

Abstract
The nuclei of differentiating cells exhibit several fundamental principles of self-organization. They are composed of many dynamical units connected physically and functionally to each other—a complex network—and the different parts of the system are mutually adapted and produce a characteristic end state. A unique cell-specific signature emerges over time from complex interactions among constituent elements that delineate coordinate gene expression and chromosome topology. Each element itself consists of many interacting components, all dynamical in nature. Self-organizing systems can be simplified while retaining complex information using approaches that examine the relationship between elements, such as spatial relationships and transcriptional information. These relationships can be represented using well-defined networks. We hypothesize that during the process of differentiation, networks within the cell nucleus rewire according to simple rules, from which a higher level of order emerges. Studying the interaction within and among networks provides a useful framework for investigating the complex organization and dynamic function of the nucleus.
Keywords: cellular differentiation, chromosomal organization, networks, reprogramming the network
Introduction
Genomes of higher eukaryotes are distributed non-randomly within the nucleus, but it has been debated whether the architecture of the nucleus itself is an important feature driving cell differentiation and maturation. More than a century ago, Rabl (1885) and then Boveri (1909) suggested that chromosomes occupy distinct regions of the nucleus. Cremer et al (1982) confirmed that interphase chromosomes are indeed organized into discrete, non-overlapping ‘territories.' Moreover, these chromosome territories adopt non-random positions within the nucleus with gene-rich chromosomes being located preferentially towards the center of the nucleus, an arrangement that is retained in many different cell types and seems to be conserved through evolution (Croft et al, 1999; Boyle et al, 2001; Cremer et al, 2001; Neusser et al, 2007). Gene activation and gene silencing events can be accompanied by dynamic movements (of up to 5 μm) of gene loci to and from chromosome territories, and such movements may determine access to the transcriptional machinery (Chuang et al, 2006; Dundr et al, 2007; Meister et al, 2010).
The three-dimensional architecture of chromosomes can compartmentalize the nucleus and reflect regional gene expression (Kosak and Groudine, 2004a; Bolzer et al, 2005; Misteli, 2007; Dekker, 2008), but analysis of nuclear architecture has been limited by methods that focus on interactions between specific loci rather than an unbiased genome-wide analysis (Dostie et al, 2006; Simonis et al, 2006; Zhao et al, 2006). However, two recently described variants of the classic 3C technique (Dekker et al, 2002) have been used to investigate nuclear organization on a more global level, either for a network of lineage-specific active loci (Schoenfelder et al, 2009) or for the whole genome (Lieberman-Aiden et al, 2009). Using an anchor-based e4c method to investigate the nuclear organization of active genes in murine fetal liver erythroid cells, Schoenfelder et al, found that lineage-specific genes colocalize within specialized transcription factories. Of particular significance, colocalization occurs not only in cis (genes within the same chromosome), but also in trans (between genes located on different chromosomes) in these factories. Using Hi-C, which probes the three-dimensional architecture of whole genomes by coupling proximity-based ligation with massively parallel sequencing, Lieberman-Aiden et al (2009) constructed spatial proximity maps of the human genome in B-cell and erythroid cell lines and confirmed the presence of chromosome territories, the spatial proximity of small, gene-rich chromosomes, and the spatial segregation of open and closed chromatin. The Hi-C approach reveals genome-wide spatial relationships, and can be used to study the relationships between global spatial architecture and global gene expression at multiple time points to capture the dynamics of nuclear organization during cell differentiation.
We have proposed that dynamic gene regulatory networks are manifested spatially at the level of chromosomal organization, with chromosomes associating according to their overall coregulated gene content (Kosak et al, 2007; Rajapakse et al, 2009). This relationship was established by defining and showing the collective similarity of two networks, the coregulated gene regulatory network and the chromosomal interaction network, in the nucleus during in vitro differentiation of murine hematopoietic progenitors (Bruno et al, 2004; Kosak et al, 2007; Rajapakse et al, 2009). A major question that can now be addressed on a global scale is whether lineage determination patterns a specific nuclear architecture to preconfigure expression of differentiation genes, or whether transcription of cell differentiation genes mediates transitions in nuclear architecture. In other words: is form a precondition for, or does form follow, function? We suggest that investigating the relationships between nuclear form and function will be critical to improve our understanding of cell fate, including missteps that can propel normal cells into an unstable state that leads to cancer. By studying disruptions in networks that globally represent the nucleus of any cell type, potentially we can predict instabilities as well as points that have the largest impact on cell fate, and ultimately redirect cells from a pathological to a benign state, or a differentiated state to a pluripotent state. In the following sections, we give a brief introduction to the principles of self-organization and the mathematics of networks. We then discuss how network theory can be used to help further our understanding of nuclear organization.
Self-organization
Self-organization in a system is a process by which the global-level pattern emerges solely from many interactions among lower-level components; the pattern is an emergent property of the system, rather than a property imposed on the system by an external ordering influence (Ashby, 1947; Camazine et al, 2003). The system tends to reach a particular state, a set of cycling states, or a small volume of their state space (attractor basins), with no external interference (Kauffman, 1984). The rules for behavior in such systems are non-linear (see Table I), and as such, cannot be analyzed by breaking them into smaller and smaller parts. In essence, the whole of a non-linear system is not simply an additive function of its parts (Anderson, 1972; Strogatz, 1994, 2001, 2003). However, a more refined view of self-organization is that the global pattern, while not in control of the local interactions, can feedback to influence those local components (Langton, 1990). Resulting changes in local behavior may then change the global pattern, and the self-organized system fine-tunes over time. Thus, self-organized systems have local to global and global to local feedback that leads to increasing order over time (Langton, 1990; Lewin, 1992). In other words, the system exhibits a continual interplay of bottom–up and top–down processes. Therefore, the coordination of the activities of individual complex elements enables a system to develop, sustain complexity at a higher level, and evolve.
Table 1. Glossary of terms.
| Adjacency matrix of a graph: Square matrix with aij=1, when there is an edge from node i to node j; otherwise aij=0. Weighted adjacency matrices with entries that are not simply zero or 1 have entries equal instead to the weights on the edges |
| Attractor: An attractor is the end-state of a dynamic system as it moves over time. Attractors may be fixed points, periodic, or chaotic and may also be stable or unstable |
| Basin of attraction: A region in phase space associated with a given attractor. The basin of attraction of an attractor is the set of all (initial) points that move toward that attractor |
| Cellular differentiation: The dynamical process of a less specialized cell transitioning into a more specialized cell type |
| Cellular reprogramming: The process of changing a mature unipotent adult cell's unique genetic and epigenetic signature, typically by manipulating signal transduction mechanisms and growth factors, so as to confer plasticity, pluripotency, or ability to differentiate into at least one other type of cell |
| Chromosomal interaction network: Nodes are chromosome or genes and the edges are computed based on proximity of chromosomes. Spatial relationships between each pair of chromosomes include distance between centroids, closest distance, and also more complex relationships such as shared volume and contact area |
| Communication: Interaction between networks. We quantify network communication by comparing the similarity between the network's corresponding weighted adjacency matrices |
| Connectivity: Interaction within a network. We quantify network connectivity using the second smallest eigenvalue of the network's Laplacian matrix |
| Dynamical system: An evolution rule that defines a trajectory as a function of a single parameter (time) on a set of states (the phase space) is a dynamical system |
| E-box: Is a regulatory DNA sequence that usually lies upstream of a gene in a promoter region, commonly bound by basic helix-loop-helix transcription factors |
| Eigenvalues: A special set of scalars associated with linear systems of equations. They are also known as characteristic roots. The decomposition of a square matrix A into eigenvalues and eigenvectors (the vectors associated with eigenvalues) is known in this work as eigen decomposition. Eigenvalues are represented by λ and eigenvectors by x. Ax=λx with x≠0 so det(A–λI)=0 |
Euclidean distance: Is the ‘ordinary' distance between two points. In a plane with p1 at (x1,y1) and p2 at (x2,y2), it is 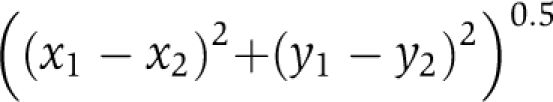
|
| Graph G: A set of n nodes connected pairwise by m edges. In a complete graph, all 0.5(n(n−1)) edges between nodes |
| Nonlinear: A function that is not linear. Most things in nature are nonlinear. This means that in a very real way, the whole is at least different from the sum of the parts |
| Spectral karyotyping (SKY): A method of visualizing all chromosomes in the genome simultaneously, with each chromosome labeled with a unique color |
| Symmetric matrix: A matrix with the lower-left half equal to the mirror image of the upper-right half |
| Trace: Let A be an n × n matrix with eigenvalues λ1,λ2,…,λn. The sum of the eigenvalues is the sum of the diagonal entries of A and is called the trace of A |
| Transcriptome network: Nodes are chromosome or genes and the edges are computed based on gene coregulation. One of the measures of gene coregulation is the relative entropy between gene profiles |
Does self-organization in biological systems arise only from stochastic events or can self-organization emerge from ordered assembly (deterministic) events (Misteli, 2001, 2007, 2008)? Evidence suggests that both may occur. Formation of Cajal bodies in the nucleus seems to be a self-organizing process that arises from stochastic events. Kaiser et al (2008) show that any constituent protein can initiate formation of Cajal bodies, and a specific order of assembly is not required. In other words, Cajal bodies can take shape without specific initial conditions. This type of self-organizing system has well-defined scaling laws that arise as a result of stochastic processes. In contrast, the deterministic world is characterized by non-stochastic processes that require specific initial conditions for a certain outcome to arise. Proteins, for example, self-organize into three-dimensional structures, but depend on specific initial conditions, or amino-acid sequence. A recent study found that by altering a small number of critical amino acids, just 5%, the structure and therefore function of a protein can change dramatically (He et al, 2008).
On a macroscopic level, groups of organisms also exhibit self-organization. Fish and birds both form highly organized and sometimes massive collective movements. Fireflies across vast distances emit light flashes absolutely synchronously (Strogatz, 2003). These events are not directed by a leader or top–down process, but occur due to individual adherence to simple rules regarding how to react to environmental signals. In mathematics, the Mandelbrot set, beautiful structures arise from simple mathematical rules (Gleick, 1987). These structures emerge as a result of the application of deterministic rules. However, they show statistical characteristics that are often indistinguishable from random events, and also have well-defined physical structures and scaling laws. In dynamical systems theory, deterministic systems can be self-organizing, but randomness is not essential (S Strogatz, personal communication). A key feature of self-organizing systems is that they converge towards global attractors (see Table I). Stochasticity accelerates the process of self-organization and improves the stability or robustness of the resulting ordered state by allowing the system to escape local basins of attraction (see Table I) and move into global ones. It should also be mentioned that processes in biological systems that are assumed to be stochastic may only seem so due to the complexity of patterns among elements, whereas in truth, deterministic rules govern their behavior.
Emergent features may arise from the interplay between the structure and function of the underlying pattern of connections. The cell is in a meta-stable state—a local attractor—and when it receives specific signals, the system reorganizes into a particular state or form that leads to the global attractor. MyoD could be such a signal for myoblasts, as subsequent to its activation, the cell commits to differentiation, initiates expression of muscle-specific genes, exits the cell cycle, and fuses with other muscle cells to form muscle fibers. During such a process, form and function must mutually evolve and adapt to reach a state where stable function, or terminal differentiation, is achieved. If form is an initiating global trigger, it precedes a functional outcome, which in turn influences form. Such a system might oscillate between form and function until a stable, optimized function emerges. We hypothesize that this process captures the mechanics of self-organization in the nucleus during differentiation. The basic mechanisms underlying self-organization in complex biological networks are still far from clear. However, as discussed below, self-organizing systems can be simplified, while retaining complex information, by deconstruction of their elements into well-defined networks.
Networks
In recent years, there has been a strong upsurge in the study of networks in many disciplines, ranging from computer science and communications to sociology and epidemiology (Newman et al., 2006). A network—a graph (see Table I) in the mathematics literature—is a collection of points (called nodes or vertices), joined by lines (called edges). The edges can be directed or undirected, and weighted or unweighted. Many—perhaps most—natural phenomena can be usefully described in network terms. Biological networks can be considered abstract representations of biological systems that capture their essential characteristics (Barabási and Oltvai, 2004). Interestingly, mathematicians have thought about networks since 1736, when Leonard Euler solved the so-called Königsberg bridge problem (seven bridges connect four land masses in Königsberg, and the question was whether any single path exists that crosses all seven bridges exactly once). Euler's method of abstracting the details of a problem, thereby representing it as a set of nodes or vertices—a graph or network—established the foundation for network theory (Newman et al, 2006). The complexity of a network depends on topological structure, network evolution, node connectivity and diversity, and dynamical evolution (Watts and Strogatz, 1998). The evolving nature of a network is determined by both the dynamical rules governing the nodes and the flow occurring along each edge. The nodes of a network are often dynamical systems evolving according to certain rules, and the edges represent their pairwise interactions. Network nodes can also have self-edges, where edges connect a node to itself (Newman, 2003, 2004). Conceptualization of complexity by representation in terms of networks can provide a general approximation for understanding, modeling, and studying of biological systems.
The behavior of a whole system arises not just from the dynamics of individual components, but also in equal measure from the rules by which the whole is assembled. The emergent property of complex interactions among these elements defines the specific characteristics of an individual cell (Misteli, 2001; Felsenfeld and Groudine, 2003; Kosak and Groudine, 2004a, 2004b). Consider the nucleus a dynamical system (see Table I) composed of many interacting elements, among them networks having variable interactions with each other, for example the networks of coregulated genes and chromosomal interactions (Rajapakse et al, 2009). Thus, the nucleus is self-organized because all interacting elements lead to a defined state, or signature, of that cell type (Misteli, 2001; Kosak et al, 2007; Rajapakse et al, 2009). Networks within the nucleus could rewire in both space and time, if for example the mutual exchange of information between the coregulated gene network and the chromosomal interaction network changes (Rajapakse et al, 2009). Defining elements within the nucleus as networks allows assignment of quantifiable values, and comparison of these values over time may then provide a framework with which to study the process of differentiation as well as how nuclear organization generally affects the properties of a cell. Gene expression data provides the basis for constructing a transcriptome network based on coregulated genes either within or between chromosomes. The Hi-C technique and spectral karyotyping (SKY) (see Table I) determine spatial relationships between whole chromosomes as well as between chromosomal compartments.
The mathematics of networks
Mathematically, a network can be represented by an adjacency matrix, denoted A (see Table I). In the simplest case A is a N × N symmetric matrix (see Table I), where N is the number of vertices (nodes) in the network (Newman, 2003). Most simple networks are binary in nature; that is, the edges between nodes are either present or not. Such networks can be represented by (0, 1) or binary matrices. Let G be a finite, undirected, simple graph with node set V(G)=(1,…,N). The adjacency matrix of G is defined as the N × N matrix AG=(Aij) in which
The matrix is symmetric, as if there is an edge between i and j there is also an edge between j and i. Therefore Aij=Aji. We may also define networks with weighted edges, or weighted adjacency matrices, where some edges represent stronger connections than others (Newman, 2004; Strang, 2009). We restrict ourselves to positive weights and the non-zero elements of the adjacency matrix can therefore be generalized to values other than one to represent stronger and weaker connections. A weighted adjacency matrix can be represented mathematically by a matrix with entries that are not simply zero or 1, but are equal instead to the weights on the edges:
A weight between two nodes can represent any desired measure, such as physical distance or amount of shared information, rather than the presence or absence of a connection. As an example, the Euclidian distance between nodes i and j is the weight between them, which in our case may represent the physical proximity between two chromosomes.
Between-network communication
We define communication between networks by using a global measure of comparing the similarity between their corresponding weighted adjacency matrices (see Table I). If X and Y are two weighted adjacency matrices (e.g. representing two different measures of interaction between pairs of the same set of nodes), and d is the number of nodes in each network, the communication between X and Y can be determined by symmetrized Stein distance (SSD):
which is invariant under both matrix scale transformations and matrix inversion (Kullback, 1959; Rajapakse et al, 2009). Note especially that SSD(X,Y)=0 if and only if X=Y, and can be extended to the case where X and/or Y is singular by using the Moore–Penrose generalized inverse (for this extension and other global measures see Rajapakse and Perlman (2010)).
For example, we define two weighted networks in the nucleus during cellular differentiation (see Table I): chromosomal interaction network (X) and the transcriptome network (Y). Elements in X are a measure of proximity of chromosomes and elements in Y are a measure of gene coregulation. Our claim is that if the overall proximity of chromosomes is related to gene coregulation during differentiation, then the two matrices X and Y are related (communicate), and the distance between X and Y approaches 0. From a statistical perspective, SSD can be used to measure the distance between two covariance matrices and thus compare the similarity between two weighted adjacency matrices (Anderson, 2003; Rajapakse and Perlman, 2010).
KL divergence is often used as a measure of the difference between two distributions (Kullback, 1959; Cover and Thomas, 2006). KL is not symmetric, implying that if two distributions, x and y, are compared, KL(x,y) is not equal to KL(y,x). This comparison therefore does not define a distance, and also requires a designation of one distribution as a reference. The symmetrized version of KL(SKL) does not require such a designation, and because SKL(x,y) is equal to SKL(y,x), this comparison yields a measure of similarity in terms of a distance. SSD is the matrix extension of symmetrized Kullback–Leibler (SKL) distance (Anderson, 2003; Rajapakse et al, 2009; Rajapakse and Perlman, 2010). Intuitively and without rigorous mathematical proof, as SSD(X,Y) decreases, the mutual information between X and Y increases, or the matrices reach high similarity. Thus, this framework captures between-network communication (Box 1).
Quantifying the dynamics of networks that capture nuclear organization during differentiation in a hypothetical example.

X and Y are simplified illustrations of chromosomal interaction and transcriptome networks. Symmetrized stein distance (SSD) is a global measure of similarity between the two networks, SSD(X,Y)t=1 at early time point 1, and SSD(X,Y)t=2 at a later time point 2 during differentiation. Algebraic connectivity (AC) is a measure of within-network connectivity, where AC(X) or (Y) and subscript t=1 or t=2 represent this measure for each of the four networks presented here. As described in the accompanying lower box, networks X and Y are more similar at time point 1 than at time point 2, as shown by shorter and longer SSD measures, respectively. AC(X)t=2, or within network connectivity of X at time point 2, is greater than the connectivity of X at time point 1. In contrast, AC(Y) or the connectivity of network Y does not change over these time points. This may indicate that changes in connectivity within network X, or changes in chromosomal interactions over time, drive divergence and therefore direct system evolution.
Within-network connectivity
Understanding dynamic changes in the nucleus using networks requires global evaluation of connectivity within each network and investigation of how it changes over time. If we have two related networks in an evolving system, an important question is whether changes in within-network connectivity in one network precede changes in the other. If one network does lead, this could imply an important global driving force behind changes in cell function.
The largest eigenvalue (see Table I) of the network adjacency matrix or the second smallest eigenvalue of the Lapalacian matrix (algebraic connectivity) have been used to characterize a variety of dynamical processes on networks (Fiedler, 1973; Newman, 2003; Restrepo et al, 2006). In non-linear oscillator models of synchronization on networks, where the Laplacian matrix arises naturally, the algebraic connectivity gives an indication of ‘synchronizability' or how easily the network will synchronize (Olfati-Saber et al, 2007). The appearance of a giant component in a certain class of directed networks depends on the largest eigenvalue of the network adjacency matrix (Vázquez and Moreno, 2003). These examples show the utility of these two measures in determining within-network connectivity or network organization.
The adjacency matrix is closely related to the Laplacian matrix (Mohar, 1992), which treats the graph as a system of masses coupled by linear springs in place of the edges. Laplacian matrices of graphs are closely related to the Laplacian operator, or the second order differential operator Δf=−div(grad(f)). This relation yields an important bilateral link between the spectral geometry of the Riemannian manifold and graph theory (Mohar, 1992). We now define the Laplacian matrix of a weighted graph, and present it in a more useful form. Given a weighted adjacency matrix A, the Laplacian is defined as the N × N matrix LG=(Lij) in which
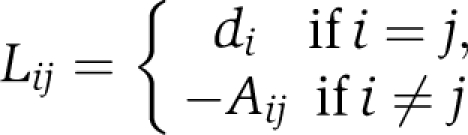 |
Here, di denotes the degree of the node i, in the case of the weighted adjacency matrix where 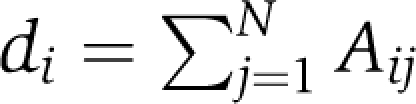 . Thus LG=DG−AG, where DG is the diagonal matrix of the degrees of G. Some features of L are immediate. L does not depend on the diagonal entries of A. It is a symmetric and positive semidefinite matrix and LG1=0, where l is the vector of all ones. Many of the properties of G can be determined from LG. Let 0=λ1⩽λ2⩽…λN be the eigenvalues (see Table I) (Strang, 2009) of LG. The second smallest eigenvalue λ2(LG) is the algebraic connectivity (Fiedler eigenvalue) of the network (Fiedler, 1973). We prefer algebraic connectivity as it does not depend on the diagonal entries of the adjacency matrix and is considered a measure of how well connected a graph is, or degree of connectivity. For one, λ2(LG) is monotonically increasing in the edge set, that is if G1=(N,E1) and G2=(N,E2) are such that
. Thus LG=DG−AG, where DG is the diagonal matrix of the degrees of G. Some features of L are immediate. L does not depend on the diagonal entries of A. It is a symmetric and positive semidefinite matrix and LG1=0, where l is the vector of all ones. Many of the properties of G can be determined from LG. Let 0=λ1⩽λ2⩽…λN be the eigenvalues (see Table I) (Strang, 2009) of LG. The second smallest eigenvalue λ2(LG) is the algebraic connectivity (Fiedler eigenvalue) of the network (Fiedler, 1973). We prefer algebraic connectivity as it does not depend on the diagonal entries of the adjacency matrix and is considered a measure of how well connected a graph is, or degree of connectivity. For one, λ2(LG) is monotonically increasing in the edge set, that is if G1=(N,E1) and G2=(N,E2) are such that 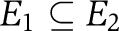 , where both graphs have the same node set with a different edge set, then
, where both graphs have the same node set with a different edge set, then 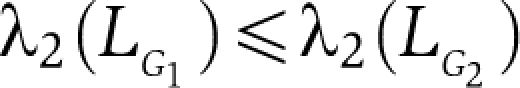 . This implies that the network corresponding to LG2 is more connected, or has greater algebraic connectivity, than the network corresponding to LG1 (Fiedler, 1973; Grone et al, 1990; Mohar, 1992; Yoonsoo and Mesbahi, 2006; Cucker and Smale, 2007; Olfati-Saber et al, 2007). The Laplacian spectrum is applicable more generally to the dynamics of coupled oscillators near the synchronized state, including the relaxation of coupled identical limit-cycle oscillators to equilibrium. When natural frequencies are the same, all oscillators will exponentially synchronize and the rate of approach to a synchronous state as well as the speed of synchronization itself is determined by λ2(LG). Other measures such as the average distance (characteristic path length) can also be used (Newman et al, 2006), and in fact the algebraic connectivity is closely related to the average distance (Mohar, 1992). In our context, we can interpret this to mean that the higher the algebraic connectivity, the higher the network organization. The rationale of using λ2(LG) to measure the network organization is as follows. The basis for constructing the transcriptome network is gene coregulation, and in the differentiated state, lineage-specific genes are more highly coregulated than in the undifferentiated state. As we can think of gene coregulation as a measure of synchronization, we can say that the differentiated state is more synchronized than the undifferentiated state with respect to the lineage-specific genes. For the chromosomal network, we can argue that the optimal spatial configuration is achieved in the differentiated state, where λ2(LG) is maximal (Box 1). Thus, during differentiation, λ2(LG) yields within-network connectivity or network organization.
. This implies that the network corresponding to LG2 is more connected, or has greater algebraic connectivity, than the network corresponding to LG1 (Fiedler, 1973; Grone et al, 1990; Mohar, 1992; Yoonsoo and Mesbahi, 2006; Cucker and Smale, 2007; Olfati-Saber et al, 2007). The Laplacian spectrum is applicable more generally to the dynamics of coupled oscillators near the synchronized state, including the relaxation of coupled identical limit-cycle oscillators to equilibrium. When natural frequencies are the same, all oscillators will exponentially synchronize and the rate of approach to a synchronous state as well as the speed of synchronization itself is determined by λ2(LG). Other measures such as the average distance (characteristic path length) can also be used (Newman et al, 2006), and in fact the algebraic connectivity is closely related to the average distance (Mohar, 1992). In our context, we can interpret this to mean that the higher the algebraic connectivity, the higher the network organization. The rationale of using λ2(LG) to measure the network organization is as follows. The basis for constructing the transcriptome network is gene coregulation, and in the differentiated state, lineage-specific genes are more highly coregulated than in the undifferentiated state. As we can think of gene coregulation as a measure of synchronization, we can say that the differentiated state is more synchronized than the undifferentiated state with respect to the lineage-specific genes. For the chromosomal network, we can argue that the optimal spatial configuration is achieved in the differentiated state, where λ2(LG) is maximal (Box 1). Thus, during differentiation, λ2(LG) yields within-network connectivity or network organization.
Determining the critical node—the most important or central node—in the network and also how perturbation of nodes or edges impacts within-network connectivity (dynamical importance) may provide useful information about network organization (Restrepo et al, 2006). The simplest of centrality measures is degree centrality. The degree di of a node i is the number of its neighbors and is defined as 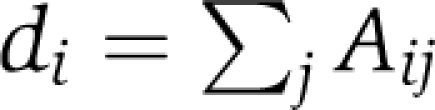 ). (Newman, 2003). Although simple, degree centrality is often a highly effective measure of the influence or importance of a node: in many settings, nodes with more connections tend to have more power (Newman, 2003). We define the dynamical importance (Restrepo et al, 2006) of the edge between nodes i and j, Iij, as
). (Newman, 2003). Although simple, degree centrality is often a highly effective measure of the influence or importance of a node: in many settings, nodes with more connections tend to have more power (Newman, 2003). We define the dynamical importance (Restrepo et al, 2006) of the edge between nodes i and j, Iij, as 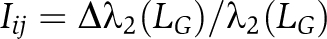 where Δλ2(LG) is the amount λ2(LG) decreases on removal of the edge Iij. Similarly,
where Δλ2(LG) is the amount λ2(LG) decreases on removal of the edge Iij. Similarly, 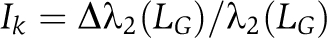 defines the dynamical importance of node k where Δλ2(LG) is the amount λ2(LG) decreases on removal of the node k or the removal of all edges into and out of node k. We can adapt this mathematical framework to identify the chromosome or genes that are most important in defining a given cell type, and quantitative characterization of their dynamical importance will be in terms of their effect on network organization during differentiation.
defines the dynamical importance of node k where Δλ2(LG) is the amount λ2(LG) decreases on removal of the node k or the removal of all edges into and out of node k. We can adapt this mathematical framework to identify the chromosome or genes that are most important in defining a given cell type, and quantitative characterization of their dynamical importance will be in terms of their effect on network organization during differentiation.
Reprogramming the network
As described in the Introduction, previously we used the principles of network theory to test the hypothesis of dynamical genomic organization (Kosak et al, 2007; Rajapakse et al, 2009). Using data sets on gene expression changes during in vitro differentiation of hematopoietic progenitors to derived erythroid and neutrophil cell types (Bruno et al, 2004), we created weighted adjacency matrices (the transcriptome network) for the following: progenitor, (the onset of differentiation), and erythroid or neutrophil (the endpoint of differentiation). For each of these conditions, we also measured the relative proximity of all chromosomes in prometaphase rosettes using SKY and confirmed these proximal relations in interphase nuclei by fluorescent in situ hybridization (Kosak et al, 2007). We used these frequencies of interaction, or how frequent one chromosome is proximal to another, to construct another set of weighted adjacency matrices (chromosomal interaction network) for each condition. On computation of the SSD (described above) between the two matrices in each lineage, the distance between them was close to zero, indicating that gene coregulation was correlated with overall chromosomal organization. This led to the suggestion that the genome—at the level of chromosomes—may self-organize to facilitate coordinate gene regulation during cellular differentiation.
We posit that local interactions (gene coregulation) lead to chromosomal associations that emerge cooperatively in a cell-specific organization of the nucleus, which in turn feeds back to strengthen the local associations. During differentiation, loci containing upregulated genes move from a repressive to an active nuclear compartment, whereas loci containing downregulated genes move in the opposite direction (Brown et al, 1997; Skok et al, 2001; Kosak et al, 2002; Ragoczy et al, 2006). On a local level, movement of loci is often accompanied by the looping of loci from their chromosome territories (Williams et al, 2006). Moreover, global reorganization of chromosome proximities also occurs during differentiation (Kim et al, 2004; Parada et al, 2004; Kosak et al, 2007). However, it is unclear whether local changes in positioning (e.g. looping of loci from chromosome territories to active or repressive compartments) drive global reorganization on the whole chromosome level, or vice versa. In this regard, it has been shown that artificially tethering a 50–100 Kb lacO array to the periphery is sufficient to relocalize the whole chromosome territory (Finlan et al, 2008).
As described in the Introduction, Hi-C (see Box 2) generates a complete map of interactions of all open active or repressed domains (as defined by histone modifications, DNase1 sensitivity, etc.) in the genome at various scales, including inter- and intra- chromosomally, globally (whole chromosome) and locally (loci specific). In the future, combination of Hi-C and interphase SKY may provide a more complete map of local and global spatial proximities with which to construct the chromosomal interaction network. Furthermore, to fully represent the relationships between spatial organization and gene coregulation, it will be critical to investigate this relationship in native gene loci over a time course throughout differentiation, as discussed below for two model systems.
Chromosomal association analysis.
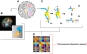
(A) A schematic representation of the three-dimensional genome. (B) Image of a murine hematopoietic progenitor nucleus labeled by spectral karyotyping (SKY). All chromosomes are labeled with a unique color to visualize their territories. Analysis of SKY data reveal spatial relationships between each pair of chromosomes, including distance between centroids and closest distance and also more complex relationships such as shared volume and contact area. Lower right insert is a theoretical magnification of two chromosomal territories. (C) The technique of Hi-C. (C1) DNA is cross-linked and digested with restriction enzymes. (C2) Ends are filled and marked with biotin before the blunt ends are ligated. (C3) In the biotin pull-down step, DNA is sheared and purified before being immunoprecipitated with avidin-conjugated beads. (C4) High-throughput sequencing (8.5 million reads) is used to determine the spatial proximity of sequences, including those on the same or different chromosomes using paired-end sequencing. (D) Both SKY and Hi-C generate spatial proximity maps for inter- and intra-chromosomal interactions.
MyoD
Studying the nucleus in terms of networks may allow us to determine whether there exists a locus or set of loci particularly important to a specific cell lineage, and whether we can predict the fate of (and eventually manipulate) nuclear organization given an understanding of the behavior of a ‘master' gene. Some genes may have global impact on genomic organization in certain cell types, thus conferring the ability to transform from one cell type to another. One candidate is Myogenic differentiation 1 (MyoD), the muscle specific basic-helix-loop-helix transcription factor that can initiate the myogenic program and by forced expression convert fibroblasts into skeletal muscle cells (Davis et al, 1987; Tapscott, 2005).
MyoD is of particular interest, as it is able to convert certain cell types (e.g. mouse embryonic fibroblasts (MEFs)), but not others (e.g. white blood cells) to skeletal muscle (Weintraub et al, 1991). Thus, the MEF regulatory networks must have unique patterns that are permissive to conversion on expression of MyoD. Application of the methods described here may provide insight into whether MyoD achieves reprogramming on a global scale through nuclear reorganization. Using MEFs, or a non-specialized cell type not committed to the myogenic lineage, the effect of forced expression of MyoD on the chromosomal topology and the transcriptome networks can be determined. We suggest that deconstructing the system into these two networks and studying their behavior over time, will reveal whether MyoD is involved in global reorganization of the genome by mathematical criteria, using network theory (Figure 1). MyoD could impose global changes in the genomic landscape through several routes. For example, it is known that MyoD binds E-boxes (see Table I) throughout the genome, in regions known to transcriptionally regulate downstream genes, as well as other E-boxes of unknown function (Tapscott, 2005). Thus, it is possible that changes in the chromosomal topology network resulting from MyoD occupancy of non-regulatory E-boxes precede changes in the MyoD regulated transcriptome network, resulting in divergence of the chromosomal and transcriptome networks. Our recent studies indicate that MyoD can have a much broader function in cell specification, and that its function as a transcription factor regulating expression of skeletal muscle genes represents only a small fraction of its activity (Cao et al, 2010). This broad influence could point to a function for MyoD in reorganization of the genome, which could lead to rewiring of networks within the nucleus, possibly changing the accessibility of additional E-boxes or affinity of MyoD for specific targets (a cooperative effect). Thus, both chromatin conformation and the spatial arrangement of chromosomes may facilitate activation of specific subsets of MyoD targets. Over time, global rewiring effects of MyoD may make a cell type amenable to skeletal muscle differentiation, given appropriate environmental cues. In this case, the transcriptome network will gradually increase connectivity to match that of the chromosomal network. Thus, the initial state or signature of the cell type involved in forced myogenic conversion via MyoD may affect its receptivity to artificial imposition of trans-differentiation (Figure 3), which could explain why some cell types convert more readily than others (Tapscott, 2005). This transition to a new MyoD-dependent pattern (or steady state) can be captured quantitatively. Divergence and convergence—network communication—between the networks can be evaluated, as shown in Figure 2, where each set of chromosomal and transcriptome networks at a given time point are represented by unweighted adjacency matrices. Measuring each network's within-network connectivity over time should indicate whether one changes first and modifies between-network communication. From this framework, whether a change in the chromosomal topology network precedes or follows that of the transcriptome network can be quantified (Figures 1F and 2A).
Figure 1.

Reprogramming: a network view. Network diagrams (A–C) are representations of collective information from chromosomal topology (D) and transcriptome (E) networks, that is the biological network, where nodes are genes or chromosomes. Thus, we define the network in (A) as a nuclear circuit, composed of the networks in (D, E) (rectangular outline). (A) A biological network representing the specific network signature of the MEF, where some genes are connected, and some are not. The connectivity is weighted, that is the strength of the connection depends on the gene pair, shown by edge thickness. For example, connectivity between genes 2 and 5 is weaker than between 2 and 3. MyoD (the red node) is part of the network but in this case has no connection with other genes as it not expressed. (B) Activated MyoD establishes connections with the rest of the genes and initiates rewiring, accompanied by an increase in MyoD binding affinity. (C) The differentiated system has unique network architecture; new edges appear, resulting in a new network. Note that between (B) and (C) other network structures exist but we have only shown a few points during the process. (D, E) (Within-network organization): the MEF network initially exists in a unique pattern before induction of MyoD. After MyoD induction, the chromosomal network begins to rewire, increasing its connectivity and indicating changes in spatial architecture. The transcriptome network lags behind but gradually increases connectivity, and reaches a network state similar to the chromosomal network (higher communication). (F) Schematic representation of the question: is form a precondition for, or does form follow, function? The fibroblast and myoblast differ in nuclear architecture. In both cell types, actively transcribed genes (red circles) will be localized in active regions of chromatin (blue circles) and show a network of physical adjacencies (edges) within other regions of active chromatin. If form follows function, then muscle gene expression will precede or coincide with localization of a gene to an active region and rearrangement of nuclear adjacencies; whereas if function follows form, then repositioning of genes to active regions and rearrangement of nuclear adjacencies will precede gene transcription.
Figure 2.

A quantitative view of how spatial arrangement of chromosomes may influence gene expression, and may precede nuclear reprogramming. (A) The blue networks on the left represent chromosomal spatial arrangement over six time points during reprogramming or differentiation, and the yellow networks represent gene expression. (B) Each connection (edge) between nodes within the networks in (A) at the first time point is assigned a value of one, and absence of an edge is assigned a value of zero in the unweighted adjacency matrices shown. This can be easily extended to the weighted case, where an edge can be assigned numerical values according to the strength of the connection. Equations for the computation of algebraic connectivity between adjacency matrices are given and are plotted in (C) for the six time points. In this case, the network organization is initially similar, but the chromosomal network precedes the expression network in organization. After the fifth time point, the two networks, possibly by an iterative communication process, converge on a unique steady state. This concept is also illustrated in Figure 3, where the initial networks at time point 1 exist within steady state 1, and the final networks at time point 6 exist within steady state 2.
GATA-1
Another ‘master regulator' is GATA-1, a zinc-finger transcription factor essential to the maintenance of the erythroid and megakaryocyte lineages (Orkin, 1992). GATA-1 may have a global impact on nuclear organization by catalyzing interactions within and between the coregulated gene and chromosome topology networks. Thus, as for MyoD, studying the effect of GATA-1 on nuclear organization and gene coregulation in terms of networks during a time course of hematopoietic stem cell differentiation would provide a convenient framework for understanding the genome-wide influence of GATA-1 in specific lineages. Cheng et al (2009) have begun to address this question using Chip-seq methods to identify the spatial distribution of cis-regulatory elements targeted by GATA-1, and they determine criteria for distinguishing between target sites that promote activation versus repression of genes during erythroid development.
Disrupting the network
Mutations in GATA-1 have been associated with development of acute megakaryoblastic leukemia (Wechsler et al, 2002; Shimizu et al, 2008). Such mutations may dysregulate the global influence of a lineage-specific transcription factor, and thereby disrupt appropriate maintenance of the lineage. Consider Figure 3, in which a critical factor such as MyoD or GATA-1 induces a state shift from one basin of attraction to another—switching steady states (Kitano, 2007; MacArthur et al, 2009)—with only transient instability in the system (i.e. differentiation of a normal cell). One possibility is that mutations in the factor alter this shift in such a way that instead of differentiation or regulated proliferation, no stable state is reached, resulting in a continuously evolving state without entering a basin of attraction, that is reflected by long term or permanent genomic instability (i.e. cancer cell). Defining the state or overall pattern of instability of this cell may give insight into how to redirect it back toward a basin of attraction. This can be described not only in terms of transcriptional networks, but also using spatial characteristics (chromosomal networks) of the unstable genome. With knowledge of critical nodes or edges in a network comes the opportunity to repair dysfunctional connections, and target high impact connections to restore a disrupted network in a disease state. Furthermore, the methods described in the within-network connectivity section can be used to design networks with specific dynamical properties and evaluate the effects of therapies that target specific nodes or edges. An existing network might be rewired through removal (knockdown), addition (overexpression), or swapping pairs of edges (translocations). This understanding could be the key to achieving global reprogramming of an abnormal cell to a normal cell.
Figure 3.

Schematic illustration for the mechanics of self-organization and differentiation. Right: a nuclear circuit demonstrating organization during cellular differentiation. Local interactions (gene coregulation) lead to chromosomal associations that emerge cooperatively in cell-specific organization of the nucleus, which in turn feeds back to strengthen the local associations, and the self-organized system fine-tunes over time. Left: the initial state of this system is at the center of steady state 1. A perturbation, such as activation of a specific signaling pathway or shown here as induction of the transcription factor MyoD, may drive the state of the system towards the boundary of the basin of attraction of stable state 1 (locally stable state). A basin of attraction is a set of initial conditions that ultimately lead to behavior that approaches a specific state (the attractor). In other words, the system approaches cell-specific organization. When it transitions to stable state 2 (globally stable state), stability is lost and the system regains its stability only in the new steady state. In the case of MyoD, induction in MEF cells leads to a state transition into the myogenic lineage. The system is considered to be robust if its functions are still intact, regardless of whether it is in stable state 1 or 2. In an extreme case, the system may continue to transition between multiple stable state points to cope with ongoing perturbations.
MyoD and GATA-1 are two model systems that offer the opportunity to distinguish whether form precedes or follows function. For example, if form precedes function, chromosomal topology changes first and as a result gene coregulation is facilitated. In this case, these two networks then initiate communication that drives the transition into a new basin of attraction and stabilization in a new steady state (terminal differentiation). It is possible that in cancer cells, disrupted chromosomal topology leads to loss of communication between networks. Control over cell fate and function is therefore disrupted, and, as explained above, the ability to smoothly transition into a new steady state is lost. The network approaches outlined here allow exploration of whether, during differentiation, cells maintain the predicted interactions at both the gene and chromosomal levels. We can also take cues from our understanding of induced pluripotent stem cells, which can be reprogrammed from fibroblasts into a less specialized state with only four factors (Nakagawa et al, 2008; Yamanaka, 2009). This can also be viewed as a transition between steady states. A quantitative framework to define nuclear steady states may provide information important for determining factors with the highest potential for changing the way an unstable cell behaves, or how genomic instability in cancer cells may be controlled.
‘Networking the nucleus' provides a unique opportunity to investigate the principles of complex processes and emergence of self-organization in biological systems. Ultimately, we may gain insight into how global genomic organization distinguishes stem or progenitors from the differentiated cell as well as a disease state. By deconstructing a system into a network, even in the simplest case, we can capture features of complex systems that linear models simply are not able to accommodate. Using these methods, we can study the dynamic connections within and between networks during cell differentiation and develop more sophisticated models of nuclear function.
Outlook
The nucleus may be best described as a self-organizing system, but it is unclear how to quantify the underlying mechanism. Deconstructing nuclear architecture into well-defined networks—networking the nucleus—and studying the connections and communication between networks provides a useful new framework for investigating complex four-dimensional nuclear organization. Studying the nucleus as a set of interconnected networks will help us to understand not only how the nucleus operates, responds to cellular cues, and adapts to environmental changes, but how networked systems behave. Evidence suggests that genes communicate with each other in space, and communication patterns rewire over time, driving specific topological organization of chromosomes that ensures efficient and coordinated expression of sets of genes (Kosak and Groudine, 2004b; Takizawa et al, 2008; Rajapakse et al, 2009). Coordination of the activities of individual dynamic elements enables such a system to develop unique patterns, sustain complexity at a higher level, and evolve. The function that the organization of the nucleus has in its function has become an increasingly important question. We believe that understanding nuclear networks will provide insight into topics ranging from the regulation of gene expression, to stem cell biology, to the basis for differentiation and cellular reprogramming. Although still in initial stages of development, integration of more sophisticated technical methods with complex network theory open new avenues for investigating topological structure and its impact on the dynamic function of the nucleus.
Acknowledgments
We thank Lindsey Muir, Joan Ritland Politz, Daniel Strongin, Tobias Ragoczy, Michael Perlman, and Jon Cooper for discussion and critical reading of the paper; Job Dekker and Nynke L van Berkam for providing Box 2 (A). IR is supported by the Mentored Quantitative Research Career Development Award (K25) from National Institutes of Health (NIH) grant 1K25DK082791-01A109, STK by a CABS award from the Burroughs Wellcome Fund, and MG by NIH grants R37 DK44746 and RO1 HL65440.
Footnotes
The authors declare that they have no conflict of interest.
References
- Anderson PW (1972) More is different. Science 177: 393–396 [DOI] [PubMed] [Google Scholar]
- Anderson TW (2003) An Introduction to Multivariate Statistical Analysis. New York: Wiley [Google Scholar]
- Ashby WR (1947) Principles of the self-organizing dynamic system. J Gen Psychol 37: 125–128 [DOI] [PubMed] [Google Scholar]
- Barabási A-L, Oltvai ZN (2004) Network biology: understanding the cell's functional organization. Nat Rev Genet 5: 101–113 [DOI] [PubMed] [Google Scholar]
- Bolzer A, Kreth G, Solovei I, Koehler D, Saracoglu K, Fauth C, Müller S, Eils R, Cremer C, Speicher MR, Cremer T (2005) Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol 3: e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri T (1909) Die blastomerenkeme von ascaris megalocephala and die theorie der chromosomenindividualitat. Arch Exp Zellforsch 3: 181–268 [Google Scholar]
- Boyle S, Gilchrist S, Bridger JM, Mahy NL, Ellis JA, Bickmore WA (2001) The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum Mol Genet 10: 211–219 [DOI] [PubMed] [Google Scholar]
- Brown K, Guest S, Smale S, Hahm K, Merkenschlager M, Fisher A (1997) Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell 91: 845–854 [DOI] [PubMed] [Google Scholar]
- Bruno L, Hoffmann R, McBlane F, Brown J, Gupta R, Joshi C, Pearson S, Seidl T, Heyworth C, Enver T (2004) Molecular signatures of self-renewal, differentiation, and lineage choice in multipotential hemopoietic progenitor cells in vitro. Mol Cell Biol 24: 741–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camazine S, Deneubourg L, Franks N, Sneyd J, Theraulaz G, Bonabeau E (2003) Self-Organization in Biological Systems. Princeton, NJ: Princeton University Press [Google Scholar]
- Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, Gentleman RC, Tapscott SJ (2010) Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev Cell 18: 662–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Wu W, Ashok Kumar S, Yu D, Deng W, Tripic T, King DC, Chen K-B, Zhang Y, Drautz D, Giardine B, Schuster SC, Miller W, Chiaromonte F, Zhang Y, Blobel GA, Weiss MJ, Hardison RC (2009) Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res 19: 2172–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang C-H, Carpenter AE, Fuchsova B, Johnson T, de Lanerolle P, Belmont AS (2006) Long-range directional movement of an interphase chromosome site. Curr Biol 16: 825–831 [DOI] [PubMed] [Google Scholar]
- Cover T, Thomas J (2006) Elements of Information Theory. New York: Wiley-Interscience [Google Scholar]
- Cremer M, von Hase J, Volm T, Brero A, Kreth G, Walter J, Fischer C, Solovei I, Cremer C, Cremer T (2001) Non-random radial higher-order chromatin arrangements in nuclei of diploid human cells. Chromosome Res 9: 541–567 [DOI] [PubMed] [Google Scholar]
- Cremer T, Cremer C, Baumann H, Luedtke EK, Sperling K, Teuber V, Zorn C (1982) Rabl's model of the interphase chromosome arrangement tested in Chinise hamster cells by premature chromosome condensation and laser-UV-microbeam experiments. Hum Genet 60: 46–56 [DOI] [PubMed] [Google Scholar]
- Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA (1999) Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol 145: 1119–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucker F, Smale S (2007) Emergent behavior in flocks. IEEE Trans Automat Contr 52: 852–862 [Google Scholar]
- Davis RL, Weintraub H, Lassar AB (1987) Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51: 987–1000 [DOI] [PubMed] [Google Scholar]
- Dekker J (2008) Gene regulation in the third dimension. Science 319: 1793–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, Kleckner N (2002) Capturing chromosome conformation. Science 295: 1306–1311 [DOI] [PubMed] [Google Scholar]
- Dostie Je, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, Green RD, Dekker J (2006) Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res 16: 1299–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr M, Ospina JK, Sung M-H, John S, Upender M, Ried T, Hager GL, Matera AG (2007) Actin-dependent intranuclear repositioning of an active gene locus in vivo. J Cell Biol 179: 1095–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenfeld G, Groudine M (2003) Controlling the double helix. Nature 421: 448–453 [DOI] [PubMed] [Google Scholar]
- Fiedler M (1973) Algebraic connectivity of graphs. Czechoslov Math J 23: 298–305 [Google Scholar]
- Finlan LE, Sproul D, Thomson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA (2008) Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet 4: e1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleick J (1987) Chaos: Making a New Science. New York: Penguin Books [Google Scholar]
- Grone R, Merris R, Sunder VS (1990) The Laplacian spectrum of a graph. SIAM J Matrix Anal Appl 11: 218–238 [Google Scholar]
- He Y, Chen Y, Alexander P, Bryan PN, Orban J (2008) NMR structures of two designed proteins with high sequence identity but different fold and function. Proc Natl Acad Sci 105: 14412–14417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser TE, Intine RV, Dundr M (2008) De novo formation of a subnuclear body. Science 322: 1713–1717 [DOI] [PubMed] [Google Scholar]
- Kauffman SA (1984) Emergent properties in random complex automata. Physica D: Nonlinear Phenomena 10: 145–156 [Google Scholar]
- Kim SH, McQueen PG, Lichtman MK, Shevach EM, Parada LA, Misteli T (2004) Spatial genome organization during T-cell differentiation. Cytogenet Genome Res 105: 292–301 [DOI] [PubMed] [Google Scholar]
- Kitano H (2007) Towards a theory of biological robustness. Mol Syst Biol 3: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosak ST, Groudine M (2004a) Form follows function: the genomic organization of cellular differentiation. Genes Dev 18: 1371–1384 [DOI] [PubMed] [Google Scholar]
- Kosak ST, Groudine M (2004b) Gene order and dynamic domains. Science 306: 644–647 [DOI] [PubMed] [Google Scholar]
- Kosak ST, Scalzo D, Alworth SV, Li F, Palmer S, Enver T, Lee JSJ, Groudine M (2007) Coordinate gene regulation during hematopoiesis is related to genomic organization. PLoS Biol 5: e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosak ST, Skok JA, Medina KL, Riblet R, Le Beau MM, Fisher AG, Singh H (2002) Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science 296: 158–162 [DOI] [PubMed] [Google Scholar]
- Kullback S (1959) Information Theory and Statistics. New York: John Wiley and Sons [Google Scholar]
- Langton CG (1990) Computation at the edge of chaos: phase transitions and emergent computation. Physica D: Nonlinear Phenomena 42: 12–37 [Google Scholar]
- Lewin R (1992) Complexity: Life at the Edge of Chaos. New York: Macmillan [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur BD, Máayan A, Lemischka IR (2009) Systems biology of stem cell fate and cellular reprogramming. Nat Rev Mol Cell Biol 10: 672–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister P, Towbin BD, Pike BL, Ponti A, Gasser SM (2010) The spatial dynamics of tissue-specific promoters during C. elegans development. Genes Dev 24: 766–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T (2001) The concept of self-organization in cellular architecture. J Cell Biol 155: 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T (2007) Beyond the sequence: cellular organization of genome function. Cell 128: 787–800 [DOI] [PubMed] [Google Scholar]
- Misteli T (2008) Cell biology: nuclear order out of chaos. Nature 456: 333–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohar B (1992) Laplace eigenvalues of graphs—a survey. Discrete Math 109: 171–183 [Google Scholar]
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S (2008) Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 26: 101–106 [DOI] [PubMed] [Google Scholar]
- Neusser M, Schubel V, Koch A, Cremer T, Müller S (2007) Evolutionarily conserved, cell type and species-specific higher order chromatin arrangements in interphase nuclei of primates. Chromosoma 116: 307–320 [DOI] [PubMed] [Google Scholar]
- Newman MEJ (2003) The structure and function of complex networks. SIAM Rev 45: 167–256 [Google Scholar]
- Newman MEJ (2004) Analysis of weighted networks. Phys Rev E 70: 056131. [DOI] [PubMed] [Google Scholar]
- Newman MEJ, Barabasi AL, Watts DJ (2006) The Structure and Dynamics of Networks. Princeton, NJ: Princeton University Press [Google Scholar]
- Olfati-Saber R, Fax JA, Murray RM (2007) Consensus and cooperation in networked multi-agent systems. Proc IEEE 95: 215–233 [Google Scholar]
- Orkin S (1992) GATA-binding transcription factors in hematopoietic cells. Blood 80: 575–581 [PubMed] [Google Scholar]
- Parada L, McQueen P, Misteli T (2004) Tissue-specific spatial organization of genomes. Genome Biol 5: R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl C (1885) Uber Zelltheilung. Morphol Jahrb 10: 214–330 [Google Scholar]
- Ragoczy T, Bender MA, Telling A, Byron R, Groudine M (2006) The locus control region is required for association of the murine Î2-globin locus with engaged transcription factories during erythroid maturation. Genes Dev 20: 1447–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse I, Perlman D (2010) A general framework for testing equality of covariance matrices. In Technical Reports. Seattle: University of Washington [Google Scholar]
- Rajapakse I, Perlman MD, Scalzo D, Kooperberg C, Groudine M, Kosak ST (2009) The emergence of lineage-specific chromosomal topologies from coordinate gene regulation. Proc Natl Acad Sci 106: 6679–6684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo JG, Ott E, Hunt B (2006) Characterizing the dynamical importance of network nodes and links. Phys Rev Lett 97: 094102. [DOI] [PubMed] [Google Scholar]
- Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, Eskiw CH, Luo Y, Wei CL, Ruan Y, Bieker JJ, Fraser P (2009) Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet 42: 53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu R, Engel JD, Yamamoto M (2008) GATA1-related leukaemias. Nat Rev Cancer 8: 279–287 [DOI] [PubMed] [Google Scholar]
- Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W (2006) Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet 38: 1348–1354 [DOI] [PubMed] [Google Scholar]
- Skok JA, Brown KE, Azuara V, Caparros M-L, Baxter J, Takacs K, Dillon N, Gray D, Perry RP, Merkenschlager M, Fisher AG (2001) Nonequivalent nuclear location of immunoglobulin alleles in B lymphocytes. Nat Immunol 2: 848–854 [DOI] [PubMed] [Google Scholar]
- Strang G (2009) Introduction to Linear Algebra. Wellesley, MA: Cambridge Press [Google Scholar]
- Strogatz S (1994) Nonlinear Dynamics and Chaos: With Applications to Physics, Biology, Chemistry, and Engineering. Cambridge: Perseus Books [Google Scholar]
- Strogatz SH (2001) Exploring complex networks. Nature 410: 268–276 [DOI] [PubMed] [Google Scholar]
- Strogatz S (2003) Sync: The Emerging Science of Spontaneous Order. New York: Hyperion [Google Scholar]
- Takizawa T, Meaburn KJ, Misteli T (2008) The meaning of gene positioning. Cell 135: 9–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ (2005) The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 132: 2685–2695 [DOI] [PubMed] [Google Scholar]
- Vázquez A, Moreno Y (2003) Resilience to damage of graphs with degree correlations. Physical Review E 67: 015101 [DOI] [PubMed] [Google Scholar]
- Watts DJ, Strogatz SH (1998) Collective dynamics of ‘small-world' networks. Nature 393: 440–442 [DOI] [PubMed] [Google Scholar]
- Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, Crispino JD (2002) Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 32: 148–152 [DOI] [PubMed] [Google Scholar]
- Weintraub H, Davis R, Tapscott S, Thayer M, Krause M, Benezra R, Blackwell TK, Turner D, Rupp R, Hollenberg S (1991) The myoD gene family: nodal point during specification of the muscle cell lineage. Science 251: 761–766 [DOI] [PubMed] [Google Scholar]
- Williams RRE, Azuara V, Perry P, Sauer S, Dvorkina M, Jorgensen H, Roix J, McQueen P, Misteli T, Merkenschlager M, Fisher AG (2006) Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J Cell Sci 119: 132–140 [DOI] [PubMed] [Google Scholar]
- Yamanaka S (2009) A fresh look at iPS cells. Cell 137: 13–17 [DOI] [PubMed] [Google Scholar]
- Yoonsoo K, Mesbahi M (2006) On maximizing the second smallest eigenvalue of a state-dependent graph Laplacian. IEEE Trans Automat Contr 51: 116–120 [Google Scholar]
- Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, Kanduri C, Lezcano M, Singh Sandhu K, Singh U, Pant V, Tiwari V, Kurukuti S, Ohlsson R (2006) Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet 38: 1341–1347 [DOI] [PubMed] [Google Scholar]