

Abstract
Degradable microparticles have the potential to protect and release drugs over extended periods and, if sized appropriately, can be passively targeted to phagocytic cells in vivo. Dendritic cells (DC) are a class of phagocytic cells known to play important roles in transplant rejection. Previously, we have demonstrated that DC treated with an immunosuppressive drug, rapamycin, have the ability to suppress transplant rejection. Herein, we describe a strategy to deliver an intracellular depot of rapamycin to DC. To achieve this, rapamycin was encapsulated into ∼3.4 μm sized poly(lactic-co-glycolic)acid (PLGA) microparticles (rapaMPs), and release behavior was examined under intra-phagosomal (pH = 5) and extracellular (pH = 7.4) conditions. It was observed that 4 days following phagocytosis of rapaMP, DC have significantly reduced ability to activate T cells, in comparison to DC treated with soluble rapamycin. Hence, we conclude that DC-specific intracellular delivery of rapamycin results in better efficacy of the drug, with respect to its ability to modulate DC function, when compared to treating DC with extracellular rapamycin.
Introduction
Currently, the greatest concerns with transplantation of tissues and organs are immune-mediated rejection and the adverse effects of systemically delivered immunosuppressive drugs[1, 2]. Rejection occurs when the body recognizes the transplant as a foreign entity and initiates immune responses against it[2]. The primary mediators of these responses are lymphocytes, a heterogeneous but well-characterized population of immune cells, which are activated by cells with antigen-presenting function. Among these antigen-presenting cells (APC), dendritic cells (DC)[3] are probably the best characterized and also the most potent. Suppression of immune responses against a transplant can be achieved by either inhibiting the proliferation of activated lymphocytes or by limiting their activation through blocking DC-mediated lymphocyte stimulation[4]. For the past 50 years, immunosuppressive drugs such as azathioprine, corticosteroids, cyclosporine, and tacrolimus have been used to target lymphocyte activation and proliferation in order to prevent rejection. To achieve this, these drugs must be administered systemically so that they come in contact with all lymphocytes.
Long-term systemic administration of immunosuppressive drugs is associated with numerous side effects including, but not limited to, increased risk of cardiovascular diseases, and enhanced risk of malignancies and opportunistic infections[5, 6]. There have also been numerous reports describing their hepatic and renal toxicities[7, 8]. Controlled release of immunosuppressive drugs has been explored as an option to limit the toxicity associated with unstable and high concentrations of drug immediately after intake[9-12]. Although controlled release vehicles can help to maintain steady blood levels of the drug, they do little to reduce toxicity associated with systemic administration. Additionally, it has been reported that these immunosuppressive agents cannot prevent chronic rejection and may, in fact, act as a barrier to the indefinite acceptance of the graft (a.k.a. tolerance)[2, 7].
Rapamycin is another important immunosuppressive drug, approved by FDA for use in renal transplantation and has low renal toxicity[13-15]. It is currently administered systemically as an oral solution or a tablet, and is believed to primarily act by blocking lymphocyte proliferation in response to cytokines and growth factors. It is possible that the highly effective immunosuppressive activity of rapamycin is not only restricted to lymphocyte inhibition, but may also extend to other targets. For instance, we have shown previously that rapamycin has the ability to inhibit DC maturation and function including their capacity to induce lymphocyte activation[16, 17]. Additionally, injection of rapamycin-treated DC into mice recipients of allogeneic heart transplants induced significant prolongation of graft survival[17, 18]. These experimental observations suggest that rapamycin can mediate immunosuppression through an effect on DC. In light of these findings, we hypothesized that a prolonged DC-specific delivery of rapamycin might potentiate the efficacy of the drug, with respect to its inhibitory action on DC. Such cell-specific delivery has the potential to minimize/eliminate toxicities associated with systemic administration of rapamycin.
Targeted delivery to DC can be achieved through polymeric encapsulation of drugs into appropriately sized microparticles[19-21]. Synthesizing microparticles of the appropriate size is important as: 1) if the particles are too small (nanoparticles) they will be taken up by many different cells of the body, 2) if they are too big (>10 μm) they will not be taken up by DC. We and others have previously shown that microparticles within the size range of ∼1-10 μm are specifically taken up by phagocytic APC, such as DC[20, 21]. Herein, we show that rapamycin can be encapsulated into ∼3.4 μm sized poly(lactic-co-glycolic)acid (PLGA) microparticles (rapaMPs), and controllably released for over 20 days. These particles are readily phagocytosed by DC, and strongly impair the ability of DC to induce lymphocyte activation. This altered function appears to be unrelated to any cytotoxicity associated with the delivery system, or intracellular delivery of the drug.
Materials and Methods
Microparticle Preparation
Microparticles were prepared using the single emulsion/evaporation technique as described[9, 22]. Briefly, 200 mg poly(lactic-co-glycolic)acid (RG504H – viscosity 0.45-0.6 dl/g, Boehringer Ingelheim Chemicals Inc., Petersburg, VA) was dissolved in 4 ml dichloromethane (oil phase). One hundred μl of a stock solution of rapamycin (LC laboratories, Woburn, MA) in Dimethyl Sulfoxide (10mg/ml) was dissolved in the oil phase, which was then homogenized at 10100 rpm for 1 minute in a 2% PVA (MW ∼ 25,000, 98% hydrolyzed; Polysciences) solution, using a homogenizer (Silverson L4RT-A). This emulsion was immediately poured into 90 ml of 1% PVA solution, and dichloromethane was allowed to evaporate. After 3 hours, the particles were centrifuged (1500g, 10 min, 4°C) and washed x4 in deionized water. Microparticles were then re-suspended in 5 ml of deionized water, frozen on dry ice and lyophilized (Virtis Benchtop K freeze dryer, Gardiner, NY; operating at 60 mTorr). Fluorescently labeled rapamycin-microparticles were prepared by adding 100 μl of 2mg/ml (in DMSO) Alexa Fluor 647 carboxylic acid, succinimidyl ester (Invitrogen) to the oil phase along with rapamycin, and following the same protocol as above.
Microparticle Characteristics
Microparticles were sized and counted using volume impedance measurements on a Beckman Coulter Counter (Multisizer 3). Average size was determined by counting at least 10,000 particles. Microparticle surface morphology and shape were examined using a scanning electron microscope (JEOL JSM-6330F, Peabody, MA). The surface charge of microparticles was determined by zeta potential measurements (ZetaPALS, Brookhaven Instruments).
Measuring encapsulation efficiency
Five mg of rapamycin-microparticles (rapaMP) were dissolved in 1 ml of acetonitrile (HPLC grade), sonicated for 5 min and left under constant vortex for 30 min. The amount of rapamycin was then determined by measuring absorbance of this solution at 278 nm using a UV-equipped plate reader (SpectraMax M5, Molecular Devices). Encapsulation efficiency was calculated as the ratio of rapamycin present inside particles to the amount of rapamycin that was initially added during microparticle preparation; and normalized based on the yield.
In vitro release studies
The in vitro release profile of rapamycin from rapaMPs was measured as described. A known amount of rapaMP was suspended in a solution of 1ml of phosphate buffered saline (DPBS, pH 7.4, GIBCO, Invitrogen), or 1 ml of 100mM phosphate buffer (pH 5) containing 0.2% Tween-80. These suspensions were placed on an end-to-end rotator, at 37 °C. At regular intervals, the particles were centrifuged, the supernatant collected, and the particles re-suspended in equal volume of the buffer. Rapamycin content in the supernatant was analyzed by measuring absorbance at 278 nm as described above. Tween-80 was added to maintain sink conditions, and prevent adhesion of rapamycin to the sides of the tubes used for measuring release.
Dendritic cell (DC) cultures and exposure to MPs
Bone marrow-derived myeloid DC were propagated from C57BL/10 (B10) mice (The Jackson Laboratory, Bar Harbor, ME) and purified as described[17]. Briefly, single cell suspensions of B10 bone marrow were cultured for 7 days with 1000 Units/ml of GM-CSF and IL-4 in complete media supplemented with 10% FCS. On day 8 non-adherent cells were collected and CD11c+ cells positively selected using anti-CD11c-cojugated Miltenyi beads (Myltenyi Biotech Auburn, CA). Where indicated, blankMPs or rapaMPs were added (0.11 mg of rapaMPs to achieve a 5:1 ratio of particles to DC) to the culture the day before harvesting, and the culture left undisturbed till subsequent experimentation. DCs exposed to 10 ng/ml soluble rapamycin (Sigma-Aldrich) were used as additional comparison group.
Live cell imaging
One day after the addition of rapaMPs, DC were purified as previously indicated, and cultured for 2 hours over glass bottom culture dishes (MatTek Corporation, Ashland, MA). Adherent DC were then imaged using differential interference contrast microscopy (inverted Olympus 1X81).
Flow cytometric analysis
Following incubation with blankMP or rapaMPs for either 1 day or 4 days, DC were collected from the cultures and viability determined by Annexin-V/7-aminoactinomycin D (7-AAD) staining. Collected cells were stained with FITC-conjugated anti-CD11c mAb (BD PharMingen), PE-conjugated AnnexinV and 7-AAD (Apoptosis Detection Kit, BD PharMingen), following the manufacturer's instructions and then analyzed with a LSR-II flow cytometer (BD Bioscience).
To determine the level of expression of cell surface stimulatory molecules, DC incubated with fluorescent-rapaMPs for 1 day were collected and stained with FITC-conjugated anti-CD11c mAb, PE-conjugated anti-MHC-II, anti-CD86, or anti-CD40 (all from BD PharMingen), following manufacturer's instructions and analyzed as above. For both the experiments FITC-conjugated anti-CD11c mAb was used to gate for DC using flow cytometry.
T cell isolation
CD4+ T cells were purified from spleen and lymph node cell suspensions by negative selection. Non-CD4+ cells were labeled with anti-CD11b, anti-TER-119, anti-Gr-1, anti-I-A/I-E, anti-CD8, anti-B220, and anti-Gr-1 mAbs (all from BD Pharmingen). Following incubation with anti-rat-IgG-Dynabeads (Dynal Biotech, Invitrogen), bead-bound cells were removed by magnetic isolation. Purity of CD4+ T cell preparation was assessed by flow cytometry and corresponded to 90%, reproducibly.
Mixed leukocyte reaction (MLR)
B10 DC (15,000), cultured for 1 day with rapaMPs (using different ratios of rapaMPs to DC), were mixed with 150,000 CD4 T cells (isolated from lymph nodes and spleen of normal BALB/c mice) in u-bottomed 96 well culture plates. The cells were co-cultured in complete RPMI 1640 (GIBCO, Carlsbad, CA), supplemented with 10% fetal calf serum, for 3 days following which tritiated thymidine was added. After 16 hrs, the amount of thymidine incorporated into the cells was measured using a Wallac 1205 Betaplate analyzer. A no treatment (NT) group consisting of DC that were not exposed to any particles, and DC cultured with blank particles (5:1 – particle to DC ratio) served as a negative controls. A DC group treated for 1 day with soluble rapamycin (10ng/ml) acted as a comparative control.
Statistics
Statistical analysis was performed using the 2-tailed Student's t-test, and all results are expressed as ± 1 SD. Standard deviation was calculated based on n=3 experiments, unless otherwise stated.
Results
Microparticle characteristics
Rapamycin-encapsulated PLGA microparticles (rapaMP) were prepared successfully using a single emulsion/evaporation technique. These particles were sized to be 3.4 ± 1.65 μm (diameter) using volume impedance measurements on a Beckman Coulter Counter. SEM images of these particles (Figure 1) show smooth surface morphology and confirm the size obtained from volume impedance measurements. The size distribution observed in the SEM images is characteristic of the microparticle synthesis procedure described above, and has been reported elsewhere [9, 23, 24]. Further, the zeta potential of these rapaMPs was determined to be -35.38 ± 4.49 mV (n=4 particle sets).
Figure 1.

SEM images of rapamycin containing PLGA particles. These images confirm that the particles exhibit surface integrity. Also, sizes are consistent with measurements via volume impedance (Avg. Vol. Dia – 3.4 μm).
Encapsulation efficiency
Encapsulation efficiency of rapaMP was determined to be 73.95 ± 5.43%. The presence of rapamycin inside microparticles was further confirmed by tandem mass spectroscopy analysis (data not shown). Based on the volume impedance measurements and encapsulation efficiencies, the amount of rapamycin in each rapaMP was calculated to be approximately 50 femtograms.
In vitro release profile
The release rate of rapamycin from rapaMPs was studied in both pH 7.4 and pH 5 buffer. It has been reported previously that DC efficiently phagocytose PLGA microparticles (3-4 μm in size) in 4-5 hrs, and that the particles may remain inside phagosomes (pH ∼ 5)[20, 25]. Hence, to replicate intra-phagosomal pH conditions in vitro, release was studied in a pH 5 buffer. There were no significant differences observed in the release rates between pH 7.4 and pH 5 buffers (Figure 2). The release appeared to follow Fickian diffusion kinetics, and the calculated values of diffusivity of the drug in the polymer matrix were found to be in the same order of magnitude, as predicted by theoretical correlations developed by us and others[26, 27]. The pH of the supernatant was measured to be stable throughout the release study (data not shown).
Figure 2.

In vitro controlled release of rapamycin from small-rapaMPs in pH 5 and pH 7.4 buffer. Percentages are based on amount encapsulated. 100% indicates ∼3.7 μg of rapamycin/mg of rapaMPs – the maximum amount that would be released if the particles degraded completely. Standard deviations are based on n = 6 samples for release at pH 7.4, and n=3 samples for release at pH 5.
Live cell imaging and apoptosis level
We (and others) have shown previously that PLGA microparticles are phagocytosed by DC and can be identified inside these cells [20, 25, 28]. Figure 3 shows images of live DC that were exposed overnight to different amounts of rapaMPs. These images show that, for any given ratio (number of particles: number of DC, adopted during the co-culture phase), a certain degree of variability in the number of particles phagocytosed by individual DC was evident. However, a general direct correlation appeared between the co-culture ratio and the average number of particles internalized. Additionally, we observed that when a particle:DC ratio of 5:1 or 10:1 was used, the majority of DCs internalized at least one particle.
Figure 3.

Representative images of live DC cultured for 1 day with rapaMPs. NT (no treatment) implies that no particles were added to these DC cultures. The ratios indicate the number of particles added to the number of DC. Scale = 50 μm.
Next, we tested whether phagocytosis of blankMPs or rapaMPs leads to DC death. Initially, we measured the level of trypan blue exclusion from cultures treated with different particle:DC ratios and observed a small percentage of DC positive for trypan blue. This number was not significantly different from the control population of non-treated DC at any ratio tested (data not shown). To further investigate the status of DC exposed to MPs, we collected the cells after 24 hours or 96 hours of co-incubation and stained them with FITC-conjugated anti-CD11c mAb, Annexin-V (AnnV) and 7-aminoactinomycin D (7-AAD). FITC-conjugated anti-CD 11c mAb was used to gate DC using flow cytometry, as CD11c is a DC-restricted marker. The combination of the other two dyes allowed the discrimination of viable DC (AnnV- 7-AAD-) from those that were either entering apoptosis (AnnV+ 7-AAD-), in late apoptosis (AnnV+ 7-AAD+), or necrotic (AnnV- 7-AAD+). As observed from the AnnV/7-AAD profile of CD11c+ cells, 1 day after co-incubation (Figure 4A and Table 1) the proportion of apoptotic cells (early and late) among rapaMP-loaded DC was not different from that of untreated and blankMP-loaded DC. After 4 days of co-incubation (Figure 4B and Table 1), the apoptotic level of untreated or blankMP-loaded DC was not significantly different. This indicated that the process of MP phagocytosis per se was not harmful to the cells. In the rapaMP group, however, a significant increase in the proportion of early apoptotic cells was evident in comparison to untreated and blankMP groups. As blankMP phagocytosis did not induce such alteration, the pro-apoptotic effect observed is attributable to intracellular release of rapamycin.
Figure 4.
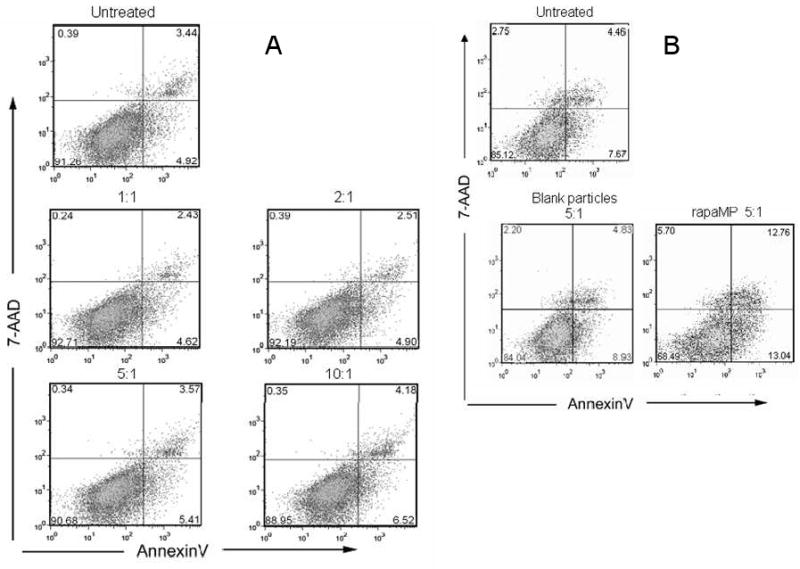
Annexin-V and 7-AAD staining of DC to examine potential toxicity of rapaMPs. Apoptosis of DC incubated for (A) - 1 day, or (B) − 4 days with different number of rapaMPs was compared to untreated DC. Flow diagrams were generated by gating on CD11c+ (DC restricted marker) cells.
Table 1.
Quantitative analysis of apoptosis levels in DC that have phagocytosed rapaMPs. Live cells represent % cells that are AnnV- 7-AAD-, early apoptotic represents % cells that are AnnV+ 7-AAD-, late apoptotic represents % cells that are AnnV+ 7-AAD+, and necrotic represents % cells that are AnnV- 7-AAD+. Both rapaMP and blank MP were added at a ratio of 5:1 (particle to DC). SD are based on n=3 experiments.
| Live cells | Early apoptotic | Late apoptotic | Necrotic | |
|---|---|---|---|---|
| NT (1 day) | 91.1 ± 1.6 | 5.2 ± 0.3 | 3.2 ± 1.0 | 0.5 ± 0.5 |
| Blank MP (1 day) | 89.2 ± 6.0 | 6.2 ± 2.4 | 3.7 ± 2.8 | 0.8 ± 0.8 |
| rapaMP (1 day) | 87.2 ± 7.2 | 7.4 ± 3.4 | 4.0 ± 3.1 | 1.3 ± 1.4 |
| NT (4 day) | 84.8 ± 0.5 | 8.2 ± 0.7 | 5.2 ± 0.1 | 1.9 ± 1.2 |
| Blank MP (4 day) | 83.7 ± 1.1 | 8.5 ± 0.6 | 6.0 ± 1.4 | 1.8 ± 0.4 |
| rapaMP (4 day) | 74.3 ± 7.0 | 12.2 ± 1.0*,** | 10.3 ± 3.1 | 3.3 ± 2.9 |
indicates comparison between rapaMP (4 day) and NT (4 day) groups - p<0.05, and
indicates comparison between rapaMP (4 day) and blank MP (4 day) groups – p<0.05.
RapaMP phagocytosis alters DC functionality
The functional modulation of DC by rapaMPs was characterized in vitro using a mixed leukocyte reaction in which DC from one mouse strain (B10) were treated overnight with different ratios of rapaMP:DC and then co-cultured with CD4+ T cells isolated from secondary lymphoid organs of a different mouse strain (BALB/c). T cell proliferation (an indicator of DC stimulatory capacity) was assessed by cellular incorporation of radioactive thymidine after 3 days of co-culture (a total of 4 days after addition of rapaMPs to the DCs). As shown in figure 5, we observed a reduction in T cell proliferation that correlated with the rapaMPs:DC ratio used. Over 5 times reduction in the ability of rapaMP-loaded DC (ratio of 5:1 and 10:1) to induce T cell proliferation (p<0.005) was observed when compared to blank particle-loaded or non-treated DC. Further, figure 5 shows that rapaMP treatment (at 5:1 and 10:1 ratio) resulted in significantly greater inhibition of T cell stimulatory function (about 2.5 times greater reduction in T cell proliferation) than that achieved with soluble rapamycin-treated DC (p<0.01). Based on the encapsulation efficiency and assuming that the rate of drug release is accurately represented by the profile described in figure 2, we calculate that the amount of rapamycin delivered to DCs by rapaMP is approximately 3 ng/ml/day, while the amount added in the soluble rapamycin-treated DC group was 10 ng/ml.
Figure 5.

A mixed leukocyte reaction indicating the amount of radioactive thymidine incorporated into responding T lymphocytes. DC were loaded with rapaMPs using the indicated MP:cell ratio and then used to stimulate allo-reactive CD4+ T cells. NT (no treatment) group was assigned the value of 100% and all other percentages are normalized based on this value. The ratio of MPs to DC in the blank particles group is 5:1. Standard deviations are based on n = 6 experiments per group. * indicates comparison between 5:1 and NT groups - p <0.005; ** indicates comparison between 5:1 and soluble Rapa treatment groups - p < 0.01.
RapaMP's lower expression of cell-surface stimulatory molecules
To investigate the mechanism behind functional modulation of DC loaded with rapaMPs, we examined the expression of cell-surface co-stimulatory molecules (activation markers). Particles were loaded with Alexa Fluor 647 (A647) to track percentage of DC that have taken up particles and also determine the co-localization of particle uptake with any differences in cellular phenotype. Flow cytometric analysis revealed that DC that had taken up fluorescently stained rapaMPs (A647 high) had lowered expression of CD86, MHC-II and CD40 (common stimulatory molecules expressed on DC) when compared to DC that were A647 low (Figure 6).
Figure 6.

Flow Cytometric Analysis of DC activation markers on cells that have taken up fluorescently labeled rapaMPs (A647+). Flow diagrams were generated by gating on CD11c+ and Alexa-fluor 647high or Alexa-fluor 647low, as described in the top-center plot. Numbers on flow diagrams indicate mean fluorescence intensity values. DC were cultured with rapaMPs at a ratio of 5:1 (rapaMPs to DC) for 24 hours, prior to flow analysis.
Discussion
Microparticles composed of the degradable polymer PLGA have been widely used for controlled release of drugs and proteins[19]. Appropriately-sized microparticles have also been used specifically for targeted delivery of drugs, antigens and DNA to phagocytic cells[20, 29]. The size of the polymeric particles has an important bearing on their ability for long-term release, total payload delivered and specific targeting to phagocytic cells. If the size is too small, as is the case with nanoparticles (∼200 nm or lower), it is widely believed that they are not suited to act as long term delivery vehicles [19, 30-32]. We have also observed that nanoparticulates are not taken up to the same extent as microparticulates, possibly due to the differences in uptake mechanisms (data not shown). Furthermore, nanoparticles are typically taken up by all cells capable of endocytosis[19], and although such systems may deliver drugs to dendritic cells if placed in a physiologic milieu with large numbers of resident dendritic cells (e.g. the dermis)[33, 34] they have no inherent targeting capabilities per se. Conversely, if particles are too large (>10 μm), they cannot be internalized by phagocytic cells[35]. Hence, for applications where long term and phagocytic cell-specific targeted delivery is desirable (such as transplantation and autoimmune disease immunotherapy, or anti-tumor immunity) it is imperative that drugs are encapsulated into micron-sized particles (∼ 1-10 μm).
One of the recently developed approaches in transplantation and autoimmune disease immunotherapy involves use of dendritic cell (DC)-based therapeutics[36, 37]. We have shown previously that DC cultured with specific agents, such as the immunosuppressive drug rapamycin, have altered function and can be used to suppress transplant rejection and promote donor-specific tolerance[18, 38]. Our long term goal was to create a system capable of specifically targeting DC in vivo and then sustaining intracellular delivery of rapamycin to produce a more pronounced effect with respect to suppression of transplant rejection and promotion of tolerance without systemic immunosuppression. Here, we tested the hypothesis that intracellular delivery of rapamycin to DC using appropriately sized microparticulates is effective at inhibiting an important initial step in the onset of transplant rejection - T cell activation.
We demonstrate that rapamycin can be encapsulated and controllably released from appropriately sized PLGA microparticles. PLGA was the polymer of choice as it does not activate DC[20], and allows for intracellular release of drug over extended periods as shown in figure 2. A Fickian release that lasts for about 3 weeks was observed, which is similar to a previously reported release profile for cyclosporine (a drug with similar mol. Wt. and hydrophobicity as rapamycin)[9]. Further, the release can be modulated as desired by changing the molecular weight of PLGA used to make the microparticles. We also demonstrate that rapaMPs are associated with a more pronounced effect on DC when compared to conventional rapamycin treatments. Previously, we have shown that DC treated with soluble rapamycin are characterized by a lower capacity to activate T cells[16, 17]. However, in comparison to soluble rapamycin-treated DC, rapaMP-loaded DC exhibit significantly higher capacity in preventing T cell activation (Figure 6). Annexin V and 7AAD staining was used to analyze if this reduction was due to an increase in DC apopotosis in the presence of rapaMPs over the 96 hour period (Figure 4 and Table 1). Figure 4A shows that annexin V and 7AAD profiles for untreated and rapaMP-loaded DC following 1 day of co-incubation are similar, confirming that phagocytosis of rapaMPs does not cause increase in DC apoptosis. After 4 days of co-incubation, a small increase in apoptosis of rapaMP-loaded DC is observed (Figure 4B and Table 1). Since the number of apoptotic cells in the untreated and blank particle-loaded DC groups is not different, the observed increase in apoptosis of rapaMP-loaded DC is most probably due to the presence of rapamycin. Importantly, it has been previously shown that the presence of soluble rapamycin in DC cultures leads to increase in apoptosis rates similar to what is depicted in figure 4B for rapaMP [39]. In light of this data, we would conclude that apoptosis alone is not responsible for the effects of rapaMP-loaded DCs when compared to soluble rapamycin.
It should be noted that the observed increase in inhibitory capacity when using rapaMPs is pronounced (Figure 5) despite the fact that the theoretical quantity of rapamycin delivered is equal to or less than the soluble rapamycin group (amounts calculated based on encapsulation efficiency and cumulative release from rapaMPs). It is likely that this superior inhibitory effect is due to greater intracellular availability of the drug, as it is known that rapamycin inhibits DC function through its action on an intracellular protein kinase[16, 17].
One possible mechanism by which the function of DC is altered upon rapaMP phagocytosis is the limited expression of certain necessary stimulatory modalities on the cell surface (Figure 6). It is well known that activation of T cells by DC requires the expression of MHC (signal 1) in the presence of other co-stimulatory molecules (signal 2). Flow cytometric analysis reveals that the expression of both MHC-II and co-stimulatory molecules (CD86 and CD40) is substantially lower in DC that have taken up rapaMPs (Figure 6). Although it cannot be ruled out that rapaMPs also affect DC in other ways, agents which specifically down-regulate co-stimulation have been identified as promising candidates for clinical transplantation immunotherapy[2] and current phase I clinical trials are underway for autoimmune Type I diabetes that utilize antisense oligonucleotides that knock down CD40 and CD86[40].
We are currently testing the influence of rapaMP-loaded DC on T cell alloreactivity and their potential to modulate the allograft rejection response in a murine heart transplant model[18]. This mode of treatment involves ex-vivo loading of donor-derived DCs, in which the DC themselves will act as the antigen via the direct antigen presentation pathway[2]. Additionally, we are also testing the possibility of using sub-cutaneous injections of rapaMPs as a mode of treatment in a murine skin transplant model. In this case, the rapaMPs will be passively targeted to the local phagocytic cell populations that are responsible for initiating immune responses against the skin transplant.
We also speculate that the obtained results may have implications for DC-specific delivery of many other immuno-modulatory agents that act intracellularly, such as aspirin, corticosteroids, and vitamin D3 analogues[41]. Thus, it is possible that appropriately-sized degradable microparticulates could be used as a modular platform technology to deliver any number of the newly-identified agents that exhibit immuno-modulatory effects through interactions with DC[41].
In conclusion, we have successfully prepared and characterized rapaMPs that can be phagocytosed by DC. We demonstrate the release of rapamycin in pH 7.4 and pH 5 buffers lasts for about 2-3 weeks. Additionally, we observe that in vitro, rapaMP-loaded DC have a much lower capacity to induce proliferation in T cells than DC exposed to soluble rapamycin. This effect may be mediated by down-regulation of surface co-stimulatory molecules. These microparticles have the potential to either be preloaded into DC ex vivo or to passively target DC in vivo as an antigen presenting cell-specific immunotherapy for transplantation and autoimmune diseases.
Acknowledgments
We thank Marc Rubin for assistance with the SEM, Jennifer Karlsson, Catherine Baty & Simon Watkins for help with the differential interference contrast microscopy. This work was supported by NIH grants AI 67541, AI 60994, KL2 RR024154-02 and the American Heart Association grant 0730081N. GR is in receipt of a research fellowship from the Transplantation Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tantravahi J, Womer KL, Kaplan B. Why hasn't eliminating acute rejection improved graft survival? Annual Review of Medicine. 2007;58:369–385. doi: 10.1146/annurev.med.58.061705.145143. [DOI] [PubMed] [Google Scholar]
- 2.Lechler RI, Sykes M, Thomson AW, Turka LA. Organ Transplantation - How much of the promise has been realized. Nature Medicine. 2005;11(6):605–613. doi: 10.1038/nm1251. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 4.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic Dendritic Cells. Annual Review of Immunology. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 5.Krämer BK, Böger C, Krüger B, Marienhagen J, Pietrzyk M, Obed A, Paczek L, Mack M, Banas B. Cardiovascular Risk Estimates and Risk Factors in Renal Transplant Recipients. Transplantation Proceedings. 2005;37:1868–1870. doi: 10.1016/j.transproceed.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Rubin RH, Ikonen T, Gummert JF, Morris RE. The therapeutic prescription for the organ transplant recipient: the linkage of imunosuppression and antimicrobial strategies. Transplant Infectious Diseases. 1999;1:29–39. doi: 10.1034/j.1399-3062.1999.10104.x. [DOI] [PubMed] [Google Scholar]
- 7.Duncan MD, Wilkes DS. Transplant-related Immunosuppression: A Review of Immunosuppression and Pulmonary Infections. Proceedings of American Thoracic Society. 2005;2:449–455. doi: 10.1513/pats.200507-073JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Libby P, Pober JS. Chronic Rejection. Immunity. 2001;14:387–397. doi: 10.1016/s1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- 9.Sanchez A, Vila-Jata JL, Alonso MJ. Development of biodegradable microspheres and nanospheres for the controlled release of cyclosporin A. International Journal of Pharmaceutics. 1993;99(2-3):263–273. [Google Scholar]
- 10.Sanchez A, Alonso MJ. Poly(D,L-lactide-co-glycolide) Micro and Nanospheres as a Way to Prolong Blood/Plasma Levels of Subcutaneously Injected Cyclosporin A. European Journal of Pharmaceutics and Biopharmaceutics. 1995;41(1):31–37. [Google Scholar]
- 11.Lamprechta A, Yamamoto H, Takeuchi H, Kawashima Y. A pH-sensitive microsphere system for the colon delivery of tacrolimus containing nanoparticles. Journal of Controlled release. 2005;104:337–346. doi: 10.1016/j.jconrel.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 12.Italia JL, Bhatt DK, Bhardwaj V, Tikoo K, Kumar MNVR. PLGA nanoparticles for oral delivery of cyclosporine: Nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral. J Control Rel. 2007;119:197–206. doi: 10.1016/j.jconrel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Kahan BD. Two-year results of multicenter phase III trials on the effect of the addition of sirolimus to cyclosporine-based immunosuppressive regimens in renal transplantation. Transplantation Proceedings. 2003;35(3):37S–51S. doi: 10.1016/s0041-1345(03)00353-1. [DOI] [PubMed] [Google Scholar]
- 14.Thliveris JA, Solez KS, Yatscoff RW. A comparison of the effects of rapamycin and cyclosporine on kidney and heart morphology in a rabbit heterotopic heart transplant model. Histology and Histopathology. 1995;10(2):417–421. [PubMed] [Google Scholar]
- 15.Thliveris JA, Yatscoff RW. Effect of rapamycin on morphological and functional parameters in the kidney of the rabbit. Transplantation. 1995;59(3):427–429. [PubMed] [Google Scholar]
- 16.Hackstein H, Taner T, Zahorchak AF, Morelli AE, Logar AJ, Gessner J, Thomson AW. Rapamycin inhibits IL-4–induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood. 2003;101:4457–4463. doi: 10.1182/blood-2002-11-3370. [DOI] [PubMed] [Google Scholar]
- 17.Taner T, Hackstein H, Wang Z, Morelli AE, Thomson AW. Rapamycin-treated, Alloantigen-pulsed host Dendritic cells induce Ag-specific T cell regulation and prolong graft survival. American Journal of Transplantation. 2005;5:228–236. doi: 10.1046/j.1600-6143.2004.00673.x. [DOI] [PubMed] [Google Scholar]
- 18.Turnquist HR, Raimondi G, Zahorchak AF, Fischer RT, Wang Z, Thomson AW. Rapamycin-Conditioned Dendritic Cells Are Poor Stimulators of Allogeneic CD4+ T Cells, but Enrich for Antigen-Specific Foxp3+ T Regulatory Cells and Promote Organ Transplant Tolerance. The Journal of Immunology. 2007;178:7018–7031. doi: 10.4049/jimmunol.178.11.7018. [DOI] [PubMed] [Google Scholar]
- 19.Kohane DS. Microparticles and Nanoparticles for Drug Delivery. Biotechnology and Bioengineering. 2007;96(2):203–209. doi: 10.1002/bit.21301. [DOI] [PubMed] [Google Scholar]
- 20.Little SR, Lynn DM, Ge Q, Anderson DG, Puram SV, Chen J, Eisen HN, Langer R. Poly-β amino ester-containing microparticles enhancethe activity of nonviral genetic vaccines. PNAS. 2004;101:9534–9539. doi: 10.1073/pnas.0403549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wischke C, Borchert HH, Zimmermann J, Siebenbrodt I, Lorenzen DR. Stable cationic microparticles for enhanced model antigen delivery to dendritic cells. J Control Rel. 2006;114:359–368. doi: 10.1016/j.jconrel.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 22.Zweers MLT, Engbers GHM, Grijpma DW, Feijen J. Release of anti-restenosis drugs from poly(ethylene oxide)-poly (DL-lactic-co-glycolic acid) nanoparticles. Journal of Controlled Release. 2006;114:317–324. doi: 10.1016/j.jconrel.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 23.Thomas TT, Kohane DS, Wang A, Langer RS. Microparticulate Formulations for the Controlled Release of Interleukin-2. Journal of Pharmaceutical Sciences. 2004;93(5):1100–1109. doi: 10.1002/jps.20009. [DOI] [PubMed] [Google Scholar]
- 24.Martínez Gómez JM, Csaba N, Fischer S, Sichelstiel A, Kündig TM, Gander B, Johansen P. Surface coating of PLGA microparticles with protamine enhances their immunological performance through facilitated phagocytosis. J Control Rel. 2008 doi: 10.1016/j.jconrel.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 25.Yoshida M, Babensee JE. Molecular aspects of microparticle phagocytosis by dendritic cells. Journal of Biomaterial Science. (Polymer) 2006;17(8):893–907. doi: 10.1163/156856206777996844. [DOI] [PubMed] [Google Scholar]
- 26.Siepmann J, Elkharraz K, Siepmann F, Klose D. How Autocatalysis Accelerates Drug Release from PLGA-Based Microparticles: A Quantitative Treatment. Biomacromolecules. 2005;6:2312–2319. doi: 10.1021/bm050228k. [DOI] [PubMed] [Google Scholar]
- 27.Rothstein SN, Federspiel WJ, Little SR. A simple model framework for the prediction of controlled release from bulk eroding polymer matrices. J Mater Chem. 2008;18:1873–1880. [Google Scholar]
- 28.Fischer S, Allmen EU, Waeckerle-Men Y, Groettrup M, Merkle HP, Gander M. The preservation of phenotype and functionality of dendritic cells upon phagocytosis of polyelectrolyte-coated PLGA microparticles. Biomaterials. 2007;28:994–1004. doi: 10.1016/j.biomaterials.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 29.Walter E, Dreherb D, Kokc M, Thiele L, Kiama SG, Gehr P, Merkle HP. Hydrophilic poly(DL-lactide-co-glycolide) microspheres for the delivery of DNA to human-derived macrophages and dendritic cells. Journal of Controlled Release. 2001;76:149–168. doi: 10.1016/s0168-3659(01)00413-8. [DOI] [PubMed] [Google Scholar]
- 30.Lecaroz C, Gamazo C, Renedo MJ, Blanco-Prieto MJ. Biodegradable micro- and nanoparticles as long-term delivery vehicles for gentamicin. Journal of Microencapsulation. 2006;23(7):782–792. doi: 10.1080/02652040600946886. [DOI] [PubMed] [Google Scholar]
- 31.Lecaroz MC, Blanco-Prieto MJ, Campanero MA, Salman H, Gamazo C. Poly(D,L-Lactide-Coglycolide) Particles Containing Gentamicin: Pharmacokinetics and Pharmacodynamics in Brucella melitensis-Infected Mice. Antimicrobial Agents and Chemotherapy. 2007;51(4):1185–1190. doi: 10.1128/AAC.00809-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee WK, Park JY, Yang EH, Suh H, Kim SH, Chung DS, Choi K, Yang CW, Park JS. Investigation of the factors influencing the release rates of cyclosporin A-loaded micro- and nanoparticles prepared by high-pressure homogenizer. Journal of Controlled Release. 2002;84:115–123. doi: 10.1016/s0168-3659(02)00239-0. [DOI] [PubMed] [Google Scholar]
- 33.Haddadi A, Elamanchili P, Lavasanifar A, Das S, Shapiro J, Samuel J. Delivery of rapamycin by PLGA nanoparticles enhances its suppressive activity on dendritic cells. J Biomed Mater Res. 2008;84A:885–898. doi: 10.1002/jbm.a.31373. [DOI] [PubMed] [Google Scholar]
- 34.Newman KD, Elamanchili P, Kwon GS, Samuel J. Uptake of poly(D,L-lactic-co-glycolic acid) microspheres by antigen-presenting cells in vivo. J Biomed Mater Res. 2002;60(3):480–486. doi: 10.1002/jbm.10019. [DOI] [PubMed] [Google Scholar]
- 35.Tabata Y, Ikada Y. Macrophage phagocytosis of biodegradable microspheres composed of L-lactic acid/glycolic acid homo- and copolymers. Journal of Biomedical Materials Research. 1988;22:837–858. doi: 10.1002/jbm.820221002. [DOI] [PubMed] [Google Scholar]
- 36.Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nature Reviews Immunology. 2007;7(8):610–621. doi: 10.1038/nri2132. [DOI] [PubMed] [Google Scholar]
- 37.Bluestone JA, Thomson AW, Shevach EM, Weiner HL. What does the future hold for cell-based tolerogenic therapy? Nature Reviews Immunology. 2007;7(8):650–654. doi: 10.1038/nri2137. [DOI] [PubMed] [Google Scholar]
- 38.McCurry KR, Colvin BL, Zahorchak AF, Thomson AW. Regulatory dendritic cell therapy in organ transplantation. Transplant International. 2006;19(7):525–538. doi: 10.1111/j.1432-2277.2006.00306.x. [DOI] [PubMed] [Google Scholar]
- 39.Woltman AM, Fijter JWD, Kamerling SWA, van der Kooij SW, Paul LC, Daha MR, Kooten CV. Rapamycin induces apoptosis in monocyte- and CD34-derived dendritic cells but not in monocytes and macrophages. Blood. 2001;98(1):174–181. doi: 10.1182/blood.v98.1.174. [DOI] [PubMed] [Google Scholar]
- 40.Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, Gillis K, Brown L, Lafreniere D, Gallo M, Knox J, Hogeland K, Trucco M, Giannoukakis N. A Microsphere-Based Vaccine Prevents and Reverses New-Onset Autoimmune Diabetes. Diabetes. 2008;57(6):1544–1555. doi: 10.2337/db07-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hackstein H, Thomson AW. Dendritic cells: Emerging pharmacological targets of immunosuppressive drugs. Nature Reviews Immunology. 2004;4(1):24–34. doi: 10.1038/nri1256. [DOI] [PubMed] [Google Scholar]