

Abstract
Background and purpose:
Diabetes mellitus (DM) causes multiple dysfunctions including circulatory disorders such as cardiomyopathy, angiopathy, atherosclerosis and arterial hypertension. Rho kinase (ROCK) and protein kinase C (PKC) regulate vascular smooth muscle (VSM) Ca2+ sensitivity, thus enhancing VSM contraction, and up-regulation of both enzymes in DM is well known. We postulated that in DM, Ca2+ sensitization occurs in diabetic arteries due to increased ROCK and/or PKC activity.
Experimental approach:
Rats were rendered hyperglycaemic by i.p. injection of streptozotocin. Age-matched control tissues were used for comparison. Contractile responses to phenylephrine (Phe) and different Ca2+ concentrations were recorded, respectively, from intact and chemically permeabilized vascular rings from aorta, tail and mesenteric arteries.
Key results:
Diabetic tail and mesenteric arteries demonstrated markedly enhanced sensitivity to Phe while these changes were not observed in aorta. The ROCK inhibitor HA1077, but not the PKC inhibitor chelerythrine, caused significant reduction in sensitivity to agonist in diabetic vessels. Similar changes were observed for myofilament Ca2+ sensitivity, which was again enhanced in DM in tail and mesenteric arteries, but not in aorta, and could be reduced by both the ROCK and PKC blockers.
Conclusions and implications:
We conclude that in DM enhanced myofilament Ca2+ sensitivity is mainly manifested in muscular-type blood vessels and thus likely to contribute to the development of hypertension. Both PKC and, in particular, ROCK are involved in this phenomenon. This highlights their potential usefulness as drug targets in the pharmacological management of DM-associated vascular dysfunction.
Keywords: arterial smooth muscles, diabetes, vascular complications, protein kinase C, Rho kinase, myofilaments, calcium sensitivity, phenylephrine
Introduction
Diabetes mellitus (DM) leads to multiple dysfunctions including cardiovascular diseases, one of the major causes of morbidity and mortality in patients with DM. Hyperglycaemia is considered to be the primary aetiological factor that initiates circulatory disorders such as cardiomyopathy, angiopathy, atherosclerosis and arterial hypertension (Sowers et al., 1993; Koya and King, 1998; Meier and King, 2000; Sowers, 2004).
The underlying mechanisms of the increased contractile responsiveness of vascular smooth muscle (VSM) in DM are generally considered to be related to either impaired endothelium-dependent vasorelaxation or to an increase in the contractility of VSM cells themselves. Indeed, numerous studies have demonstrated that DM impairs vascular function via the inhibition of the endothelium-dependent vasodilatation, accompanied by selective impairment of the nitric oxide (NO)- or prostaglandin-dependent component of vasodilatation (Okon et al., 2003; Sowers, 2004; Fitzgerald et al., 2005). For example, in the diabetic retina there is down-regulation of constitutive nitric oxide synthase (cNOS), which impairs retinal blood flow, an early sign of diabetes (Chakravarthy et al., 1998). However, other studies have shown that DM also enhances vascular contractility in an endothelium-independent manner (Speirs et al., 2006).
VSM contractile responses are primarily triggered by an increase in intracellular free Ca2+ concentration ([Ca2+]i). Thus, both increased Ca2+ influx and Ca2+ release have been proposed to develop in DM and to account for the enhanced vascular reactivity. However, one controversy here is that voltage- and store-operated Ca2+ entry channels can be, in fact, even inhibited in DM (Wang et al., 2000; Curtis et al., 2003). Alternatively, the Ca2+-sensitization of the contractile proteins has been proposed as a more general mechanism of elevated DM-associated vascular contractility. One striking example supporting this view is the enhanced contractile response to noradrenaline in mesenteric arteries from diabetic rats which is not associated with the enhancement of the corresponding [Ca2+]i responses (Chow et al., 2001).
The increase in [Ca2+]i activates calmodulin-dependent myosin light chain (MLC) kinase (MLCK) which catalyzes the phosphorylation of MLC leading to actin-myosin interaction and VSM contraction. In addition to this primary regulatory pathway, several modulatory pathways exist in smooth muscle that can alter the magnitude of force that is developed for any given level of [Ca2+]i (Somlyo and Somlyo, 2003). Alterations in myofilament Ca2+ sensitivity can be either positive or negative depending on the pathways stimulated. For increases in myofilament Ca2+ sensitivity, two primary hypotheses have been proposed: G-protein dependent activation of Rho kinase (ROCK) (Fu et al., 1998; Janssen et al., 2001) and protein kinase C (PKC) (De Witt et al., 2001; Salamanca and Khalil, 2005). These pathways converge to phosphorylate an inhibitory protein of smooth muscle myosin phosphatase, CPI-17 (Koyama et al., 2000; Somlyo and Somlyo, 2003). In addition, ROCK directly phosphorylates myosin phosphatase targeting subunit 1 (MYPT1) (Kimura et al., 1996; Feng et al., 1999) that also consequently inhibits the phosphatase activity. The inhibition of the MLC phosphatase results in a greater level of MLC phosphorylation for any given level of [Ca2+]i and activity of the MLCK. This phenomenon, which is known as calcium sensitization of smooth muscle, may thereby maintain vascular contraction (Somlyo and Somlyo, 2003). Notably, ROCK has been shown to modulate smooth muscle contraction, even in the absence of an agonist, through both Ca2+-dependent (involving ion channels) and Ca2+-independent mechanisms (e.g. involving calcium store depletion, MLC or iPLA2 activity; Ayman et al., 2003; Shabir et al., 2004; Maeda et al., 2006). The opposing effect, calcium desensitization, is mediated by cGMP-kinase but plays a relatively minor role in the regulation of VSM contractility, although it can be more important in vascular hyporeactivity elicited by haemorrhagic and septic shock (Xu and Liu, 2005).
An increase in ROCK activity, enhanced production of diacylglycerols (DAG) and activation of PKC have all been shown to be associated with various vascular abnormalities, including arterial hypertension in DM (Inoguchi et al., 1992; Soloviev and Bershtein, 1992; Soloviev et al., 1998; 2005; Koya and King, 1998; Meier and King, 2000; Idris et al., 2001; Wettschureck and Offermanns, 2002; Curtis et al., 2003; Guo et al., 2003; Seko et al., 2003; Curtis and Scholfield, 2004; Ward et al., 2004; Salamanca and Khalil, 2005; Loirand et al., 2006; Das Evcimen and King, 2007). Taken together, these data suggest that the activation of ROCK and/or PKC may be causally linked to myofilaments Ca2+ sensitization in the DM-associated hypertensive state. Thus, the aim of this study was twofold: (i) to investigate whether Ca2+ sensitization occurs in arteries isolated from streptozotocin-induced diabetic rats; and (ii) to address the roles of PKC and ROCK in these processes.
Methods
Experimental animals and induction of diabetes
Experiments were performed on isolated vascular rings obtained from thoracic aorta, tail artery and mesenteric artery of male Sprague–Dawley rats weighing 300–400 g (obtained from Biological Research Unit, Queen's University, Belfast). Protocols were approved by local Animal Welfare and ethics committee and carried out in accordance with UK Animals (Scientific Procedures) Act 1986. Two groups of animals were used: diabetic rats injected with streptozotocin (60–65 mg·kg−1, i.p.) at 8 weeks and maintained for a further 12–14 weeks, and age-matched controls. Diabetes development was verified by the presence of hyperglycaemia (plasma glucose higher than 20 mM) 1 week after streptozotocin injection, and on the day of experimentation, all diabetic groups of animals (blood glucose 26.7 ± 1.2 mM, n= 30) were compared with control rats (blood glucose 5.0 ± 0.2 mM, n= 30; two-tailed t-test P < 0.0001). A blood sample was obtained from a tail tip sample and analysed using a glucose meter (Glucotrend, Boeringer, Mannheim, Germany).
Intact tissue isometric force measurement
Vessels were cut into 5–8 mm rings and mounted on fine stainless steel hooks in 4 mL organ baths perfused with Krebs–Hansleit (composition in mM: NaCl, 118.4; KCl, 4.75; KH2PO4 1.18; NaHCO3 25; D-glucose 5; MgSO4 1.18; CaCl2 1.9) 2 mL·min−1 at 37°C, gassed with 95% O2 and 5% CO2 to maintain pH at 7.4. Before the rings were mounted, the endothelium was removed by gently rotating the hooks and rubbing the lumen sides. For rings from diabetic animals, the concentration of glucose was increased to 25 mM. The resting tension was gradually increased to 1 g for the aorta and 0.75 g for tail and mesenteric arteries. After 1 hour equilibration, each vascular ring was exposed twice to KCl (60 mM) to assess its viability and reproducibility of contractions.
Permeabilized (‘skinned’) tissue isometric force measurement
The tail artery (proximal 5 cm), mesenteric artery and thoracic aorta were isolated and cleaned of adipose and connective tissues. Vessels were placed into relaxing solution of the following composition (in mM): Na2ATP 3.3; KCl 130, MgCl2 2.5, Tris 20, imidasole 20, EGTA 4, phosphocreatine 12; creatine phosphokinase 15 u·mL−1; pH 6.7 at 21°C. After equilibration in this solution for 30 min, the tissue was permeabilized (skinned) using a mix of cholesterol-precipitated compounds, saponin (0.05%) and β-escin (60 µM) in relaxing solution for 18 min at room temperature (Soloviev et al., 2005). As receptor agonists were not used, no GTP was added to this solution (e.g. compared with Croxton et al., 1998). Vessel segments were then mounted on stainless steel hooks in 1 mL organ baths perfused by Ca2+-free solution and attached to isometric force transducers (Dynamometer UFI, Pioden Controls Ltd, Canterbury, UK) connected to a data acquisition system (Micro1401 lab interface and Spike 2 software, CED, Cambridge, UK). Different free Ca2+ levels, expressed as pCa (−log [Ca2+]), were obtained by addition of appropriate concentrations of CaCl2 into relaxing solution (calculated using the Maxchelator software (http://maxchelator.stanford.edu). The initial resting tension of permeabilized aortic, tail artery and mesenteric artery preparations was gradually increased to 1, 0.75 and 0.75 g, respectively, and tissue was allowed to equilibrate for a period of 1 h. All contractile responses were normalized to the maximal Ca2+-dependent force attained in each vascular preparation at the highest [Ca2+] of 7.2 µM.
Data analysis and statistical procedures
Data are presented as mean ± SEM with n indicating the number of vascular preparations tested. Contractile responses were expressed as a percentage of the maximum response produced by 1 mM phenylephrine for intact preparations or 7.2 µM Ca2+ for skinned preparations. The sensitivity to the agonist was expressed as pD2 (negative logarithm of the agonist concentration required for half-maximum response). The calcium sensitivity was expressed as pCa50 (negative logarithm of Ca2+ concentration required for half-maximum response). In order to determine these parameters, data points were fitted using the Origin 7.5 software (OriginLab Corporation, Northampton, MA, USA) with the ‘DoseResp’ function in the following form:
| (1) |
where T is normalized tension (expressed as % of maximal contraction) at the agonist or Ca2+ concentration x (expressed as negative logarithm), x0 is the midpoint of the concentration-effect curve (e.g. T= 50%) and p is the slope factor of the curve. Mean values were obtained by averaging pD2 or pCa50 values obtained from individual preparations, although for illustration purposes mean data points at each agonist or calcium concentration are shown instead and fitted by the same function. Mean values for maximal tension (Emax) are given in Table 1. Statistical comparisons were made using Student's unpaired two-tailed t-test for two groups and anova, Kruskal–Wallis' test followed by a Dunn's multiple comparison post hoc test for three or more groups. Differences for P < 0.05 were considered to be statistically significant.
Table 1.
Mean pD2, pCa50 and Emax values obtained from individual preparations of intact or chemically permeabilized tail artery and aorta from control and diabetic rats in the absence or presence of chelerythrine (1 µM) or HA1077 (10 µM)
|
Tail artery | ||||
|---|---|---|---|---|
|
Phenylephrine |
Ca2+ |
|||
| pD2 | Emax, mN | pCa50 | Emax, mN | |
| Control | 5.662 ± 0.069 (n= 22) | 14.32 ± 2.19 (n= 22) | 5.959 ± 0.009 (n= 10)† | 6.02 ± 0.70 (n= 10) |
| Control + Chelerythrine | 5.260 ± 0.145 (n= 9) | 20.44 ± 2.52 (n= 9) | 5.935 ± 0.003 (n= 10) | 4.15 ± 0.58 (n= 10) |
| Control + HA1077 | 5.237 ± 0.102 (n= 12) | 21.14 ± 2.72 (n= 12) | 5.944 ± 0.037 (n= 6) | 3.44 ± 0.46 (n= 6) |
| Diabetes | 6.417 ± 0.163 (n= 11)**† | 14.18 ± 2.18 (n= 11) | 6.070 ± 0.018 (n= 9)**†† | 6.79 ± 1.00 (n= 9) |
| Diabetes + Chelerythrine | 5.974 ± 0.063 (n= 8) | 16.86 ± 2.18 (n= 8) | 5.891 ± 0.030 (n= 10)†† | 5.30 ± 0.49 (n= 10) |
| Diabetes + HA1077 | 4.915 ± 0.204 (n= 8)†† | 5.31 ± 1.53 (n= 8)* | 5.926 ± 0.012 (n= 8)†† | 4.04 ± 0.64 (n= 8)* |
|
Aorta | ||||
|---|---|---|---|---|
|
Phenyleprine |
Ca2+ |
|||
| pD2 | Emax, mN | pCa50 | Emax, mN | |
| Control | 6.959 ± 0.176 (n= 19) | 12.28 ± 0.89 (n= 19) | 6.138 ± 0.013 (n= 8) | 1.52 ± 0.19 (n= 8) |
| Control + Chelerythrine | 5.763 ± 0.226 (n= 6)† | 8.94 ± 1.50 (n= 6) | 6.133 ± 0.011 (n= 6) | 1.20 ± 0.18 (n= 6) |
| Control + HA1077 | 6.460 ± 0.133 (n= 8) | 4.35 ± 0.18 (n= 8)* | ||
| Diabetes | 7.463 ± 0.171 (n= 10) | 11.05 ± 0.73 (n= 19) | 6.137 ± 0.014 (n= 8) | 1.37 ± 0.16 (n= 8) |
| Diabetes + Chelerythrine | 6.889 ± 0.208 (n= 8) | 13.10 ± 0.92 (n= 8) | 6.138 ± 0.012 (n= 9) | 1.39 ± 0.22 (n= 9) |
| Diabetes + HA1077 | 5.499 ± 0.298 (n= 6)†† | 3.85 ± 1.66 (n= 6)* | ||
P < 0.05,
P < 0.01 by unpaired Student's two-tailed t-test for two groups.
P < 0.05,
P < 0.01 by anova for three or more groups.
Chemicals and drugs
All salts for physiological salt solution, relaxing/activating solutions for experiments with the skinned preparations, glucose, EGTA, DTE, phenylephrine, chelerythrine, imidasole, Tris (hydroxymethyl aminomethane), β-escin, saponin, phosphocreatine disodium salt, creatine phosphokinase, Na2ATP and streptozotocin were purchased from Sigma-Aldrich (Poole, UK) or BDH Chemicals (AnalaR grade, Poole, UK). HA1077 (fasudil hydrochloride) was obtained from Tocris Bioscience (Bristol, UK).
The abbreviations used conform to BJP's Guide to Receptors & Channels (Alexander et al., 2008), and to the IUPHAR guidelines, as published in Pharmacological Reviews.
Results
Intact vascular preparations
In control tail artery and aortic vascular rings isolated from non-diabetic rats, phenylephrine (Phe), a selective α1-adrenoceptor agonist, applied at ascending concentrations (0.1 nM–1 mM) evoked concentration-dependent contractions with a maximal response typically attained at 0.1–1 mM Phe (Figure 1A, top trace). A plateau response was obtained after the addition of each test Phe concentration before the addition of a subsequent dose. In tail artery isolated from diabetic rats, but not in aorta, the responsiveness to phenylephrine was enhanced as the diabetic vessels typically responded to lower concentrations of the agonist and also developed maximal contractions at the low micromolar range of Phe concentrations (Figure 1A, bottom trace).
Figure 1.
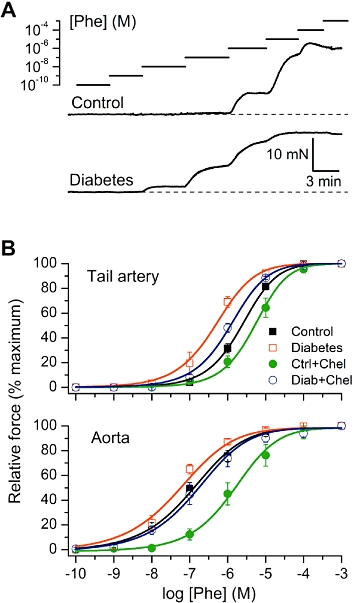
The changes in sensitivity to phenylephrine (Phe) of VSM from control and diabetic rats. (A) Original traces illustrating agonist concentration-response experiments in intact control (top) and diabetic (bottom) tail artery preparations. For illustration purposes, to facilitate direct comparison between control and diabetes, some small trace segments have been deleted (these gaps are not shown for clarity) in order to align the moments of Phe application at the same concentrations. (B) Mean data show normalized amplitude of the contractile responses plotted versus Phe concentration in control, diabetes and after treatment of the tissues with chelerythrine (Chel) (1 µM) in tail artery (top) and aorta (bottom). Data points were fitted by Equation 1 (see Methods section) as shown by the smooth continuous lines.
To quantify these differences, normalized agonist concentration-effect data were plotted and fitted by the dose-response function (Eq. 1) as shown by the smooth lines in Figures 1B and 2. Increased agonist sensitivity of tail artery from diabetic animals was evident from a higher mean pD2 value (Table 1, P < 0.0001). In contrast, aortic rings showed no significant difference in the sensitivity to Phe between non-diabetic control and diabetic rats (Table 1, P= 0.076 and Figure 1B). There were no significant differences between the maximum contractions with the exception of the effect the ROCK inhibitor (see below) in diabetic tail artery (Table 1).
Figure 2.
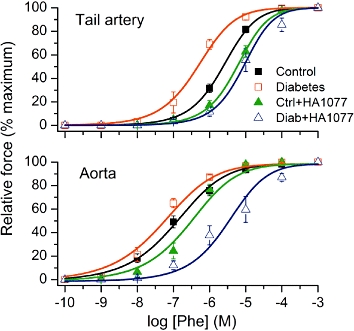
Effects of the Rho kinase inhibitor HA1077 on the sensitivity of tissues from diabetic or control rats to phenylephrine (Phe). Mean data show normalized amplitude of the contractile responses plotted versus Phe concentration in tissues from control and diabetic rats in the absence and presence of HA1077 (10 µM for tail artery and 1 µM for aorta). Data points were fitted by Equation 1 (see Methods section) as shown by the smooth continuous lines.
To investigate the role of PKC in these agonist responses, vascular tissues were pre-treated for 20 min with a potent cell-permeable PKC inhibitor, chelerythrine (1 µM). This resulted in a rightward shift of the concentration-response curves for Phe (Figure 1B and Table 1).
In the next series of experiments, vascular preparations were pre-treated with the relatively selective ROCK inhibitor HA1077 (Davies et al., 2000) for 20 min (Figure 2). Again, this resulted in a significant reduction in sensitivity to agonist (see statistical test below), which was especially pronounced in diabetic vessels (Table 1).
There were significant differences between the groups means in both the tail artery and aorta (anova, P < 0.0001). Specifically, pD2 values in tail artery, but not in aorta, were different between the control and diabetic group (P < 0.01). PKC inhibition generally had no significant effect on the responses to Phe (with the exception of aorta from the control group, P < 0.05), while in both types of vessel ROCK inhibition caused significant desensitization to the action of Phe in the diabetic group (P < 0.001), but not in the control group.
Chemically permeabilized vascular preparations: role of PKC and ROCK in myofilament Ca2+ sensitivity
These experiments were designed to test the hypothesis that in VSM diabetes can induce myofilament Ca2+-sensitization via the activation of PKC and/or ROCK. Chemically permeabilized (‘skinned’) vascular rings are ultimately suitable for this purpose as they allow direct access of exogenously buffered calcium to contractile proteins. Permeabilized vascular rings from control and diabetic animals were subjected to the cumulative ascending calcium concentrations in the range from 0.1 to 7.2 µM. Similar to the Phe experiments, a plateau response was obtained before the addition of a higher calcium concentration. There were no significant differences between the maximum contractions elicited in control and diabetic tail arteries and aortae (Table 1).
Compared with control, diabetic tail artery rings responded to much lower calcium concentrations, as illustrated in Figure 3A. Analysis of the pCa-tension relationships revealed a significant difference in the Ca2+ sensitivity between control and diabetic animals (Table 1; t-test, P < 0.0001) (Figure 3B, top panel). In contrast, there was no significant shift in the pCa-tension relationship associated with diabetes in aorta (Figure 3B, bottom panel; t-test, P= 0.96). These results demonstrate that increased myofilament Ca2+ sensitivity develops in diabetes, specifically in the tail artery but not in aortic smooth muscles. This is consistent with the observed enhanced Phe sensitivity, specifically in the tail artery, of diabetic rats (Figure 1B).
Figure 3.
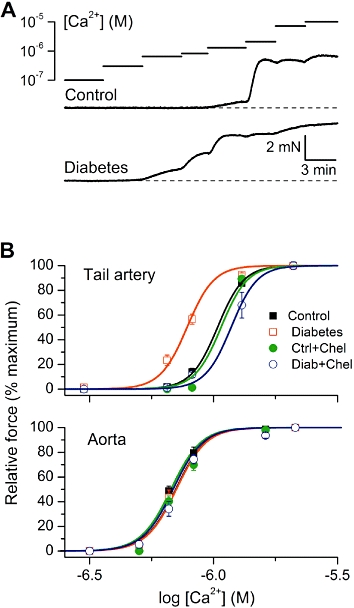
Altered myofilament Ca2+ sensitivity in diabetic rat vascular smooth muscle. (A) Original traces showing typical examples of force development in chemically permeabilized tail artery from control (top) and diabetic animals (bottom) as a function of calcium concentration. The traces were aligned as described for Figure 1A. (B) Mean normalized amplitude of the contractile responses plotted versus [Ca2+] in tissues from control and diabetic rats before and after treatment with chelerythrine (Chel) (1 µM). Data points were fitted by Equation 1 (see Methods section) as shown by the smooth continuous lines.
Moreover, in the tail artery, pre-treatment with chelerythrine (1 µM) caused a rightward shift of the pCa-tension relationship curve both in control rings and in diabetic vessels when compared with untreated vessels (Figure 3B, top panel and Table 1). Whereas PKC inhibition by chelerythrine had no effect on the pCa50 values in either the control or diabetic aortic rings (Table 1).
Because the development of myofilament Ca2+ sensitization in diabetes appeared to be specific to the tail artery, we also investigated this phenomena in another muscular type of blood vessel, the mesenteric artery. Permeabilized mesenteric rings from diabetic animals also had significantly different pCa50 values for Phe compared with those from control animals: 6.168 ± 0.036 (n= 13) in diabetic vessels versus 6.023 ± 0.034 (n= 8) in control segments (t-test, P= 0.0135), thus again demonstrating the phenomenon of enhanced VSM Ca2+ sensitivity in diabetes. Similar to the tail artery, 20 min treatment with chelerythrine 1 µM shifted the pCa-tension relationship curve to the right, both in control and diabetic mesenteric artery segments. Mean pCa50 values were in control arteries 5.916 ± 0.044 (n= 4, t-test, P= 0.092 compared with untreated tissues) and in diabetic arteries 5.929 ± 0.018 (n= 4, t-test P= 0.003; compared with untreated tissues). This, again, demonstrated an increased contribution of PKC activation to the development of higher Ca2+-sensitivity of the contractile machinery in diabetic blood vessels.
As ROCK showed the most pronounced effects on the agonist sensitivity, especially in diabetic vessels (Figure 2), we further investigated its possible role in the increased myofilament Ca2+ sensitivity using its inhibitor HA1077. Pre-treatment (20 min) of tail artery rings with HA1077 10 µM caused a marked shift of the pCa-tension relationship curve, reversing the increased sensitivity to Ca2+, in blood vessels from diabetic, but not control rats (Figure 4 and Table 1). It should be noted that these effects refer to the basal activity of ROCK because no receptor agonists were used in the experiments on skinned preparations.
Figure 4.
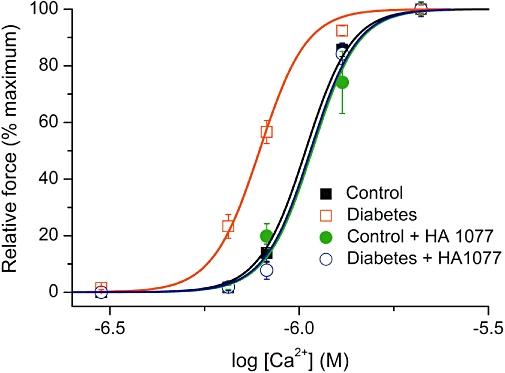
Calcium sensitivity of tail artery from control and diabetic rats before and after tissue pre-treatment with HA1077 (10 µM). Mean normalized force amplitude was plotted against the logarithm of Ca2+ concentration and data point fitted by Equation 1 (see Methods section) as shown by the smooth continuous lines.
Overall, there were significant differences between the group means in tail artery (anova, P < 0.0001), but not in aorta (P= 0.99). Specifically, in tail artery, pCa50 values were different between the control and diabetic tissues (P < 0.01). Neither PKC nor ROCK inhibition caused any significant effect in control preparations, but the inhibition of either enzyme caused significant desensitization to calcium in the diabetic vessels (P < 0.001).
Discussion and conclusions
In diabetes, significant changes in vascular function occur which are associated with increased frequency and severity of vascular disease, such as atherosclerosis and arterial hypertension (Sowers et al., 1993; Idris et al., 2001; Lagaud et al., 2001; Curtis and Scholfield, 2004; Sowers, 2004). The main novel finding in this study is that STZ-induced experimental DM in rats leads to enhanced Ca2+ sensitivity of contractile myofilaments. Moreover, we found that this effect was specific to arteries of the muscular type, such as tail and mesenteric arteries, as it was not observed in aorta. Furthermore, both PKC and ROCK were clearly contributing to the enhanced Ca2+ sensitivity of contractile myofilaments in diabetic vessels.
The data obtained from the skinned preparations indicate that the increased calcium sensitivity observed in tail and mesenteric arteries from diabetic animals is reflected in intact artery responses to Phe (Figure 1A,B). In addition, there was no increase in the agonist sensitivity in aorta from the diabetic group, consistent with no change in myofilament calcium sensitivity in aorta during DM (Figure 3B).
We have observed consistent effects of ROCK inhibition on both agonist sensitivity (Figure 2) and myofilament calcium sensitivity (Figure 4), and both were specific to diabetic vessels. Notably, although the calcium sensitivity in DM was ‘normalized’ by the ROCK blocker, sensitivity to Phe was further reduced compared with control vessels. However, it should be noted that proteins like ROCK and PKC are multifunctional enzymes, as apart from myofilament calcium sensitivity they regulate a number of signalling pathways interposed between receptor stimulation and VSM contractions. Therefore, any change of myofilament calcium sensitivity may be counteracted in non-permeabilized tissues by, for example, defective calcium signalling (e.g. down-regulation of voltage- and store-operated Ca2+ entry channels, or Ca2+ release in DM: Wang et al., 2000; Curtis et al., 2003). Such changes might be considered as compensatory and therefore it is perhaps not surprising that particularly when ROCK is blocked in DM, this not just ‘normalizes’ the agonist sensitivity, but reveals an additional substantial rightward shift of the agonist curve (Figure 2).
As for PKC, it obviously contributes to the enhanced myofilament calcium sensitivity in diabetic tail artery, but not in aorta (Figures 1B and 3B). However, PKC inhibition did not significantly alter agonist sensitivity in intact preparations (except in control aorta), probably due to the reasons discussed previously. In fact, the change in the agonist pD2 values due to PKC inhibition was similar in control and diabetic tail artery (0.402 vs. 0.443 pD2 unit, respectively), while in the aorta the change was even greater (and statistically significant) in control compared with diabetic preparations (1.196 vs. 0.574, respectively). It should be noted here that different studies have described enhanced, unchanged or even attenuated vascular reactivity to α1-adrenoceptor agonists in DM. The reasons for these discrepancies are not clear, but may include differences in the species, vascular beds or even duration of experimental DM (Xavier et al., 2003). In addition, the role of PKC in the contractile responses to Phe in diabetes is also uncertain; for example, Okon et al. (2003) reported no effect of PKC inhibition in a diabetic mouse model. The controversy surrounding the role of PKC probably reflects the above-mentioned, multifunctional nature of this enzyme. It is well known that in VSM, PKC regulates the activity of l-type voltage-dependent Ca2+ channels, Ca2+-activated K+ channels, ATP-sensitive K+ channels and non-selective cation classical transient receptor potential channels (Crozatier, 2006). Thus, PKC inhibition may have complex related effects on agonist sensitivity, in addition to the more direct effect on myofilament calcium sensitivity. Apart from ion channel effects, high glucose and increases in PKC activity in VSM have been shown to regulate not only the phosphorylation of myofibrillar proteins, but also the G protein-coupled receptor density, expression of Gq/11α and PLCβ proteins, and intracellular pH (Idris et al., 2001; Descorbeth and Anand-Srivastava, 2008).
Nevertheless, our present results show that at least in the muscular-type arteries, elevated myofilament Ca2+ sensitivity (pCa-tension relationship) may be one of the major factors affecting vascular contractility in DM. The results are consistent with those of Chow et al. (2001) who showed enhanced contractile responses to noradrenaline in mesenteric arteries from diabetic rats which were dissociated from changes in [Ca2+]i, thus suggesting a similar mechanism. With regard to the molecular mechanism of these changes, our results show that the increased sensitivity to Ca2+ in DM is mediated by pathways dependent on both PKC and ROCK. The effect of ROCK can be explained by its involvement in phosphorylation of MYPT1 (Kimura et al., 1996; Feng et al., 1999) and CPI-17 leading to inhibition of smooth muscle myosin phosphatase, which is well known to regulate VSM calcium sensitivity in various pathophysiological conditions (Amano et al., 2000; Koyama et al., 2000; Janssen et al., 2001; Vanbavel et al., 2001; Wettschureck and Offermanns, 2002; Seko et al., 2003; Loirand et al., 2006) that, according to our present results, also includes DM.
The finding that DM and hyperglycaemia cause elevated DAG production and associated activation of PKC (especially, but not exclusively the PKC-β isoform) is well documented and this key enzyme is one of the main elements in sensitizing myofilaments to Ca2+ (Koya and King, 1998; Meier and King, 2000; Idris et al., 2001; Guo et al., 2003; Curtis and Scholfield, 2004; Mueed et al., 2005; Das Evcimen and King, 2007). We have previously investigated the role of PKC in Ca2+-sensitization during hypoxic pulmonary hypertension, genetically determined hypertension and hypertension evoked by ionizing irradiation. In all cases, up-regulation of PKC-mediated myofilament Ca2+-sensitivity was found to play a key role (Soloviev and Bershtein, 1992; Soloviev et al., 1998; 2005;).
Based on our results, the list of pathological conditions in which changes in calcium sensitivity are involved can be extended to include DM. In the vasculature, PKC and ROCK are the two key regulatory enzymes involved in the signal transduction of multiple cellular functions such as growth, differentiation, metabolism and smooth muscle contractility (Amano et al., 2000; Meier and King, 2000; Wettschureck and Offermanns, 2002; Seko et al., 2003; Stanton et al., 2004; Ward et al., 2004; Soloviev et al., 2005; Loirand et al., 2006). In particular, ROCK- and PKC-dependent sensitization of the contractile apparatus to Ca2+ have been suggested to play an important role in the signal transduction events leading to VSM contraction and, incidentally, in the development of hypertensive conditions (Fu et al., 1998; Soloviev et al., 1998; 2005; Meier and King, 2000; De Witt et al., 2001; Janssen et al., 2001; Ward et al., 2004; Salamanca and Khalil, 2005).
In conclusion, the present study shows that PKC and especially ROCK are involved in DM-induced Ca2+ sensitization in VSM but the roles of various isoforms of these enzymes remain to be established.
Acknowledgments
This work was supported by The Royal Society International Joint Project Grant and School of Medicine, Dentistry and Biomedical Sciences, Queen's University, Belfast. We thank Mervyn Merdock and Stewart Davidson for the construction of vital equipment.
Glossary
Abbreviations:
- [Ca2+]i
intracellular free calcium concentration
- DAG
diacylglycerol
- DM
diabetes mellitus
- HA1077
fasudil hydrochloride
- MLC
myosin light chain
- NO
nitric oxide
- NOS
nitric oxide synthase
- Phe
phenylephrine
- PKC
protein kinase C
- ROCK
Rho kinase
- VSM
vascular smooth muscle
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano M, Fukata Y, Kaibuchi K. Regulation and functions of Rho-associated kinase. Exp Cell Res. 2000;261:44–51. doi: 10.1006/excr.2000.5046. [DOI] [PubMed] [Google Scholar]
- Ayman S, Wallace P, Wayman CP, Gibson A, McFadzean I. Receptor-independent activation of Rho-kinase-mediated calcium sensitisation in smooth muscle. Br J Pharmacol. 2003;139:1532–1538. doi: 10.1038/sj.bjp.0705394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy U, Hayes RG, Stitt AW, McAuley E, Archer DB. Constitutive nitric oxide synthase expression in retinal vascular endothelial cells is suppressed by high glucose and advanced glycation end products. Diabetes. 1998;47:945–952. doi: 10.2337/diabetes.47.6.945. [DOI] [PubMed] [Google Scholar]
- Chow WL, Zhang L, MacLeod KM. Noradrenaline-induced changes in intracellular Ca2+ and tension in mesenteric arteries from diabetic rats. Br J Pharmacol. 2001;134:179–187. doi: 10.1038/sj.bjp.0704221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxton TL, Lande B, Hirshman CA. Role of G proteins in agonist-induced Ca2+ sensitization of tracheal smooth muscle. Am J Physiol. 1998;275:L748–L755. doi: 10.1152/ajplung.1998.275.4.L748. [DOI] [PubMed] [Google Scholar]
- Crozatier B. Central role of PKCs in vascular smooth muscle cell ion channel regulation. J Mol Cell Cardiol. 2006;41:952–955. doi: 10.1016/j.yjmcc.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Curtis TM, Scholfield CN. The role of lipids and protein kinase Cs in the pathogenesis of diabetic retinopathy. Diabetes Metab Res Rev. 2004;20:28–43. doi: 10.1002/dmrr.431. [DOI] [PubMed] [Google Scholar]
- Curtis TM, Major EH, Trimble ER, Scholfield CN. Diabetes-induced activation of protein kinase C inhibits store-operated Ca2+ uptake in rat retinal microvascular smooth muscle. Diabetologia. 2003;46:1252–1259. doi: 10.1007/s00125-003-1178-5. [DOI] [PubMed] [Google Scholar]
- Das Evcimen N, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res. 2007;55:498–510. doi: 10.1016/j.phrs.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Witt BJ, Kaye AD, Ibrahim IN, Bivalacqua TJ, D'Souza FM, Banister RE, et al. Effects of PKC isozyme inhibitors on constrictor responses in the feline pulmonary vascular bed. Am J Physiol Lung Cell Mol Physiol. 2001;280:L50–L57. doi: 10.1152/ajplung.2001.280.1.L50. [DOI] [PubMed] [Google Scholar]
- Descorbeth M, Anand-Srivastava MB. High glucose increases the expression of Gq/11α and PLC-β proteins and associated signaling in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2008;295:H2135–H2142. doi: 10.1152/ajpheart.00704.2008. [DOI] [PubMed] [Google Scholar]
- Feng J, Ito M, Ichikawa K, Isaka N, Nishikawa M, Hartshorne DJ, et al. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem. 1999;274:37385–37390. doi: 10.1074/jbc.274.52.37385. [DOI] [PubMed] [Google Scholar]
- Fitzgerald SM, Kemp-Harper BK, Tare M, Parkington HC. Role of endothelium-derived hyperpolarizing factor in endothelial dysfunction during diabetes. Clin Exp Pharmacol Physiol. 2005;32:482–487. doi: 10.1111/j.1440-1681.2005.04216.x. [DOI] [PubMed] [Google Scholar]
- Fu X, Gong MC, Jia T, Somlyo AV, Somlyo AP. The effects of the Rho-kinase inhibitor Y-27632 on arachidonic acid-, GTPγS-, and phorbol ester-induced Ca2+-sensitization of smooth muscle. FEBS Lett. 1998;440:183–187. doi: 10.1016/s0014-5793(98)01455-0. [DOI] [PubMed] [Google Scholar]
- Guo M, Wu MH, Korompai F, Yuan SY. Upregulation of PKC genes and isozymes in cardiovascular tissues during early stages of experimental diabetes. Physiol Genomics. 2003;12:139–146. doi: 10.1152/physiolgenomics.00125.2002. [DOI] [PubMed] [Google Scholar]
- Idris I, Gray S, Donnelly R. Protein kinase C activation: isozyme-specific effects on metabolism and cardiovascular complications in diabetes. Diabetologia. 2001;44:659–673. doi: 10.1007/s001250051675. [DOI] [PubMed] [Google Scholar]
- Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci USA. 1992;89:11059–11063. doi: 10.1073/pnas.89.22.11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen LJ, Lu-Chao H, Netherton S. Excitation-contraction coupling in pulmonary vascular smooth muscle involves tyrosine kinase and Rho kinase. Am J Physiol Lung Cell Mol Physiol. 2001;280:L666–L674. doi: 10.1152/ajplung.2001.280.4.L666. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- Koyama M, Ito M, Feng J, Seko T, Shiraki K, Takase K, et al. Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett. 2000;475:197–200. doi: 10.1016/s0014-5793(00)01654-9. [DOI] [PubMed] [Google Scholar]
- Lagaud GJL, Masih-Khan E, Kai S, van Breemen C, Dubq GP. Influence of type II diabetes on arterial tone and endothelial function in murine mesenteric resistance arteries. J Vasc Res. 2001;38:578–589. doi: 10.1159/000051094. [DOI] [PubMed] [Google Scholar]
- Loirand G, Guerin P, Pacaud P. Rho Kinases in cardiovascular physiology and pathophysiology. Circ Res. 2006;98:322–334. doi: 10.1161/01.RES.0000201960.04223.3c. [DOI] [PubMed] [Google Scholar]
- Maeda A, Ozaki Y, Sivakumaran S, Akiyama T, Urakubo H, Usami A, et al. Ca2+ -independent phospholipase A2-dependent sustained Rho-kinase activation exhibits all-or-none response. Genes Cells. 2006;11:1071–1083. doi: 10.1111/j.1365-2443.2006.01001.x. [DOI] [PubMed] [Google Scholar]
- Meier M, King GL. Protein kinase C activation and its pharmacological inhibition in vascular disease. Vasc Med. 2000;5:173–185. doi: 10.1177/1358836X0000500307. [DOI] [PubMed] [Google Scholar]
- Mueed I, Zhang L, MacLeod KM. Role of the PKC//CPI-17 pathway in enhanced contractile responses of mesenteric arteries from diabetic rats to α-adrenoceptor stimulation. Br J Pharmacol. 2005;146:972–982. doi: 10.1038/sj.bjp.0706398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okon EB, Szado T, Laher I, McManus B, van Breemen C. Augmented contractile response of vascular smooth muscle in a diabetic mouse model. J Vasc Res. 2003;40:520–530. doi: 10.1159/000075238. [DOI] [PubMed] [Google Scholar]
- Salamanca DA, Khalil RA. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem Pharmacol. 2005;70:1537–1547. doi: 10.1016/j.bcp.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, et al. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res. 2003;92:411–418. doi: 10.1161/01.RES.0000059987.90200.44. [DOI] [PubMed] [Google Scholar]
- Shabir S, Borisova L, Wray S, Burdyga T. Rho-kinase inhibition and electromechanical coupling in rat and guinea-pig ureter smooth muscle: Ca2+-dependent and -independent mechanisms. J Physiol. 2004;560:839–855. doi: 10.1113/jphysiol.2004.070615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soloviev AI, Bershtein SA. The contractile apparatus in vascular smooth muscle cells of spontaneously hypertensive rats possess increased calcium sensitivity: the possible role of protein kinase C. J Hypertens. 1992;10:131–136. doi: 10.1097/00004872-199202000-00004. [DOI] [PubMed] [Google Scholar]
- Soloviev AI, Parshikov AV, Stefanov AV. Evidence for the involvement of protein kinase C in depression of endothelium-dependent vascular responses in spontaneously hypertensive rats. J Vasc Res. 1998;35:325–331. doi: 10.1159/000025602. [DOI] [PubMed] [Google Scholar]
- Soloviev AI, Tishkin SM, Zelensky SN, Ivanova IV, Kizub IV, Pavlova AA, et al. Ionizing radiation alters myofilament calcium sensitivity in vascular smooth muscle: potential role of protein kinase C. Am J Physiol Regul Integr Comp Physiol. 2005;289:R755–R762. doi: 10.1152/ajpregu.00748.2004. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Sowers JR. Insulin resistance and hypertension. Am J Physiol Heart Circ Physiol. 2004;286:H1597–H1602. doi: 10.1152/ajpheart.00026.2004. [DOI] [PubMed] [Google Scholar]
- Sowers JR, Standley PR, Ram JL, Jacober S, Simpson L, Rose K. Hyperinsulinemia, insulin resistance, and hyperglycemia: contributing factors in the pathogenesis of hypertension and atherosclerosis. Am J Hypertens. 1993;6:260S–270S. doi: 10.1093/ajh/6.7.260s. [DOI] [PubMed] [Google Scholar]
- Speirs L, Donnelly A, Lynch J, Scholfield CN, Johnson C. ATP and norepinephrine contributions to sympathetic vasoconstriction of tail artery are altered in streptozotocin-diabetic rats. Am J Physiol. 2006;291:H2327–H2333. doi: 10.1152/ajpheart.01298.2005. [DOI] [PubMed] [Google Scholar]
- Stanton MC, Delaney D, Zderic SA, Moreland RS. Partial bladder outlet obstruction abolishes the receptor- and G protein-dependent increase in calcium sensitivity in rabbit bladder smooth muscle. Am J Physiol Renal Physiol. 2004;287:F682–F689. doi: 10.1152/ajprenal.00117.2004. [DOI] [PubMed] [Google Scholar]
- VanBavel E, van der Meulen ET, Spaan JA. Role of Rho-associated protein kinase in tone and calcium sensitivity of cannulated rat mesenteric small arteries. Exp Physiol. 2001;86:585–592. doi: 10.1113/eph8602217. [DOI] [PubMed] [Google Scholar]
- Wang R, Wu Y, Tang G, Wu L, Hanna ST. Altered l-type Ca2+ channel currents in vascular smooth muscle cells from experimental diabetic rats. Am J Physiol Heart Circ Physiol. 2000;278:H714–H722. doi: 10.1152/ajpheart.2000.278.3.H714. [DOI] [PubMed] [Google Scholar]
- Ward JPT, Knock GA, Snetkov VA, Aaronson PI. Protein kinases in vascular smooth muscle tone – role in the pulmonary vasculature and hypoxic pulmonary vasoconstriction. Pharmacol Ther. 2004;104:207–231. doi: 10.1016/j.pharmthera.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Wettschureck N, Offermanns S. Rho/Rho-kinase mediated signaling in physiology and pathophysiology. J Mol Med. 2002;80:629–638. doi: 10.1007/s00109-002-0370-2. [DOI] [PubMed] [Google Scholar]
- Xavier FE, Davel AP, Rossoni LV, Vassallo DV. Time-dependent hyperreactivity to phenylephrine in aorta from untreated diabetic rats: role of prostanoids and calcium mobilization. Vascul Pharmacol. 2003;40:67–76. doi: 10.1016/s1537-1891(02)00315-4. [DOI] [PubMed] [Google Scholar]
- Xu J, Liu L. The role of calcium desensitization in vascular hyporeactivity and its regulation after hemorrhagic shock in the rat. Shock. 2005;23:576–581. [PubMed] [Google Scholar]