

Abstract
Background and purpose:
Suppression of the renin-angiotensin-aldosterone system can prevent atrial fibrillation (AF) by attenuating atrial structural remodelling but the role of aldosterone in AF prevention has not been investigated thoroughly. We explored whether the aldosterone antagonist, spironolactone, could improve atrial structural remodelling in long-term rapid pacing-induced AF.
Experimental approach:
Three groups of dogs were used, sham-operated, control and spironolactone-treated groups. Dogs in the control and spironolactone groups had right atrial pacing for 6 weeks. The spironolactone group was given spironolactone 1 week before and during the atrial pacing. After 6 weeks of pacing, atrial structural and functional changes were assessed by echocardiography, haemodynamic parameters by cardiac catheterization, histopathological changes by light and electron microscopy and cardiomyocyte apoptosis by TUNEL. Caspase-3, Bcl-2, bax, calpain I, calpastatin, matrix metalloproteinase (MMP)-9 and tissue inhibitors of metalloproteinase (TIMP)-1 were analysed by immunohistochemistry and Western blotting. The inducibility and duration of AF were measured by atrial burst pacing.
Key results:
After atrial pacing, the proportion of TUNEL positive cells, myolysis, atrial fibrosis and dilatation were all significantly increased and these changes were inhibited by spironolactone. Spironolactone treatment reversed the increased expression of caspase-3, bax, calpain I and MMP-9 and the decreased level of Bcl-2, calpastatin and TIMP-1, induced by chronic atrial pacing. Also spironolactone prevented the increased inducibility and duration of AF, induced by tachypacing.
Conclusions and implications:
Treatment with spironolactone prevented myocardial apoptosis, myolysis, atrial fibrosis and dilatation, suggesting a possible beneficial effect of aldosterone antagonism on atrial structural remodelling in AF.
This article is commented on by Lendeckel et al., pp. 1581–1583 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2010.00675.x
Keywords: atrial fibrillation, structural remodelling, aldosterone, spironolactone
Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia in clinical practice, but still difficult to treat because of its complex pathological properties (Nattel, 2002). AF by itself induces electrical and structural remodelling in the atrium favoring its recurrence and persistence (Schoonderwoerd et al., 2005). Atrial structural abnormalities include severe apoptosis, myolysis, interstitial fibrosis and atrial enlargement, which in turn promote the occurrence or maintenance of AF (Allessie et al., 2002). After decades of focus on the electrical aspects of AF with unsatisfactory results, current interest has shifted to the underlying structural substrate.
Activation of the renin-angiotensin-aldosterone system (RAAS) correlates directly with atrial structural remodelling and its blockade by angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin type 1 (AT1) receptor antagonists is beneficial (Ehrlich et al., 2006). However, long-term treatment with ACEIs or AT1 receptor antagonists does not reliably suppress aldosterone production, a well-documented phenomenon known as ‘aldosterone escape’ (Struthers, 2004). Indeed, aldosterone is a potentially important component of RAAS that has not been well investigated in the context of AF prevention. Serum aldosterone levels are elevated in patients with persistent AF and decline after sinus rhythm is restored (Goette et al., 2001). Patients with primary hyperaldosteronism show a 12-fold greater AF risk compared with controls (Milliez et al., 2005b). A chronic increase in the plasma concentration of aldosterone has also been shown to contribute to the pathophysiology of AF (Dixen et al., 2007). As an end-product of RAAS, aldosterone is one element of neurohormonal activation associated with both chronic heart failure (CHF) and AF. Aldosterone participates in cardiac remodelling and exerts deleterious effects including vascular inflammation and damage, endothelial dysfunction, noradrenaline release, collagen production and interstitial fibrosis, independent of its effects on blood volume and pressure (Delcayre and Swynghedauw, 2002;). Therefore, we hypothesized that atrial structural abnormalities caused by an activated aldosterone system might be involved in the mechanisms of AF maintenance.
It is well-known that aldosterone antagonists, such as spironolactone and eplerenone, remarkably improve the prognosis of heart failure (Cicoira et al., 2002; Pitt et al., 2003);. Furthermore, aldosterone antagonists are beneficial across a broad spectrum of cardiovascular diseases. Spironolactone has been shown to protect against damage to target organs not only in CHF, but also in AF (Yang et al., 2008). A raised aldosterone level in AF due to RAAS activation indicates the blockade of aldosterone receptors by spironolactone has great therapeutic promises for preventing AF, but a clear effect of spironolactone in attenuating atrial structural remodelling has not yet been described. Therefore, the purpose of the present study was to investigate the effects of spironolactone on the improvement of atrial structural remodelling in a rapid atrial pacing model in dogs.
Methods
Preparation of animals
All animal care and experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. 21 mongrel dogs (15–25 kg) of either sex, were randomly divided into three groups: sham-operated (n= 7), control (n= 7) and spironolactone (n= 7). All dogs were anaesthetized with pentobarbital sodium (25 mg·kg−1 i.v.). After intubation and mechanical ventilation, medial thoracotomy was performed. Followed by radiofrequency ablation to create complete atrioventricular block, a unipolar ventricular-pacing lead was inserted into the right ventricular apex under fluoroscopy and attached to a pacemaker (Shanghai Fudan University, Shanghai, China) implanted in a subcutaneous pocket of the neck, programmed to 100 beats per minute throughout the study. One thin silicon plaque containing four pairs of electrodes (electrode diameter, 1 mm; interelectrode distance, 1 mm; distance between electrode pairs, 10 mm) was sutured to each atrium. The other ends of the electrode cables were tunneled subcutaneously and exposed at the back of the dogs where they were used for the pacing and electrophysiological measurements in the chronic phase. The pacemaker was attached to a screw-in epicardial lead in the right atrial appendage (Li et al., 2007). After 1 week for recovery, dogs in the control and spironolactone groups were paced at 400 beats per minute for 6 weeks. In the spironolactone group, oral administration of spironolactone (20 mg·kg−1·day−1) was started 1 week before and continued during the rapid atrial pacing, while dogs in the control group received placebo (starch, 20 mg·kg−1·day−1) simultaneously.
Echocardiography
The structure and function of left atrium (LA), left atrial appendage (LAA) and left ventricle (LV) were assessed by transthoracic and transoesophageal echocardiographic examinations (GE VIVID5, Fairfield, CT, USA) (Li et al., 2007). LA maximal volume (LAVmax), LAA maximal volume (LAAVmax), LA minimal volume (LAVmin), LAA minimal volume (LAAVmin), LV maximal volume (LVVmax) and LV minimal volume (LVVmin) were measured. LA ejection fraction (LAEF) was calculated as (LAVmax − LAVmin)/LAVmax. LAA ejection fraction (LAAEF) was calculated as (LAAVmax − LAAVmin)/LAAVmax. LV ejection fraction (LVEF) was calculated as (LVVmax − LVVmin)/LVVmax.
Haemodynamic study
Right atrial pressure (RAP), mean pulmonary artery pressure (MPAP) and pulmonary capillary wedge pressure (PCWP) were monitored by a pulmonary arterial balloon catheter (Swan-Ganz catheter) through a 6-F sheath placed in the femoral vein. Aortic systolic and diastolic blood pressure were measured by a pigtail catheter through a 6F sheath placed in the femoral artery.
AF inducibility and duration assessment
Atrial fibrillation inducibility and duration were measured before and after 6 week of rapid atrial pacing in all groups respectively. Ten bursts of atrial pacing lasting for 10 s each at a pacing cycle length of 100 ms was used to assess the inducibility and duration of AF. AF lasting more than 30 min was terminated by direct current electrical cardioversion and 30 min was allowed before the experiment continued.
Morphological evaluation
At the completion of the protocol, the animal was anaesthetized and the beating heart was quickly excised. Atrial-free wall and appendage samples were rapidly frozen in liquid nitrogen and separately stored at −80°C for further analysis. One segment from the atrium was fixed with 4% paraformaldehyde at 4°C and embedded in paraffin for morphological evaluation. From each zone, tissue blocks were collected following longitudinal and transverse planes.
Light microscopy was performed using sections (5 µm) stained with haematoxylin-eosin (HE) and Masson trichrome solution. To quantify the degree of myolysis in the cardiomyocytes, at least two sections per atrial site were examined and at least 200 cells per section were analysed. The extent of cell change was evaluated only in cells in which the nucleus was present in the plane of the section. The myolytic area of the cardiomyocytes was measured with a digital imaging system (Motic Images Advanced, Richmond, BC, Canada). Myocytes were scored as mildly myolytic if myolysis involved 10% to 25% of the cytosol and as severely myolytic if >25% of the sarcomeres were absent. Connective tissue was differentiated on the basis of its color and expressed as a percentage of the reference tissue area. Blood vessels and perivascular interstitial cells were excluded from the connective tissue quantification. Interstitial collagen volume fraction (CVF) in the atrium was determined by quantitative morphometry with an image analyser (HPISA-1000, Olympus, Japan).
For electron microscopy, ultrathin sections (50–100 nm) were cut from each sample, counterstained with uranium acetate and lead citrate, and examined with a transmission electron microscope (Philips 201, Hillsboro, OR, USA) by two observers unaware of the treatments.
TUNEL staining
Tissue specimens were embedded in paraffin and cut to 7 µm thickness. Then, tissue was dewaxed and rehydrated according to standard protocols. Preparation of tissue for TUNEL staining was performed following the protocol provided by Roche Applied Science. Tissue was placed on glass slides and incubated in a plastic jar containing 200 mL 0.1 M citrate buffer and 0.1% Triton, pH 6.0. The sections were exposed to 350 W microwave irradiation for 5 min, cooled rapidly by adding 80 mL distilled water (20–25°C) and rinsed with PBS at 15–25°C. Afterwards, 50 µL of TUNEL reaction mixture (Roche Applied Science) was added to the sections, and glass slides were incubated for 60 min at 37°C. Finally, slides were rinsed three times in PBS. Negative controls were sections incubated with label solution alone, and sections incubated with DNAse I (Sigma Aldrich) served as positive controls. Following the staining procedure, samples were analysed for TUNEL-positive cells using a fluorescent microscope. The percentage of TUNEL positive nuclei was calculated as followed: 10 randomly chosen fields per section corresponding to approximately 700 cells were examined at high magnification (×400).
Western blotting
In total, 50 µg of total protein from homogenized atrial tissue solubilized for 5 min at 95°C in one volume loading buffer (1% SDS, 30% glycerol, 0.8 mM dithiothreitol, 1 mM Tris-HCl pH 6.8, 2% bromophenol-blue) was loaded onto a 10% SDS−PAGE gel and then transferred to PVDF membranes. The membranes were blocked with 5% nonfat dry milk in PBST (containing 0.05% Tween 20), and incubated overnight at 4°C with the primary antibodies (monoclonal rabbit anti-caspas-3, anti-Bcl-2, anti-bax, anti-MMP-9, anti-TIMP-1, anti-calpain I and anti-calpastatin antibodies; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA; used at 1:200), washed in PBST, incubated with horseradish peroxidase (HRP)-conjugated second antibody for 2 h at 37°C, and revealed by Immun-Star HRP Substrate.
Statistical analysis
Quantitative data were presented as mean ± standard deviation. Comparisons between the quantitative data were made using the t-test, whereas those for qualitative data were tested with X2 test. Differences in continuous variables were examined by anova. spss 13.0 was used in the statistical analysis. P < 0.05 was considered statistically significant.
Materials
Spironolactone was obtained from Minsheng Pharm (Hangzhou, Jiangsu, China). Pentobarbital sodium and general chemicals were obtained form Sigma-Aldrich (St Louis, MO, USA). HE and Masson trichrome stains were purchased from Maxin Company (Fuzhou, Fujian, China).
Results
Changes of atrial structure and function
There was no significant difference in LVVmax, LVVmin and LVEF among three groups (Table 1). Compared with sham-operated dogs, LAVmax, LAVmin, LAAVmax and LAAVmin increased significantly in the control and spironolactone groups, while LAEF and LAAEF decreased after termination of the 6-week rapid pacing. Compared with the control group, the LA and LAA volume decreased, whereas the LAEF and LAAEF increased in the spironolactone group, suggesting that spironolactone could improve LA function and prevent atrial structural dilatation in the canine pacing model.
Table 1.
Changes of echocardiographic indices before and after rapid atrial pacing
|
Sham-operated group |
Control group |
Spironolactone group |
||||
|---|---|---|---|---|---|---|
| Preoperation | 7 weeks post operation | Baseline | 6-week pacing | Baseline | 6-week pacing | |
| LAVmax (cm3) | 12.9 ± 2.4 | 14.5 ± 3.4 | 12.4 ± 2.7 | 23.1 ± 6.4##* | 12.7 ± 2.1 | 17.3 ± 3.3*†# |
| LAVmin (cm3) | 6.3 ± 1.5 | 6.9 ± 1.2 | 7.0 ± 1.2 | 15.3 ± 3.5##** | 6.5 ± 1.1 | 10.7 ± 2.5**††# |
| LAEF (%) | 51.1 ± 10.9 | 50.8 ± 9.8 | 48.7 ± 8.8 | 32.6 ± 7.4##** | 50.5 ± 8.5 | 41.1 ± 6.9**††# |
| LAAVmax (cm3) | 2.1 ± 0.5 | 2.2 ± 0.4 | 1.9 ± 0.3 | 3.2 ± 0.7##** | 1.9 ± 0.4 | 2.3 ± 0.7**††# |
| LAAVmin (cm3) | 0.8 ± 0.2 | 0.9 ± 0.4 | 0.9 ± 0.4 | 2.0 ± 0.5##** | 0.8 ± 0.3 | 1.2 ± 0.4*††# |
| LAAEF (%) | 55.3 ± 9.3 | 54.6 ± 9.8 | 54.9 ± 8.9 | 33.1 ± 8.0##** | 55.1 ± 8.5 | 42.0 ± 6.7**††# |
| LVVmax (cm3) | 34.7 ± 4.3 | 35.5 ± 4.1 | 36.6 ± 5.1 | 38.1 ± 6.4 | 35.4 ± 4.8 | 36.1 ± 4.3 |
| LVVmin (cm3) | 13.3 ± 2.6 | 13.9 ± 3.2 | 12.9 ± 2.2 | 14.3 ± 2.7 | 13.7 ± 2.9 | 13.6 ± 2.7 |
| LVEF (%) | 62.5 ± 5.9 | 60.8 ± 5.8 | 64.3 ± 6.8 | 63.0 ± 7.2 | 62.1 ± 4.5 | 62.3 ± 6.9 |
P < 0.05,
P < 0.01, compared with baseline;
P < 0.05,
P < 0.01, compared with sham-operated group;
P < 0.05,
P < 0.01, compared with control group.
LAAVmax, LAA maximal volume; LAAVmin, LAA minimal volume; LAAEF, LAA ejection fraction; LAEF, LA ejection fraction; LAVmax, LA maximal volume; LAVmin, LA minimal volume; LVEF, LV ejection fraction; LVVmax, LV maximal volume; LVVmin, LV minimal volume.
Haemodynamic changes
Treatment with spironolactone also prevented the increase of RAP, MPAP and PAWP induced by 6-week atrial pacing (Table 2). Compared with the control group, spironolactone decreased systolic and diastolic blood pressure.
Table 2.
Haemodynamic parameters in the sham-operated, control and spironolactone groups (mmHg)
| SBP | DBP | RAP | MPAP | PCWP | |
|---|---|---|---|---|---|
| Sham-operated group | |||||
| Pre-operation | 111 ± 11 | 69 ± 8 | 5 ± 1 | 15 ± 4 | 7 ± 3 |
| 7 weeks post operation | 109 ± 14 | 71 ± 9 | 6 ± 2 | 16 ± 4 | 8 ± 3 |
| Control group | |||||
| Baseline | 114 ± 14 | 71 ± 8 | 6 ± 2 | 16 ± 5 | 8 ± 2 |
| 6-week pacing | 112 ± 13 | 69 ± 6 | 12 ± 3**## | 23 ± 5*# | 16 ± 4**## |
| Spironolactone group | |||||
| Baseline | 110 ± 15 | 70 ± 8 | 6 ± 1 | 15 ± 3 | 7 ± 3 |
| 6-week pacing | 103 ± 12† | 65 ± 7† | 7 ± 2† | 18 ± 4† | 9 ± 3† |
P < 0.05,
P < 0.01, compared with baseline;
P < 0.05,
P < 0.01, compared with the sham-operated group;
P < 0.05, compared with the control group.
DBP, diastolic blood pressure; MPAP, mean pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; RAP, right atrial pressure; SBP, systolic blood pressure.
Changes in the properties of AF
The baseline inducibility, that is, before any intervention, of AF (Table 3) in all three groups was low and was not changed 7 weeks after operation in the sham-operated group. In the control group after 6-week pacing, inducibility was markedly increased and this increased inducibility was reduced after treatment with spironoloactone. Spironolactone also shortened AF duration, compared with that in the control group.
Table 3.
Changes in the inducibility and duration of A before and after rapid atrial pacing
| AF cases | AF times | AF inducibility (%) | Mean AF duration(s) | |
|---|---|---|---|---|
| Sham-operated group | ||||
| Preoperation | 2 | 8 | 11.4 | 47.3 ± 14.1 |
| 7-week postoperation | 2 | 9 | 12.9 | 49.1 ± 16.4 |
| Control group | ||||
| Baseline | 1 | 8 | 11.4 | 58.7 ± 15.7 |
| 6-week pacing | 7*# | 66**## | 94.3**## | 1450 ± 513**## |
| Spironolactone group | ||||
| Baseline | 2 | 9 | 12.9 | 52.1 ± 15.5 |
| 6-week pacing | 7#* | 41**††## | 58.6**††## | 588 ± 277**††## |
‘AF cases’ refers to the number of dogs successfully induced to develop AF during electrophysiological study in each group, after each dog was induced 10 times; ‘AF times’ refers to the total number of times AF was induced during electrophysiological study in all dogs within a group (with the understanding that AF may have been repeatedly induced in the same dog within the scope of its 10 inductions). AF inducibility is calculated as (AF times)/(induction times, i.e. 70).
P < 0.05,
P < 0.01, compared with baseline;
P < 0.05,
P < 0.01, compared with the sham-operated group;
P < 0.01, compared with the control group.
Morphological changes
Representative histological sections of atrial myocardium from each group are shown in Figures 1 and 2. In the sham-operated group, myofibrils were uniformly and compactly distributed throughout the cell, and levels of myolysis were low (Figure 1). After 6 weeks of rapid atrial pacing, considerable numbers of myocytes in both LA and RA were clearly suffering myolysis (Figure 1). In the paced group given spironolactone, the number of both mildly and severely affected myocytes decreased dramatically, although still higher than those in the sham-operated group. The myolysis was seen at all different atrial sites in the control group, but was found in fewer areas in the spironolactone group. Spironolactone thus decreased the area and degree of myolysis in atrial myocardium, induced by pacing.
Figure 1.
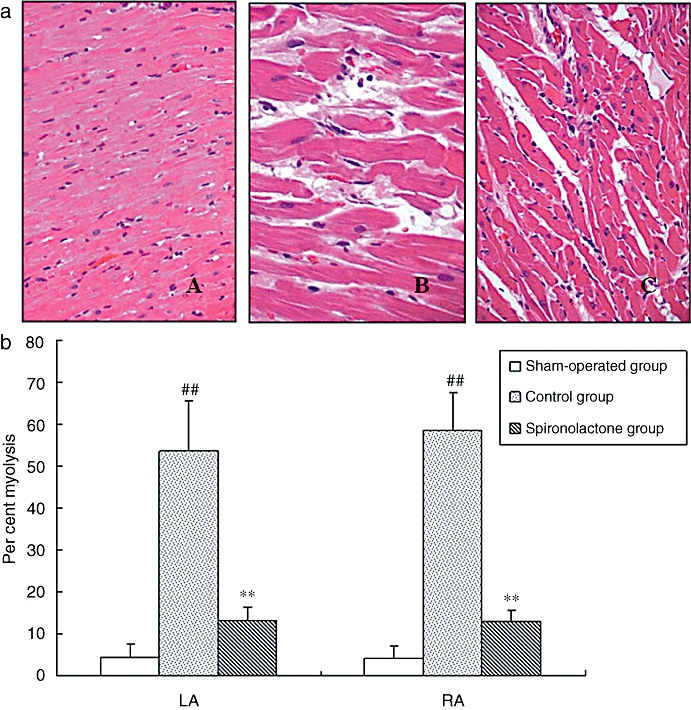
(a) Representative histological staining of atrial myocardium. A: structurally normal atrial myocardium in sham-operated group; B: in control group, severe myolysis was found; C: in spironolactone group, these myolysis changes were mostly inhibited (HE, A, C ×100; B ×400). (b) The percentage of myolysis in atrial myocardium. After 6-week rapid atrial pacing, considerable numbers of atrial myocytes were typically affected by myolysis. With the treatment of spironolactone, the number of both mildly and severely affected myocytes decreased dramatically, although still higher than that in sham-operated group. ##P < 0.01, compared with the sham-operated group; **P < 0.01, compared with the control group. HE, haematoxylin-eosin.
Figure 2.
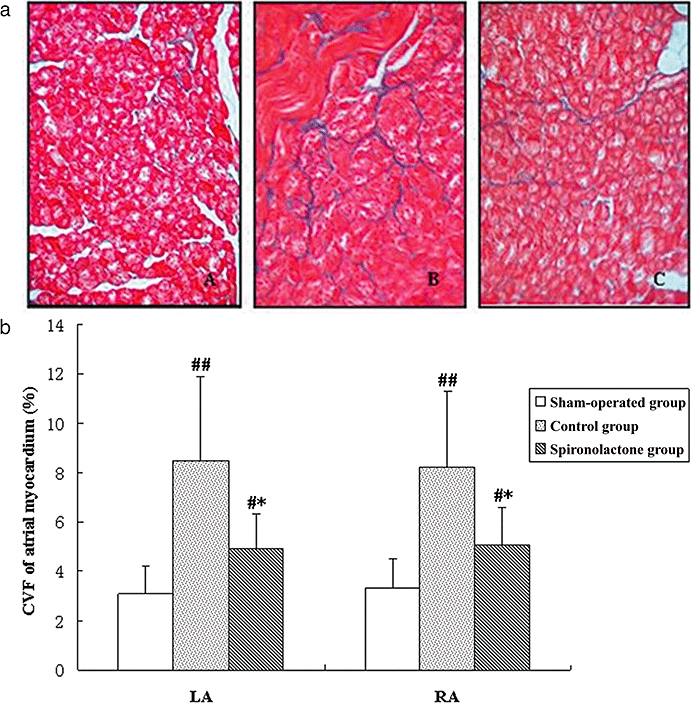
(a) Representative Masson's trichrome staining of atrial myocardium. Red areas represent myocytes and blue areas represent collagen. A: the sham-operated group; B: the control group; C: the spironolactone group. In sham-operated group (A) almost no interstitial fibrosis was present. Extensive fibrosis was especially visible around myocytes of paced atria (B), but attenuated in the spironolactone group (C) (Masson trichrome staining, original magnification ×400). (b) Collagen volume fractions (CVF) of atrial myocardium. After rapid atrial pacing, CVF in control group increased significantly compared with sham-operated group. Spironolactone treatment decreased CVF of LA and RA compared with the control group. #P < 0.05, ##P < 0.01, compared with the sham-operated group; *P < 0.05, compared with the control group. LA, left atrium; RA, right atrium.
In Figure 2, staining for collagen showed a normal intercellular space in the samples from sham-operated dogs, with CVF values of about 3% and this was significantly increased in both LA and RA of the control (pacing only) group (Figure 2b). Spironolactone treatment of the paced dogs clearly decreased CVF, compared with the control group. There were no significant differences between LA and RA in CVF among these groups (P > 0.05).
Ultrastructure
Atrial myocardial ultrastructure was examined by electron microscopy (Figure 3). Atrial myocytes from the sham-operated dogs showed a highly organized sarcomeric structure throughout the cytoplasm, with rows of uniformly sized mitochondria between them and nuclei with clustered heterochromatin. Several ultrastructural abnormalities were observed after 6-week atrial pacing, which were characterized by severe disintegration of myofilaments, partially destroyed and indistinct sarcoplasmic reticulum and mitochondrial swelling with a decrease in the density and organization of the cristae. Pyknotic nuclei with redistribution of heterochromatin were also observed, indicating cell apoptosis. In contrast, the chronic tachypacing-induced ultrastructural changes were dramatically suppressed by spironolactone.
Figure 3.
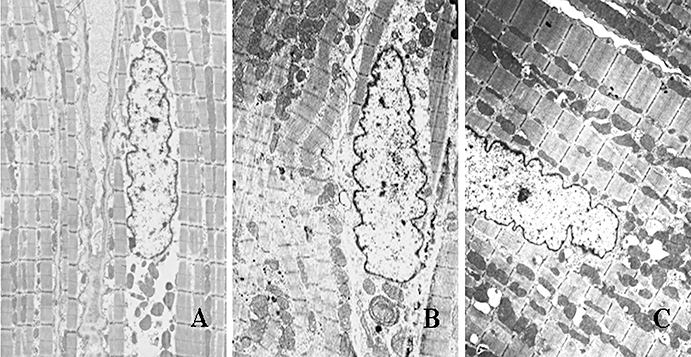
Typical ultrastructural changes by transmission electron microscopy. A: the sham-operated group; B: the control group; C: the spironolactone group. Atrial myocytes from sham-operated dogs (A) had regular sarcomere organization, uniformly sized mitochondria and a normal nucleus. Samples of atrial tissue taken from control group (B) showed abnormal ultrastructure: severe disintegration of myofilaments and loss of banding pattern and integrity of contractile elements, mitochondrial swelling with a decrease in the density and organization of the cristae, karyopyknosis with chromatin margination to nuclear membrane indicating cell apoptosis. These chronic pacing-induced ultrastructural changes were markedly attenuated by treatment with spironolactone (C) (magnification: A: ×6000; B and C: ×5000).
TUNEL assay
The representative TUNEL staining and the proportion of cells undergoing apoptosis are shown in Figure 4. Compared with sham-operated dogs, the percentage of TUNEL-positive cells was markedly increased in LA and RA of the control group, after 6 weeks of atrial pacing, but was reduced significantly by spironolactone, compared with the control group.
Figure 4.
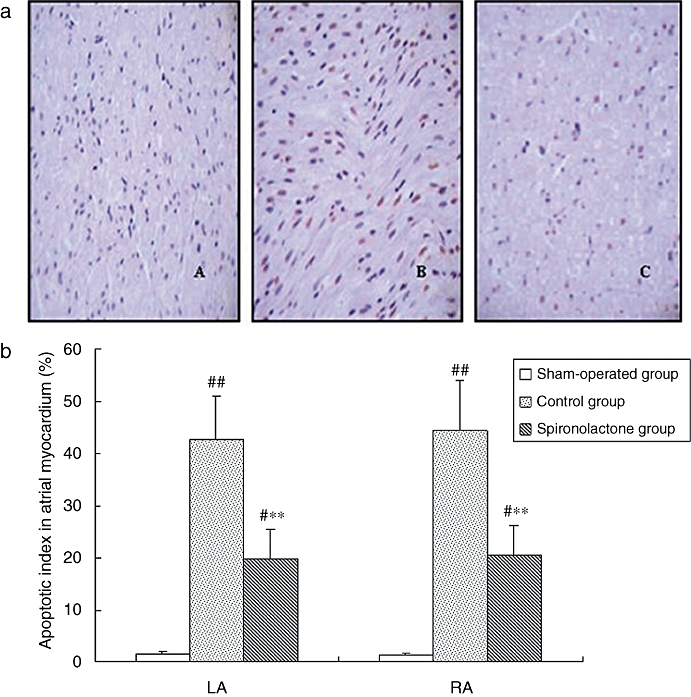
(a) Apoptosis detection with TUNEL staining in atrial myocardium. TUNEL-positive nuclei were stained red, and nuclei of normal cells were stained blue. A: the sham-operated group; B: the control group; C: the spironolactone group. (b) The percentage of TUNEL-positive cells in atrial myocardium. TUNEL-positive cells were scarce in sections from the sham-operated group. After 6 weeks of atrial pacing, the percentage of TUNEL-positive cells was markedly increased. However, this was reduced significantly by spironolactone compared with control group. #P < 0.05, ##P < 0.01, compared with the sham-operated group; **P < 0.01, compared with the control group (magnification ×400).
Levels of caspase-3, Bcl-2 and bax
As demonstrated in Figure 5, quantitative Western blotting analysis showed that the levels of caspase-3 and bax were significantly increased, whereas the level of Bcl-2 decreased in the control group in comparison with the sham-operated group. Spironolactone treatment markedly suppressed the up-regulation of caspase-3 and bax, and the down-regulation of Bcl-2 expression, compared with the control group.
Figure 5.
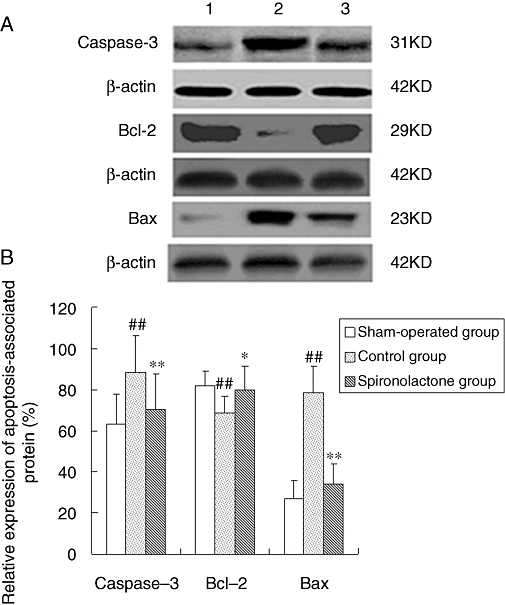
(A) Representative Western blots of apoptosis-associated proteins. (B) Quantitative analysis of expression for apoptosis-associated proteins. The Western blotting analysis showed that the levels of caspase-3 and bax were significantly increased, while the level of Bcl-2 was significantly decreased in the control group, compared with the sham-operated dogs. Spironolactone treatment significantly reduced the up-regulation of caspase-3 and bax, and the down-regulation of Bcl-2 compared with control group. ##P < 0.01, compared with the sham-operated group; *P < 0.05, **P < 0.01, compared with the control group.
Levels of calpain I and calpastatin
As shown in Figure 6, Western blotting showed that the level of calpain I was increased, while the level of calpastatin decreased after 6 weeks of atrial pacing. These changes in the levels of calpain I and calpastatin were significantly attenuated by spironolactone. Furthermore, calpain protein expression in all groups was significantly correlated with the degree of myolysis in all groups (r= 0.91, P < 0.05).
Figure 6.
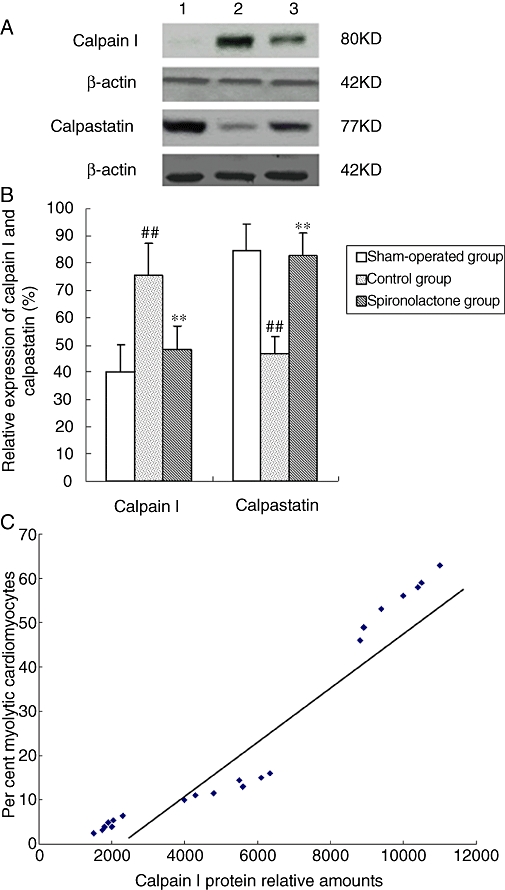
(A) Expression of calpain I and calpastatin analysed by Western blot. (B) Bars represent relative densitometric calpain I and calpastatin protein ratios. Arial myocardium subjected to chronic pacing for 6 weeks revealed a significant increase in calpain I and a decrease in calpastatin expression, compared with sham-operated group. The increase of calpain I and the decrease of calpastatin were significantly attenuated by spironolactone. ##P < 0.01, compared with the sham-operated group; **P < 0.01, compared with the control group. (C) The relationship between the expression of calpain I and percentages of myolytic myocytes. The myolytic cardiomyocytes were the total cardiomyocytes with mild and severe myolysis. Tissue calpain protein amounts correlated with the degree of myolysis in the sham-operated group, control group and spironolactone group (r= 0.91, P < 0.05).
Levels of matrix metalloproteinase (MMP)-9 and tissue inhibitors of metalloproteinase (TIMP)-1
As illustrated in Figure 7, Western blotting analysis showed that MMP-9 protein expression was increased significantly, while the level of TIMP-1 was significantly decreased in the control group compared with the sham-operated group (P < 0.01 for both). Spironolactone inhibited the up-regulation of MMP-9 and the down-regulation of TIMP-1 that was induced by 6 weeks of atrial pacing (P < 0.01 for MMP-9, P < 0.05 for TIMP-1).
Figure 7.
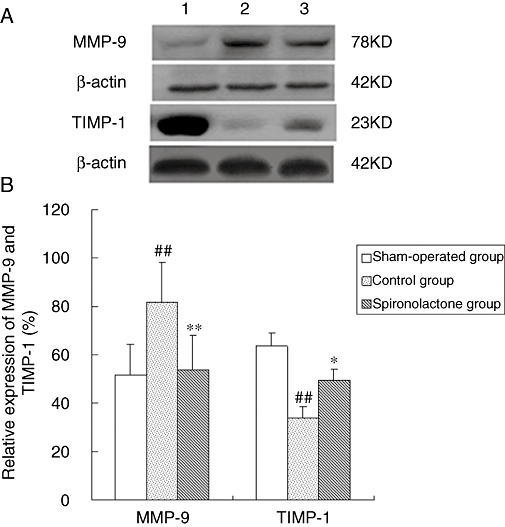
(A) MMP-9 and TIMP-1 protein expression detected by Western blot analysis. (B) Bar graph shows quantitative comparison of LA MMP-9 and TIMP-1 proteins. The expression of MMP-9 was significantly increased, while the level of TIMP-1 was significantly decreased in the control group compared with the sham-operated group. The changes of MMP-9 and TIMP-1 were significantly attenuated by spironolactone compared with control group. ##P < 0.01, compared with the sham-operated group; *P < 0.05, **P < 0.01, compared with the control group. LA, left atrium; MMP, matrix metalloproteinase; TIMP, tissue inhibitors of metalloproteinase.
Discussion
The present data demonstrated that rapid atrial pacing resulted in increased apoptosis and myolysis in atrial myocytes, together with increased fibrosis and atrial dilatation. When spironolactone was used, the improvement of atrial function was associated with a significant attenuation of atrial structural alterations. Thus, our results indicated these cellular and structural changes may be the mechanism underlying the atrial remodelling process. Furthermore, spironolactone exerted beneficial effects on the changes observed during atrial structural remodelling and thereby improved atrial dysfunction and prevented induction and promotion of AF.
Effects of spironolactone on atrial apoptosis
The accumulated data suggest that apoptosis may have a profound impact on the development and progression of AF. In fibrillating and dilated atria, apoptosis and myolysis contribute to cellular remodelling and myocardial dysfunction which may not be entirely reversible (Aimé-Sempéet al., 1999). In the present study, an obvious increase in apoptosis and altered levels of proteins involved in the regulation (Bcl-2 and bax) and execution (caspase-3) of apoptosis in the atria was also described. Long-term treatment with spironolactone significantly attenuated apoptosis and regulated the expressions of apoptosis-associated proteins. Apoptosis is regulated by the Bcl-2 family proteins: Bcl-2 can block cell death, while bax is a promoter of apoptosis. Our results suggested a change of the Bcl-2/bax ratio may promote cell apoptosis, following an apoptotic stimulus. The increase of calpain I and the decrease of calpastatin expression were also significantly attenuated by spironolactone. Calpain is known to activate the downstream protease caspase-3 whose activity has been reported to increase in chronic AF patients. Caspase-3 is one of the major caspases that is involved in the final executional step of apoptosis and are responsible for the cleavage of numerous proteins. In addition, caspase-3 causes a decrease of Bcl-2 proteins, while Bcl-2 inhibits caspsae-3. Thus, the up-regulation of calpain I may promote the execution of apoptotic cascades during AF. The inhibition of calpain and caspase activation by spironolactone is a potential approach for the prevention of apoptosis.
Aldosterone has been recently shown to detrimentally drive and accelerate apoptosis in cardiomyocytes (Mano et al., 2004). As many of the downstream signaling intermediaries of mineralocorticoid receptor (MR) activation are recognized apoptotic stimuli, we may thus hypothesize that aldosterone activity is another potential apoptosis-modulating mechanism in AF remodelling. Prior intervention with spironolactone significantly prevented aldosterone-induced apoptosis in both the heart and skeletal muscles (Burniston et al., 2005). Therefore, some of the positive outcomes observed in the spironolactone group may be due to its ability to protect the atrium from aldosterone-induced apoptosis.
Effects of spironolactone on myolysis
Atrial fibrillation usually results in myolysis (Ausma et al., 1997), which should be responsible for part of atrial contractile dysfunction (Schotten et al., 2001) and AF progression (Ausma et al., 2003). The degree of myolysis has been confirmed as a significant predictor for the development of postoperative AF (Ad et al., 2001). Atrial pacing induced ultrastructural changes, typical of myolysis, and were significantly reversed by spironolactone. Furthermore, the protein expression of calpain I increased and was positively correlated with the extent of myolysis, which suggested that myolysis was mediated by activation of calpain. Calpain activation is indeed a key molecular switch in the AF-related myolysis processes. Previously, a significant increase in calpain activity correlated well with the degree of structural changes in patients with AF (Brundel et al., 2002). It has been further confirmed that inhibiting calpain activity may prevent the myolysis processes and improve atrial function in dogs after atrial pacing (Brundel et al., 2004; Xue et al., 2008). Calpains are intracellular, strongly Ca2+-dependent, neutral cysteine proteases (Goll et al., 2003) and are readily activated by elevated cellular calcium levels, while calcium overload plays a key role in the pathogenesis of AF (Nattel, 2002). Calpain activation can degrade cytoskeletal, contractile and ion channel proteins that are involved in atrial remodelling. Furthermore, we found calpain I was predominantly localized in the nucleus, intercalated discs and cytoplasm, which may be vital for the action potential conduction, excitation-contraction coupling and structural integrity of the atrial myocytes. In summary, the persistent Ca2+ overload-induced activation of calpain might result in myolysis. Angiotensin II also could aggravate cytosolic Ca2+ overload, and angiotensin II increases the intracellular Ca2+ concentration significantly more in atrial myocytes than in ventricular myocytes. The rise of intracellular Ca2+ in atrial myocytes has been considered to be a pivotal event in the activation of calpains. Spironolactone prevents the effects of aldosterone on the rise of T-type and L-type calcium channels (Bénitah and Vassort, 1999; Martin-Fernandez et al., 2009) and modifies the activity of Na-K-ATPase (Ikeda et al., 1991), therefore inhibiting intracellular calcium overload. Our present data revealed that spironolactone attenuated atrial myolysis, partially depending on preventing intracellular calcium overload caused by RAS activation and then inhibiting activation of calpain.
Effects of spironolactone on atrial fibrosis and dilatation
In our present study, dogs with atrial pacing developed significant atrial fibrosis and dilatation with a marked increase in atrial conduction heterogeneity and AF vulnerability. Spironolactone treatment prevented arrhythmogenic artial dilatation and reversed atrial fibrosis, with reduced AF vulnerability.
It is generally accepted that AF and atrial dilatation may be mutually dependent and constitute a vicious circle. LA dilatation has been identified as an independent risk factor for the development of AF. AF results in progressive atrial dilatation, which in turn, might contribute to the self-perpetuating nature of the arrhythmia. Atrial dilatation is due to an increase in atrial compliance caused by atrial contractile dysfunction during AF (Schotten et al., 2003). An increase in atrial size by holding more numbers of wavelets, will facilitate the development of wavelet reentry. Furthermore, an elevated intra-atrial pressure will increase atrial wall stress, which may result in heterogeneities in conduction (Eijsbouts et al., 2003). In addition, atrial dilatation may promote focal arrhythmias that trigger self-perpetuating AF or maintain irregular atrial activation by mechano-electric feedback (Schotten et al., 2003;). The increased inhomogenity in atrial electrophysiological properties during atrial dilatation contributes to the inducibility of AF (Huang et al., 2003). According to these data, interventions targeting a reduction of LA size may prevent AF. Our study also revealed the use of spironolactone was indenpendently associated with reduction in atrial size.
Increased atrial diameters and volumes significantly correlate with the degree of atrial fibrosis. Atrial fibrosis is a hallmark of arrhythmogenic structural remodelling. Most importantly, both atrial dilatation and fibrosis are only incompletely reversible after restoration of sinus rhythm (Allessie et al., 2002; Ausma et al., 2003; Schotten et al., 2003). Increased fibrosis has been observed in the atrial myocardium during AF in both animal models and human biopsies (Frustaci et al., 1997; Lin et al., 2007), which is consistent with the findings of our study. Atrial fibrosis impairs cell-to-cell coupling, causing inhomogeneities in intra-atrial and interstitial conduction, thus increasing atrial susceptibility to AF and providing a discontinuous and branching morphological substrate susceptible for multiple wavelet reentry. Thus, advanced atrial fibrosis would predict an impairment of atrial conduction and create a vulnerable substrate to promote AF (Spach, 2004; Everett and Olgin, 2007).
Our results suggested spironolactone could prevent the occurrence and maintenance of AF through its antifibrotic properties. Previous studies have shown that spironolactone has a marked effect on the attenuation of ventricular fibrosis in patients and rats with myocardial infarction. Similarly, atrial fibrosis can be reduced by spironolactone treatment in CHF (Milliez et al., 2005a; Yang et al., 2008). Aldosterone is a potent stimulus for cardiac fibrosis and may play a crucial role in cardiac collagen deposition (Milliez et al., 2005a; Lin and Pan, 2008). Circulating or locally produced aldosterone stimulates cardiac collagen synthesis and fibroblast proliferation directly via mineralocorticoid receptors or, indirectly, modifying the number and/or function of angiotensin II receptors and enhancing local ACE expression. Several studies suggest the use of ACEIs or AT1 receptor antagonists may delay progression of atrial fibrosis with improvement in AF susceptibility (Burstein and Nattel, 2008). Spironolactone can prevent ‘aldosterone escape’ and therefore attenuate fibrosis more effectively than ACEI and ARB via a more complete blockade of aldosterone's fibrogenic effect. In addition, the induction of inflammation and oxidative stress by aldosterone are prevented by spironolactone, which can also attribute to its antifibrotic properties (Aragno et al., 2007; Young et al., 2007).
Turnover of atrial extracellular matrix during AF is regulated by a balance between MMPs and TIMPs (Lin and Pan, 2008). In particular, many reports have demonstrated that MMP-9 plays a critical role in AF-induced myocardial matrix remodelling and could be a target for the prevention or treatment of AF (Nakano et al., 2004; Chen et al., 2008). MMP-9 is mainly responsible for degrading structural or fibrillar collagens, gelatin, elastin as well as fibronectin. We observed an extracellular matrix remodelling manifested by the dominant up-regulation of MMP-9 along with the down-regulation of TIMP-1 in the atrium after 6-week pacing. Thus, the lack of TIMP-1 up-regulation, will bring the MMP-9/TIMP-1 balance further in the direction of increased MMP-9 activity, which would potentially favor extracellular matrix degradation and atrial dilatation. In addition, increased MMP activity has often been observed in profibrotic states, and long-term MMP inhibition has been shown to suppress fibrosis. Moreover, the MMP-9/TIMP-1 ratio was significantly correlated with CVF, suggesting this dysregulation in MMP-9/TIMP-1 expression may also contribute to the development of atrial fibrosis.
Matrix metalloproteinase and TIMP genes contain AP-1 and NF-kB binding sites in their promoter regions, and angiotensin II can influence MMP transcription via AP-1 and NF-kB signaling pathways (Deschamps and Spinale, 2006). Angiotensin-II-mediated intracellular Ca2+ overload may also be involved in altered protein expression of MMP-9 and TIMP-1 (Zhang et al., 2008). Therefore, in our present study, the effects of spironolactone on normalizing the balance between MMP-9 and TIMP-1 expression may be due to its suppression of angiotensin-II. These effects blunted the alteration in MMP/TIMP stoichiometry, which helped to restore atrial extracellular matrix homeostasis, resulting in less atrial fibrosis and dilatation. Besides, enlarged myocytes with myolysis in dilated atria appear to be an important source of MMP-9 (Boixel et al., 2003).
In our experiments, we only used spironolactone as a representative of the aldosterone antagonists, but its activity is relatively non-selective. Therefore, we need to explore effects of other more selective aldosterone antagonists, such as eplerenone, on atrial structural remodelling, The doses of spironolactone in the present study were higher than those used in clinical practice and whether clinically used doses of spironolactone are able to prevent atrial structural alterations and AF promotion in humans is not known. This issue should be investigated in the future.
In conclusion, the effects of spironolactone on atrial structural remodelling were evaluated in a long-term rapid pacing model in dogs. We found spironolactone suppressed atrial structural remodelling by preventing apoptosis, myolysis, fibrosis and atrial dilatation, and by regulating the expression of apoptosis-associated proteins, calpain and the MMP/TIMP system. The attenuation of atrial structural and functional remodelling contributed to the efficacy of spironolactone in preventing AF promotion. Hence, the inhibition by spironolactone of atrial structural remodelling may provide a promising novel approach to AF therapy.
Acknowledgments
This study was supported by the National Basic Research Program of China (No. 2007CB512000), the Key Science and Technology Program of Heilongjiang Province (No. GB07C32401), the Program for New Century Excellent Talents in University from the Department of Education of Heilongjiang Province (No. 1152-NCET-011) and by grants from the National Natural Science Foundation of China (No. 30871063).
Glossary
Abbreviations:
- ACEI
angiotensin-converting enzyme inhibitor
- AF
atrial fibrillation
- AT1
angiotensin type 1 receptor
- CHF
chronic heart failure
- CVF
collagen volume fraction
- DBP
diastolic blood pressure
- LA
left atrium
- LAA
left atrial appendage
- LAAVmax
LAA maximal volume
- LAAVmin
LAA minimal volume
- LAAEF
LAA ejection fraction
- LAEF
LA ejection fraction
- LAVmax
LA maximal volume
- LAVmin
LA minimal volume
- LV
left ventricle
- LVEF
LV ejection fraction
- LVVmax
LV maximal volume
- LVVmin
LV minimal volume
- MMP
matrix metalloproteinase
- MPAP
mean pulmonary artery pressure
- MR
mineralocorticoid receptor
- PCWP
pulmonary capillary wedge pressure
- RA
right atrium
- RAAS
renin–angiotensin–aldosterone system
- RAP
right atrial pressure
- SBP
systolic blood pressure
- TIMP
tissue inhibitors of metalloproteinase
Conflict of interest
No conflict of interest was declared.
References
- Ad N, Snir E, Vidne BA, Golomb E. Histologic atrial myolysis is associated with atrial fibrillation after cardiac operation. Ann Thorac Surg. 2001;72:688–693. doi: 10.1016/s0003-4975(01)02882-x. [DOI] [PubMed] [Google Scholar]
- Aimé-Sempé C, Folliguet T, Rücker-Martin C, Krajewska M, Krajewska S, Heimburger M, et al. Myocardial cell death in fibrillating and dilated human right atria. J Am Coll Cardiol. 1999;34:1577–1586. doi: 10.1016/s0735-1097(99)00382-4. [DOI] [PubMed] [Google Scholar]
- Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc Res. 2002;54:230–246. doi: 10.1016/s0008-6363(02)00258-4. [DOI] [PubMed] [Google Scholar]
- Aragno M, Mastrocola R, Alloatti G, Vercellinatto I, Bardini P, Geuna S, et al. Oxidative stress triggers cardiac fibrosis in the heart of diabetic rats. Endocrinology. 2007;149:380–388. doi: 10.1210/en.2007-0877. [DOI] [PubMed] [Google Scholar]
- Ausma J, van der Velden HM, Lenders MH, van Ankeren EP, Jongsma HJ, Ramaekers FC, et al. Reverse structural and gap-junctional remodeling after prolonged atrial fibrillation in the goat. Circulation. 2003;107:2051–2058. doi: 10.1161/01.CIR.0000062689.04037.3F. [DOI] [PubMed] [Google Scholar]
- Ausma J, Wijffels M, Thoné F, Wouters L, Allessie M, Borgers M. Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation. 1997;96:3157–3163. doi: 10.1161/01.cir.96.9.3157. [DOI] [PubMed] [Google Scholar]
- Bénitah JP, Vassort G. Aldosterone upregulates Ca2+ current in adult rat cardiomyocytes. Circ Res. 1999;85:1139–1145. doi: 10.1161/01.res.85.12.1139. [DOI] [PubMed] [Google Scholar]
- Boixel C, Fontaine V, Rücker-Martin C, Milliez P, Louedec L, Michel JB, et al. Fibrosis of the left atria during progression of heart failure is associated with increased matrix metalloproteinases in the rat. J Am Coll Cardiol. 2003;42:336–344. doi: 10.1016/s0735-1097(03)00578-3. [DOI] [PubMed] [Google Scholar]
- Brundel BJ, Ausma J, van Gelder IC, Van der Want JJ, van Gilst WH, Crijns HJ, et al. Activation of proteolysis by calpains and structural changes in human paroxysmal and persistent atrial fibrillation. Cardiovasc Res. 2002;54:380–389. doi: 10.1016/s0008-6363(02)00289-4. [DOI] [PubMed] [Google Scholar]
- Brundel BJ, Kampinga HH, Henning RH. Calpain inhibition prevents pacing-induced cellular remodeling in a HL-1 myocyte model for atrial fibrillation. Cardiovasc Res. 2004;62:521–528. doi: 10.1016/j.cardiores.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51:802–809. doi: 10.1016/j.jacc.2007.09.064. [DOI] [PubMed] [Google Scholar]
- Burniston JG, Saini A, Tan LB, Goldspink DF. Aldosterone induces myocyte apoptosis in the heart and skeletal muscles of rats in vivo. J Mol Cell Cardiol. 2005;39:395–359. doi: 10.1016/j.yjmcc.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Chen CL, Huang SK, Lin JL, Lai LP, Lai SC, Liu CW, et al. Upregulation of matrix metalloproteinase-9 and tissue inhibitors of metalloproteinases in rapid atrial pacing-induced atrial fibrillation. J Mol Cell Cardiol. 2008;45:742–753. doi: 10.1016/j.yjmcc.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Cicoira M, Zanolla L, Rossi A. Long-term, dose-dependent effects of spironolactone on left ventricular function and exercise tolerance in patients with chronic heart failure. J Am Coll Cardiol. 2002;40:304–310. doi: 10.1016/s0735-1097(02)01965-4. [DOI] [PubMed] [Google Scholar]
- Delcayre C, Swynghedauw B. Molecular mechanisms of myocardial remodeling. The role of aldosterone. J Mol Cell Cardiol. 2002;34:1577–1584. doi: 10.1006/jmcc.2002.2088. [DOI] [PubMed] [Google Scholar]
- Deschamps AM, Spinale FG. Pathways of matrixmetalloproteinase induction in heart failure: bioactive molecules and transcriptional regulation. Cardiovasc Res. 2006;69:666–676. doi: 10.1016/j.cardiores.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Dixen U, Ravn L, Soeby-Rasmussen C, Paulsen AW, Parner J, Frandsen E, et al. Raised plasma aldosterone and natriuretic peptides in atrial fibrillation. Cardiology. 2007;108:35–39. doi: 10.1159/000095671. [DOI] [PubMed] [Google Scholar]
- Ehrlich JR, Hohnloser SH, Nattel S. Role of angiotensin system and effects of its inhibition in atrial fibrillation: clinical and experimental evidence. Eur Heart J. 2006;27:512–518. doi: 10.1093/eurheartj/ehi668. [DOI] [PubMed] [Google Scholar]
- Eijsbouts SC, Majidi M, van Zandvoort M, Allessie MA. Effects of acute atrial dilation on heterogeneity in conduction in the isolated rabbit heart. J Cardiovasc Electrophysiol. 2003;14:269–278. doi: 10.1046/j.1540-8167.2003.02280.x. [DOI] [PubMed] [Google Scholar]
- Everett TH, 4th, Olgin JE. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. 2007;4:S24–S27. doi: 10.1016/j.hrthm.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 1997;96:1180–1184. doi: 10.1161/01.cir.96.4.1180. [DOI] [PubMed] [Google Scholar]
- Goette A, Hoffmanns P, Enayati W, Meltendorf U, Geller JC, Klein HU. Effect of successful electrical cardioversion on serum aldosterone in patients with persistent atrial fibrillation. Am J Cardiol. 2001;88:906–909. doi: 10.1016/s0002-9149(01)01905-1. [DOI] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- Huang JL, Tai CT, Chen JT, Ting CT, Chen YT, Chang MS, et al. Effect of atrial dilatation on electrophysiologic properties and inducibility of atrial fibrillation. Basic Res Cardiol. 2003;98:16–24. doi: 10.1007/s00395-003-0385-z. [DOI] [PubMed] [Google Scholar]
- Ikeda U, Hyman R, Smith TW, Medford RM. Aldosterone mediated regulation of Na, K-ATPase gene expression in adult and neonatal rat cardiocytes. J Biol Chem. 1991;266:12058–12066. [PubMed] [Google Scholar]
- Li Y, Li WM, Gong YT, Li BX, Liu W, Han W, et al. The effects of cilizapril and valsartan on the mRNA and protein expression of atrial calpains and atrial myolysis in atrial fibrillation dogs. Basic Res Cardiol. 2007;102:245–256. doi: 10.1007/s00395-007-0641-8. [DOI] [PubMed] [Google Scholar]
- Lin CS, Lai LP, Lin JL, Sun YL, Hsu CW, Chen CL, et al. Increased expression of extracellular matrix proteins in rapid atrial pacing-induced atrial fibrillation. Heart Rhythm. 2007;4:938–949. doi: 10.1016/j.hrthm.2007.03.034. [DOI] [PubMed] [Google Scholar]
- Lin CS, Pan CH. Regulatory mechanisms of atrial fibrotic remodeling in atrial Fibrillation. Cell Mol Life Sci. 2008;65:1489–1508. doi: 10.1007/s00018-008-7408-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mano A, Tatsumi T, Shiraishi J, Keira N, Nomura T, Takeda M, et al. Aldosterone directly induces myocyte apoptosis through calcineurin dependent pathways. Circulation. 2004;110:317–323. doi: 10.1161/01.CIR.0000135599.33787.CA. [DOI] [PubMed] [Google Scholar]
- Martin-Fernandez B, Miana M, De Las Heras N, Ruiz-Hurtado G, Fernandez-Velasco M, Bas M. Cardiac L-type calcium current is increased in a model of hyperaldosteronism in the rat. Exp Physiol. 2009;94:675–683. doi: 10.1113/expphysiol.2009.047688. [DOI] [PubMed] [Google Scholar]
- Milliez P, Deangelis N, Rucker-Martin C, Leenhardt A, Vicaut E, Robidel E, et al. Spironolactone reduces fibrosis of dilated atria during heart failure in rats with myocardial infarction. Eur Heart J. 2005a;26:2193–2199. doi: 10.1093/eurheartj/ehi478. [DOI] [PubMed] [Google Scholar]
- Milliez P, Girerd X, Plouin PF, Blacher J, Safar ME, Mourad JJ. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol. 2005b;45:1243–1248. doi: 10.1016/j.jacc.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Niida S, Dote K, Takenaka S, Hirao H, Miura F, et al. Matrix metalloproteinase-9 contributes to human atrial remodeling during atrial fibrillation. J Am Coll Cardiol. 2004;43:818–825. doi: 10.1016/j.jacc.2003.08.060. [DOI] [PubMed] [Google Scholar]
- Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415:219–226. doi: 10.1038/415219a. [DOI] [PubMed] [Google Scholar]
- Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al. Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- Schoonderwoerd BA, Van Gelder IC, Van Veldhuisen DJ, Van den Berg MP, Crijns HJ. Electrical and structural remodeling: role in the genesis and maintenance of atrial fibrillation. Prog Cardiovasc Dis. 2005;48:153–168. doi: 10.1016/j.pcad.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Schotten U, Ausma J, Stellbrink C, Sabatschus I, Vogel M, Frechen D, et al. Cellular mechanisms of depressed atrial contractility in patients with chronic atrial fibrillation. Circulation. 2001;103:691–698. doi: 10.1161/01.cir.103.5.691. [DOI] [PubMed] [Google Scholar]
- Schotten U, Neuberger HR, Allessie MA. The role of atrial dilatation in the domestication of atrial fibrillation. Prog Biophys Mol Biol. 2003;82:151–162. doi: 10.1016/s0079-6107(03)00012-9. [DOI] [PubMed] [Google Scholar]
- Spach MS. Mounting evidence that fibrosis generates a major mechanism for atrial fibrillation. Circ Res. 2004;101:743–745. doi: 10.1161/CIRCRESAHA.107.163956. [DOI] [PubMed] [Google Scholar]
- Struthers AD. The clinical implications of aldosterone escape in congestive heart failure. Eur J Heart Fail. 2004;6:539–545. doi: 10.1016/j.ejheart.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Xue HJ, Li WM, Li Y, Gong YT, Yang BF, Jin CL, et al. Calpain I inhibition prevents atrial structural remodeling in a canine model with atrial fibrillation. Chin Med J (Engl) 2008;121:32–37. [PubMed] [Google Scholar]
- Yang SS, Han W, Zhou HY, Dong G, Wang BC, Huo H, et al. Effects of spironolactone on electrical and structural remodeling of atrium in congestive heart failure dogs. Chin Med J (Engl) 2008;121:38–42. [PubMed] [Google Scholar]
- Young MJ, Lam EY, Rickard AJ. Mineralocorticoid receptor activation and cardiac fibrosis. Clin Sci (Lond) 2007;112:467–475. doi: 10.1042/CS20060275. [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhong M, Yang GR, Li JP, Guo C, Wang Z, et al. Matrix metalloproteinase-9/tissue inhibitors of metalloproteinase-1 expression and atrial structural remodeling in a dog model of atrial fibrillation: inhibition with angiotensin-converting enzyme. Cardiovasc Pathol. 2008;17:399–409. doi: 10.1016/j.carpath.2008.02.008. [DOI] [PubMed] [Google Scholar]