taming platelets with cyclic nucleotides. Biochem Pharmacol. 2001;62:1153–1161. doi: 10.1016/s0006-2952(01)00760-2. [DOI] [PubMed] [Google Scholar]
- Shah CM, Locke IC, Chowdrey HS, Gordge MP. Rapid S-nitrosothiol metabolism by platelets and megakaryocytes. Biochem Soc Trans. 2003;31:1450–1452. doi: 10.1042/bst0311450. [DOI] [PubMed] [Google Scholar]
- Shah CM, Bell SE, Locke IC, Chowdrey HS, Gordge MP. Interactions between cell surface protein disulphide isomerase and S-nitrosoglutathione during nitric oxide delivery. Nitric Oxide. 2007;16:135–142. doi: 10.1016/j.niox.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Simon DI, Stamler JS, Jaraki O, Keaney JF, Osborne JA, Francis SA, et al. Antiplatelet properties of protein S-nitrosothiols derived from nitric oxide and endothelium-derived relaxing factor. Arterioscler Thromb. 1993;13:791–799. doi: 10.1161/01.atv.13.6.791. [DOI] [PubMed] [Google Scholar]
- Sliskovic I, Raturi A, Mutus B. Characterization of the S-denitrosation activity of protein disulfide isomerase. J Biol Chem. 2005;280:8733–8741. doi: 10.1074/jbc.M408080200. [DOI] [PubMed] [Google Scholar]
- Sogo N, Magid KS, Shaw CA, Webb DJ, Megson IL. Inhibition of human platelet aggregation by nitric oxide donor drugs: relative contribution of cyclic GMP-independent mechanisms. Biochem Biophys Res Commun. 2000;279:412–419. doi: 10.1006/bbrc.2000.3976. [DOI] [PubMed] [Google Scholar]
- Staab CA, Alander J, Morgenstern R, Grafstrom RC, Hoog JO. The Janus face of alcohol dehydrogenase 3. Chem Biol Interact. 2009;178:29–35. doi: 10.1016/j.cbi.2008.10.050. [DOI] [PubMed] [Google Scholar]
- Stamler JS. S-nitrosothiols in the blood: roles, amounts, and methods of analysis. Circ Res. 2004;94:414–417. doi: 10.1161/01.RES.0000122071.55721.BC. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Jaraki O, Osborne J, Simon DI, Keaney J, Vita J, et al. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci USA. 1992a;89:7674–7677. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Simon DI, Jaraki O, Osborne JA, Francis S, Mullins M, et al. S-nitrosylation of tissue-type plasminogen activator confers vasodilatory and antiplatelet properties on the enzyme. Proc Natl Acad Sci USA. 1992b;89:8087–8091. doi: 10.1073/pnas.89.17.8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffel J, Luscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 2006;113:722–731. doi: 10.1161/CIRCULATIONAHA.105.567297. [DOI] [PubMed] [Google Scholar]
- Tsikas D, Ikic M, Tewes KS, Raida M, Frolich JC. Inhibition of platelet aggregation by S-nitroso-cysteine via cyclic GMP-independent mechanisms: evidence of inhibition of thromboxane A2 synthesis in human blood platelets. FEBS Lett. 1999;442:162–166. doi: 10.1016/s0014-5793(98)01633-0. [DOI] [PubMed] [Google Scholar]
- Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol. 2002;193:154–163. doi: 10.1002/jcp.10172. [DOI] [PubMed] [Google Scholar]
- Vadseth C, Souza JM, Thomson L, Seagraves A, Nagaswami C, Scheiner T, et al. Pro-thrombotic state induced by post-translational modification of fibrinogen by reactive nitrogen species. J Biol Chem. 2004;279:8820–8826. doi: 10.1074/jbc.M306101200. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf) 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- Versteeg HH, Ruf W. Tissue factor coagulant function is enhanced by protein-disulfide isomerase independent of oxidoreductase activity. J Biol Chem. 2007;282:25416–25424. doi: 10.1074/jbc.M702410200. [DOI] [PubMed] [Google Scholar]
- Vilahur G, Baldellou MI, Segales E, Salas E, Badimon L. Inhibition of thrombosis by a novel platelet selective S-nitrosothiol compound without hemodynamic side effects. Cardiovasc Res. 2004;161:806–816. doi: 10.1016/j.cardiores.2003.11.034. [DOI] [PubMed] [Google Scholar]
- Walsh GM, Leane D, Moran N, Keyes TE, Forster RJ, Kenny D, et al. S-Nitrosylation of platelet alphaIIbbeta3 as revealed by Raman spectroscopy. Biochemistry. 2007;46:6429–6436. doi: 10.1021/bi0620712. [DOI] [PubMed] [Google Scholar]
- Wang GR, Zhu Y, Halushka PV, Lincoln TM, Mendelsohn ME. Mechanism of platelet inhibition by nitric oxide: in vivo phosphorylation of thromboxane receptor by cyclic GMP-dependent protein kinase. Proc Natl Acad Sci USA. 1998;95:4888–4893. doi: 10.1073/pnas.95.9.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B, Smith JW. A redox site involved in integrin activation. J Biol Chem. 2000;275:39964–39972. doi: 10.1074/jbc.M007041200. [DOI] [PubMed] [Google Scholar]
- Zai A, Rudd MA, Scribner AW, Loscalzo J. Cell-surface protein disulfide isomerase catalyzes transnitrosation and regulates intracellular transfer of nitric oxide. J Clin Invest. 1999;103:393–399. doi: 10.1172/JCI4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Spencer NY, Hogg N. Metabolism of S-nitrosoglutathione by endothelial cells. Am J Physiol Heart Circ Physiol. 2001;281:H432–H439. doi: 10.1152/ajpheart.2001.281.1.H432. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Hogg N. The mechanism of transmembrane S-nitrosothiol transport. Proc Natl Acad Sci USA. 2004;101:7891–7896. doi: 10.1073/pnas.0401167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hogg N. S-Nitrosothiols: cellular formation and transport. Free Radic Biol Med. 2005;38:831–838. doi: 10.1016/j.freeradbiomed.2004.12.016. [DOI] [PubMed] [Google Scholar]
- Zhu J, Li S, Marshall ZM, Whorton AR. A cystine-cysteine shuttle mediated by xCT facilitates cellular responses to S-nitrosoalbumin. Am J Physiol Cell Physiol. 2008;294:C1012–C1020. doi: 10.1152/ajpcell.00411.2007. [DOI] [PubMed] [Google Scholar]