

Abstract
BACKGOUND
Previous studies have shown increased expression of AChR alpha (AChR-α) subunit transcripts in myasthenia gravis (MG) and experimental MG (EAMG) but have not examined the functional properties of this overexpression.
METHODS
We examined the mRNA and protein expression of AChR-α, as well as the pattern of α-bungarotoxin labeling in muscle tissue from EAMG mice with varying disease severity.
RESULTS
AChR-α expression was considerably increased in endplates from mice with severe EAMG, but it was distinct and greatly in excess of α-bungarotoxin labeling. This “aberrant expression” occurred in mice with morphologic endplate damage, and the pattern of complement and immunoglobulin deposition in muscle from these mice appeared to mirror the pattern of AChR-α expression.
CONCLUSIONS
The loss of functional AChR in severe MG increases transcription of AChR-α mRNA, but the expressed protein is “functionally inert,” failing to compensate for loss of AChR. This enhanced expression of AChR may play a role in driving the ongoing autoimmune response.
Keywords: Myasthenia gravis, experimental autoimmune myasthenia gravis, acetylcholine receptor, acetylcholine receptor alpha subunit
Introduction
Myasthenia gravis (MG) and its animal model, experimental autoimmune MG (EAMG), are caused by autoantibodies directed against the acetylcholine receptor on the postsynaptic muscle membrane.22 Anti-AChR antibodies bind to the AChR to cause receptor internalization and degradation, as well as complement-mediated lysis of the postsynaptic membrane and consequent loss of functional receptors.7,22 Previous studies have indicated that there may be a compensatory increase in expression of acetylcholine receptor (AChR) mRNA in response to antibody-mediated AChR loss in EAMG and MG.1,2,10,11 The consequences of this enhanced expression of AChR are not known, and we do not know whether it effectively compensates for the loss of functional receptor.
In this study, we confirm enhanced expression of AChR-alpha (AChR-α) subunit in mice with severe EAMG. We proceed to hypothesize that while the enhanced expression of the AChR-α subunit is likely a compensatory response to AChR loss, the expressed protein does not allow for effective AChR function and binding of acetylcholine or other agonists. To test this hypothesis we examined the co-localization of AChR-α expression and AChR ligand-binding using radiolabelled alpha-bungarotoxin (α-BTX) in muscle tissue from mice with EAMG. We found that the expression of AChR-α frequently occurred separately from α-BTX-labeling. This suggests that the enhanced expression results in “functionally inert” AChR, since these receptors were unable to effectively bind cholinergic agonist. While the functional consequence of this upregulated expression of functionally inert AChR proteins on the course of MG is not clear, a possible effect on the ongoing autoimmune response is hypothesized.
Methods
Induction of EAMG
Torpedo AChR (tAChR) was purified from the electric organs of Torpedo californica by affinity chromatography using a conjugate of neurotoxin coupled to agarose, as previously described.6,25 Purity of the isolated product was tested by SDS-PAGE. The purified tAChR was used to induce EAMG as detailed below.
Eight-week old female C57BL6/J mice (Jackson Laboratories, Bar Harbor, ME) were immunized with 40 μg of tAChR/CFA, 200ul, subcutaneously, and boosted with 20μg of tAChR emulsified in IFA in 200 μl volume injected in the flanks and tail base every 30 days. Control mice received an equal volume of PBS in CFA or IFA. Mice were observed and scored every other day after initial priming and two booster immunizations (day 60). At this point the mice were divided into three groups consisting of equal numbers of mice (at least three mice per group). Group 1 include CFA control mice; group 2 included mice with mild disease (score 1); and group 3 included mice with severe disease (score ≥2).
For clinical evaluation of disease severity, mice were scored as described previously.18,19 Briefly, they were observed on a flat platform for a total of two minutes. They were then exercised by gentle dragging while they were suspended by the base of the tail across a cage top grid repeatedly (20–30 times) as they attempted to grip the grid. Mice were then placed on a flat platform for two minutes and again observed for signs of EAMG. Clinical muscle weakness was graded as follows: grade 0, mouse with normal posture, muscle strength, and mobility at baseline and after exercise; grade 1, normal at rest but with muscle weakness characteristically shown by a hunchback posture, restricted mobility, and difficulty in raising the head after exercise; grade 2, grade 1 symptoms without exercise during observation period; grade 3, severely impaired mobility, paralysis, dehydration; and grade 4, moribund. We also measured the serum concentration of anti-mouse AChR Abs by ELISA using affinity-purified mouse AChR as antigen, as previously described.18,19 All animal studies were carried out as per the protocol approved by the animal care and use committee of the University of Illinois at Chicago.
RT-PCR
Mouse muscle RNA was extracted from limb muscle homogenates or from the C2C12 mouse cell line using TRIzol®, and cDNA synthesized using Thermoscript RT-PCR kit (Invitrogen). RNA from human skeletal muscle was purchased from BD Bioscience. The first set of primers for mouse AChR-α1 subunit (NM_007389) were designed as follows: sense primer [5′ CCTACCTGGACATCACCTAC 3′]; anti-sense [5′ ATGGACGCAATGACAAAGAC 3′]; the PCR product length is 277bp. The mouse primer spanning the P3A exon of mouse AChR-α subunit (NM_007389) was designed as follows: sense primer [5′ATGGAGCTCTCGACTGTTCT3′], antisense primer [TCACTTTCCGGGTTAATGGC] with an expected product length of 548 bp. Finally human primer spanning the P3A exon of human AChR-α subunit (NM_001039523) was designed as follows: sense primer: [5′TCCTCCTGCTCTTTAGCCTTTGCT3′], antisense primer [5′CCAGCTTCATGCTGCAGTTCTGTT3′]; the PCR products are 558bp and 483bp. Amplification was performed by denaturation at 95°C for 30s, annealing at 50 °C for 20s, and extension at 72°C for 30s for 35 cycles. All samples were subjected to electrophoresis using a 1.5–2% agarose gel to confirm the specificity of the PCR, and relative quantification of the resulting bands was assessed as the ratio of the densitometric values of bands containing the AChR-α subunit transcript compared to the housekeeping gene, GAPDH. Three mice per group were used in most experiments, which were repeated three times to ensure reproducibility.
Immunohistochemical Staining and Confocal microscopy
Mice were anesthetized with 50 mg/kg sodiumpentobarbital and sacrificed on day 60. Mouse tibialis anterior muscle samples were collected from mice in each of the three experimental groups: CFA-immunized controls, tAChR-immunized mice with mild disease, and tAChR-immunized mice with severe disease (total 12 mice). Muscle samples were frozen in liquid nitrogen and stored at −80°C. Sections (10uM) were taken and fixed in cold acetone. After blocking, the slides were incubated with tetramethylrhodamine-conjugated α-BTX (Molecular Probes, Inc. Eugene, OR) (1/500 dilution) at room temperature to label functional AChR at the NMJ. Slides were then incubated with goat polyclonal antibody against AChR-α subunit (C-18, 1: 200 dilution) and donkey anti-goat IgG-FITC (1:200) (Santa Cruz Biotechnology, Inc) for detection of AChR-α subunit expression in mouse muscle. The corresponding slides were stained with anti-mouse IgG FITC and anti-mouse C3 FITC as previously described.19 The sections were washed with PBS and viewed using a fluorescence microscope (Zeiss LSM510 Laser scanning microscopy). Endplate areas were identified as regions of tetramethylrhodamine-conjugated α-BTX staining. A minimum of 15 sites per muscle with 1–3 endplates per site were evaluated. All sections were stained and processed in parallel to avoid inter-assay variations. Magnification for all images is 63×10×2.5; bars are 20 μm.
Electron Microscopy
Tibialis anterior musclesamples from 3 mice per group was removed and fixed using 2.5% glutaraldehyde in0.1 M phosphate buffer, pH 7.4 in 4C. The samples were sectioned and post-fixed with 1% osmium tetroxidein 0.1 M phosphate buffer, pH 7.4, dehydrated through a gradedethanol series and embedded in epoxy resin. Endplates were located in toluidineblue-stained 1 micron semi-thin sections from the central region of eachmuscle. Ultra-thin sections (Leica ultracut UCT, Leica Microsystems, Wetzlar, Germany) from selected areas were contrastedwith uranyl acetate and lead citrate and viewed with an electron microscope (JEM-1220 Electron Microscope, Jeol USA, Inc., Peabody, MA). A minimum of 31 endplate regions with clearly defined nerve terminals and postsynaptic membranes were evaluated per muscle. Endplates were graded qualitatively as “normal” “mildly abnormal” or “severely abnormal”. The criteria for an “abnormal” endplate required a readily visible simplification of the postsynaptic membrane structure with reduction or loss of the normal postsynaptic folding pattern in a photographed endplate region with a clearly defined nerve terminal and postsynaptic membrane. Utilizing Image J software, quantitative assessments of neuromuscular junction morphology were made by measuring postsynaptic membrane lengths (PostSM) and presynaptic membrane lengths (PreSM) and calculating the ratio: (PostSM)/(PreSM) according to the method of Engel and Santa.9 Digital micrographs were taken with Gatan slow-scan CCD camera.
Results
AChR-α mRNA is over-expressed in muscle from mice with moderate to severe MG
We used RNA extracted from mouse muscle homogenates and performed RT-PCR using AChR-α-specific primers as described in the Materials and Methods section. Analyses were performed in a total of twenty mice with EAMG and five control animals.
Figure 1A shows representative data illustrating the increased expression of AChR-α transcripts in muscle from EAMG mice compared to controls. Figure 1B shows that this increased expression is most prominent in mice with severe EAMG.
Figure 1.
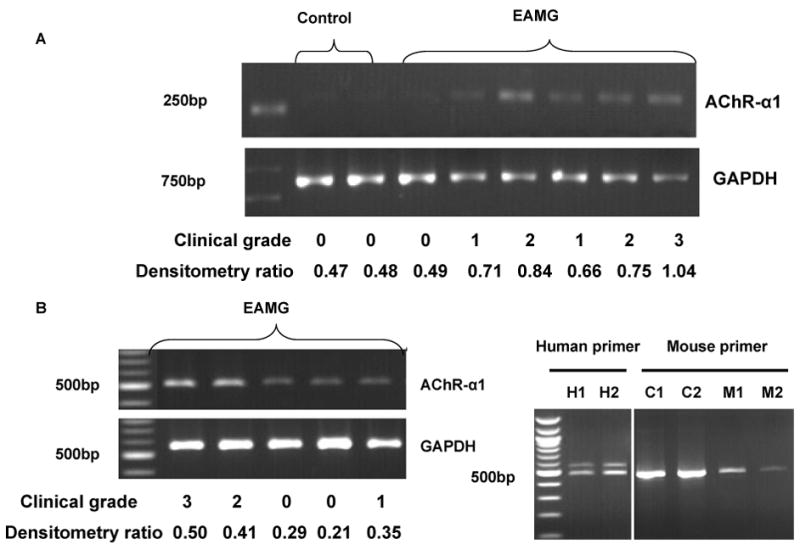
RT-PCR results of mRNA isolated from limb muscles of mice with EAMG. A. Increased mRNA expression of AChR-α1 in muscle homogenates from EAMG mice compared to control animals. B. Enhanced expression of AChR-α1 transcript is most prominent in mice with more severe EAMG. C. RT-PCR results using primers designed to detect the presence of the P3A exon. Two AChR-α1 isoforms are clearly detected in human muscle tissue (H1, H2). However, a single isoform is expressed in the C2C12 mouse skeletal muscle cell line (C1, C2) and in muscle tissue from two mice with EAMG (M1, M2).
In human muscle the AChR-α subunit is found in two isoforms, one being homologous to that expressed in other species, and the other containing an exon termed P3A, which has not been clearly identified in other species.4 The latter isoform is not integrated into functional AChRs and does not bind α-BTX.15 We specifically looked for the expression of the P3A+ isoform using primers designed to detect the presence of the P3A exon based on both mouse and human sequences. We found that the over-expression observed in EAMG muscle could not be accounted for by enhanced expression of the P3A+ isoform.
Over-expressed AChR-α in severe EAMG does not effectively bind α-BTX
To analyze the functional properties of over-expression of the AChR-α subunit, we performed immunohistochemical staining of mouse tibialis anterior muscle to examine the expression of AChR-α subunit using labeled anti-AChR-α antibodies. We simultaneously determined the ligand-binding capacity of intact AChRs using labeled α-BTX. As shown in figure 2, in control animals, the expression of AChR-α subunit and labeling of AChR binding sites by α-BTX were completely co-localized, as one would expect. In muscle from EAMG mice, the expression of AChR-α subunit occurred separate from α-BTX labeling. This aberrant over-expression was most prominent in mice with severe EAMG, in which AChR-α expression appeared to outline the periphery of muscle fibers in some cases (figure 2).
Figure 2.
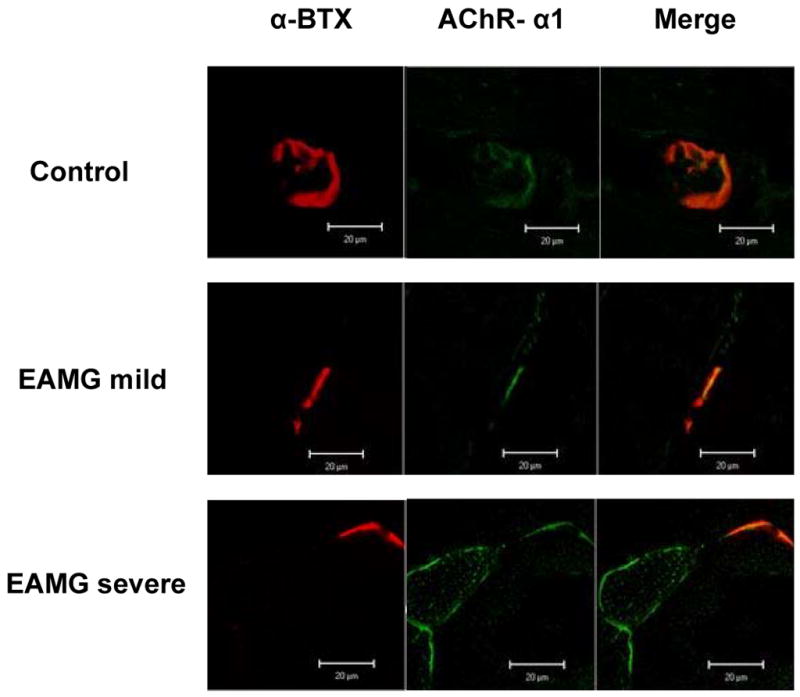
Immunohistochemical studies of AChR-α expression and ligand-binding ability at the mouse muscle endplate. Cryosections of mouse limb muscle were double-stained with tetramethylrhodamine-conjugated α-BTx (first column) and goat polyclonal antibody against AChR-α1 (second column); merge on the right. Representative data from each experimental group (Control, EAMG mild, and EAMG severe) are shown. Mice with severe EAMG show a high level of aberrant AChR protein expression (i.e., expression separate from functional ACh binding as determined by absence of α-BTx binding). Mice with mild EAMG either have normal (expression only in association with α-BTx binding) or very low levels of aberrant AChR protein expression at the NMJ. The immunofluorescence data shown represent one of at least 15 sites analyzed for each experimental group with approximately 2–3 endplates visualized per site. Magnification for all images is 63×10×2.5; bars are 20 μm. All images were captured under similar conditions.
Upregulation of AChR-α occurs in EAMG mice with severe disease and prominent morphologic damage to the postsynaptic membrane
We examined the ultrastructure of the endplates in EAMG mice by performing electron microscopic observations and morphologic analyses in three mice with severe EAMG, three mice with mild EAMG, and three control mice. In the animals with severe EAMG, marked over-expression of AChR-α had been demonstrated, as described above. In the mild EAMG group, there was minimal or no expression of AChR-α apart from that associated with the α-BTX labeling. In control animals and those with mild EAMG, the structure of the NMJ was largely maintained in most examined regions, with the majority of examined endplates graded as ‘normal’ or ‘mildly abnormal’. In contrast, in severe EAMG mice, the post-synaptic membrane was severely damaged with reduced post-synaptic folds or even complete loss of post-synaptic folding in virtually all examined regions (figure 3A). Correspondingly, (PostSM/PreSM) ratios were reduced in mild EAMG minimally but were markedly reduced in severe EAMG (figure 3B).
Figure 3.

A. Electron microscopic examination of postsynaptic folds of motor endplates in EAMG mice. Representative NMJ from a control animal shows normal morphology with characteristic “folding” of the postsynaptic membrane. This morphology is largely preserved in mice with mild EAMG, as illustrated in a representative NMJ from a mouse with mild EAMG. Mice with severe disease (and upregulation of AChR-α) showed morphological abnormalities characterized by simplification of the membrane structure and reduction or loss of the postsynaptic folding pattern. B. Quantitative analysis of the ultrastructure of neuromuscular junctions in muscle tissue from normal mice, mild EAMG, and severe EAMG. Postsynaptic membrane length (PostSM) to presynaptic membrane length (PreSM) ratios were calculated in the three experimental groups and average ratio values are shown in the graph, indicating a markedly reduced (PostSM)/(PreSM) ratio in mice with severe EAMG.
IgG and complement is deposited in EAMG muscle in a pattern correlating with upregulation of AChR-α
We performed immunofluorescence studies to detect deposition of IgG and complement at the endplate on forelimb muscles isolated from EAMG mice with mild and severe disease, and in control animals. Endplates from mild EAMG mice showed strong staining for C3 and IgG which co-localized to the endplate regions as defined by staining with tetramethylrhodamine-conjugated α-BTx, while control animals showed no staining. In mice with severe EAMG, we detected significant deposition of both IgG and C3 apart from the endplate region in patterns similar to those observed for expression of AChR-α. Representative images are shown in figure 4.
Figure 4.

Depostion of IgG (left) and C3 (right) at the postsynaptic endplate. Endplates from control animals show no significant deposition of IgG or C3 (top rows). Mice with mild EAMG show strong staining for IgG and C3 which co-localizes to endplate regions as defined by staining with tetramethylrhodamine-conjugated α-BTX (middle rows). Mice with severe EAMG show staining for IgG and C3 which appears to target the nonjunctional muscle fiber, failing to co-localize to the endplate region (bottom rows).
Discussion
Pathogenic anti-AChR antibodies are the primary cause of post-synaptic membrane dysfunction in approximately 85% of patients with autoimmune MG,8 and the majority of these antibodies are directed against the α-subunit of the skeletal muscle AChR. However, the serum concentration of anti-AChR antibodies varies widely among patients with similar degrees of weakness, and it cannot reliably predict the severity of the disease in individual patients.13 Previous observations have suggested that intrinsic differences in the expression of endplate proteins may have an effect on the susceptibility to EAMG.12 Autoimmune diseases, including MG, occur in patients treated with D-penicillamine, and this “induced autoimmunity” occurs by the mechanism of antigenic alteration of the AChR.5 Furthermore, during the course of MG, it is likely that changes in the expression of post-synaptic proteins may have an effect on the ongoing immune response to the AChR.24
In this study, we found increased expression of AChR-α subunit transcripts in muscle homogenates from animals with EAMG, as has been previously reported.1,2 The expression levels were most pronounced in mice with more severe disease. Although this finding confirms previous observations, it also appeared to be in direct conflict with reports that show reduction in total concentration of muscle AChR in EAMG (utilizing assays that measure binding to α-BTX) and correlating with increasing clinical disease severity.25,26
We surmised that that this apparent discrepancy could be explained by the idea that, while enhanced expression of the AChR-α subunit was indeed a compensatory response to AChR loss, the expressed protein did not allow for effective AChR function and binding of acetylcholine. We tested this possibility by simultaneously examining the expression of AChR-α subunit as well as the ligand-binding capabilities of AChR as determined by α-BTX labeling in our experimental animals. We found a profound increase in expression of AChR-α subunit in mice with more severe disease, and this expression was very often separate and greatly in excess of α-BTX-labeled sites. In the severe cases, the expression of AChR-α appeared to outline the course of the muscle membrane in selected muscle fibers.
The human AChR-α subunit exists as two isoforms that are generated by alternative splicing of the P3A exon located between exons P3 and P4.4,14 Two isoforms are found in an approximately 1:1 ratio in the human muscle cell line TE671, but the isoform in which P3A is included (P3A+) is not integrated into functional AChRs and does not bind labeled α-BTX.15 In contrast to a previous report,17 we did not find expression of two AChR-α isoforms in mouse limb muscle, and we were not able to demonstrate the presence of the P3A+ isoform in mice with enhanced expression of AChR-α. This is consistent with other reports of failure to confirm the presence of the P3A+ isoform in non-human species.4,21 However, this finding does not rule out the possibility that enhanced expression of P3A+ underlies the altered expression of AChR-α previously demonstrated in human patients with MG.10,11 Investigation of this possibility will be the subject of future studies utilizing human tissue.
We hypothesize that there is a compensatory mechanism in EAMG responding to loss of functional AChR protein. It increases transcription of AChR subunit mRNA, which is enhanced in more severe disease with greater losses of functional AChR. However, the resulting expressed AChR protein is “functionally inert,” failing to compensate for loss of AChR binding sites. Possible explanations for this observation include: 1) complement-mediated lysis of the endplate alters its morphology so that it does not allow for the insertion of normal numbers of intact AChR; 2) disruption of the endoplasmic reticulum resulting from lysis of the endplate region impairs assembly of AChR protein; 3) expression of AChR-α protein is altered (post-translationally modified, aberrantly spliced, etc). Alternatively, the anti-AChR-α antibodies may be binding to partially degraded AChRs that have lost their ability to bind α-BTX. An argument against this possibility is that upregulation of AChR-α is most prominent in late stage disease (probably not when one would expect the highest degree of active AChR degradation), and levels of mRNA expression are markedly increased in these animals consistent with compensatory upregulation.
It is known that alteration of self-protein expression can elicit, and in some cases, enhance, autoimmune responses.3,16 Evidence suggesting that changes in the target autoantigen (such as altered structure and enhanced expression) can regulate and shape the immune response, has been noted in autoimmune myositis.20 Upregulation of expression of these proteins may occur outside the endplate region, as is suggested by expression patterns that completely outline the muscle membrane in some cases. This may also expose epitopes that are normally sheltered from immune recognition, and they can then become immunogenic and targets of immune effector functions. In severely affected EAMG mice, the finding of IgG and complement deposition unassociated with ligand (acetylcholine) binding sites, in a pattern similar to the enhanced expression pattern of AChR-α, is supportive of the concept that this expression is mechanistically important in the ongoing immune response. As enhanced expression of AChR-α has been reported in human MG,10,11 it may contribute to progression and maintenance of autoimmunity, particularly in patients with more severe or refractory disease. Further study of the role of autoantigens in driving autoimmune diseases like MG may elucidate potential targets for therapeutic manipulation.
Supplementary Material
Acknowledgments
This work was supported by the NIH (National Institute of Neurologic Disorders and Stroke, K08NS058800-01, MNM; and National Institute of Allergy and Infectious Diseases, RO1 AI 058190-01, BSP); and by the Myasthenia Gravis Foundation of America – Postdoctoral Fellowship Award to JRS.
Abbreviations
- MG
myasthenia gravis
- EAMG
experimental autoimmune myasthenia gravis
- AChR
acetylcholine receptor
- α-BTX
alpha-bungarotoxin
References
- 1.Asher O, Neumann D, Fuchs S. Increased levels of acetylcholine receptor α-subunit mRNA in experimental autoimmune myasthenia gravis. FEBS Lett. 1988;233:277–281. doi: 10.1016/0014-5793(88)80442-3. [DOI] [PubMed] [Google Scholar]
- 2.Asher OD, Neumann D, Witzemann V, Fuchs S. Acetylcholine receptor gene expression in experimental autoimmune myasthenia gravis. FEBS Lett. 1990;267:231–235. doi: 10.1016/0014-5793(90)80932-9. [DOI] [PubMed] [Google Scholar]
- 3.Atassi MZ, Casali P. Molecular mechanisms of autoimmunity. Autoimmunity. 2008;41:123–132. doi: 10.1080/08916930801929021. [DOI] [PubMed] [Google Scholar]
- 4.Beeson D, Morris A, Vincent A, Newsom-Davis J. The human muscle nicotinic acetylcholine receptor α-subunit exists as two isoforms: a novel exon. EMBO J. 1990;9:2101–2106. doi: 10.1002/j.1460-2075.1990.tb07378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bever CT, Jr, Chang HW, Penn AS, Jaffe IA, Bock E. Penicillamine-induced myasthenia gravis. Effects of penicillamine on acetylcholine receptor. Neurology. 1982;32:1077–1082. doi: 10.1212/wnl.32.10.1077. [DOI] [PubMed] [Google Scholar]
- 6.Christadoss P, Poussin M, Deng C. Animal models of myasthenia gravis. Clin Immunol. 2000;94:75–87. doi: 10.1006/clim.1999.4807. [DOI] [PubMed] [Google Scholar]
- 7.Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present and future. J Clin Invest. 2006;116:2843–2854. doi: 10.1172/JCI29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeBaets M, Stassen MH. The role of antibodies in myasthenia gravis. J Neurol Sci. 2002;202:5–11. doi: 10.1016/s0022-510x(02)00200-9. [DOI] [PubMed] [Google Scholar]
- 9.Engel AE, Santa T. Histometric analysis of the ultrastructure of the neuromuscular junction in myasthenia gravis and in the myasthenic syndrome. Ann NY Acad Sci. 1971;183:46–63. doi: 10.1111/j.1749-6632.1971.tb30741.x. [DOI] [PubMed] [Google Scholar]
- 10.Guyon T, Levasseur P, Truffault F, Cottin C, Gaud C, Berrih-Aknin S. Regulation of acetylcholine receptor α subunit variants in human myasthenia gravis. J Clin Invest. 1994;94:16–24. doi: 10.1172/JCI117302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guyon T, Wakkach A, Poea S, Mouly V, Klingel-Schmitt I, Levaseur P, Beeson D, Asher O, Tzartos S, Berrih-Aknin S. Regulation of acetylcholine receptor gene expression in human myasthenia gravis muscles. J Clin Invest. 1998;102:249–263. doi: 10.1172/JCI1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoedemaekers A, Bessereau JL, Graus Y, Guyon T, Changeux JP, Berrih-Aknin S, Van Breda Vriesman P, DeBaets MH. Role of the target organ in determining susceptibility to experimental myasthenia gravis. J Neuroimmunol. 1998;89:131–141. doi: 10.1016/s0165-5728(98)00126-x. [DOI] [PubMed] [Google Scholar]
- 13.Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis: prevalence, clinical correlates, and diagnostic value. Neurology. 1976;26:1054–59. doi: 10.1212/wnl.26.11.1054. [DOI] [PubMed] [Google Scholar]
- 14.Morris A, Beeson D, Jacobson L, Baggi F, Vincent A, Newsom-Davis J. Two isoforms of the muscle acetylcholine receptor alpha-subunit are translated in the human cell line TE671. FEBS Lett. 1991;295:116–118. doi: 10.1016/0014-5793(91)81399-s. [DOI] [PubMed] [Google Scholar]
- 15.Newland CF, Beeson D, Vincent A, Newsom-Davis J. Functional and non-functional isoforms of the human acetylcholine receptor. J Physiol. 1995;489:767–778. doi: 10.1113/jphysiol.1995.sp021090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Notkins AL, Onodera T, Prabhakar BS. Virus-induced autoimmunity. In: Notkins AL, Oldstone MBA, editors. Concepts in Viral Pathogenesis. New York: Springer-Verlag; 1984. pp. 210–215. [Google Scholar]
- 17.Saini SS, Tuzun E, Christadoss P. The cDNA of mouse skeletal muscle transcribe for both isoforms 1 and 2 of acetylcholine receptor alpha subunit. J Neuroimmunol. 2005;169:177–179. doi: 10.1016/j.jneuroim.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 18.Sheng JR, Li L, Ganesh BB, Prabhakar BS, Meriggioli MN. Regulatory T cells induced by GM-CSF suppress ongoing experimental myasthenia. Clin Immunol. 2008;128:172–180. doi: 10.1016/j.clim.2008.03.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheng JR, Li LC, Ganesh BB, Vasu C, Prabhakar BS, Meriggioli MN. Suppression of experimental autoimmune myasthenia gravis (EAMG) by Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) is associated with an expansion of FoxP3+ regulatory T cells. J Immunol. 2006 Oct 15;177:5296–306. doi: 10.4049/jimmunol.177.8.5296. [DOI] [PubMed] [Google Scholar]
- 20.Suber TL, Casciola-Rosen L, Rosen A. Mechanisms of disease: autoantigens as clues to the pathogenesis of myositis. Nat Clin Pract Rheumatol. 2008;4:201–209. doi: 10.1038/ncprheum0760. [DOI] [PubMed] [Google Scholar]
- 21.Talib S, Okarma TB, Lebkowski JS. Differential expression of human nicotinic acetylcholine receptor alpha subunit variants in muscle and non-muscle tissues. Nucleic Acids Res. 1993;21:233–237. doi: 10.1093/nar/21.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vincent A. Unravelling the pathogenesis of myasthenia gravis. Nat Rev Immunol. 2002;2:797–804. doi: 10.1038/nri916. [DOI] [PubMed] [Google Scholar]
- 23.Vincent A. Immunology of disorders of neuromuscular transmission. Acta Neurol Scand. 2006;113 (Suppl 183):1–7. doi: 10.1111/j.1600-0404.2006.00605.x. [DOI] [PubMed] [Google Scholar]
- 24.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated disease: implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 25.Wu B, Goluszko E, Christadoss P. Experimental autoimmune myasthenia gravis in the mouse. In: Coligan JE, Shevach EM, Strober W, editors. Current Protocols of Immunology. Vol. 3. New York: John Wiley & Sons; 1997. pp. 8.1–8.16. [Google Scholar]
- 26.Yang H, Goluszko E, David C, Okita DK, Conti-Fine B, Chan TS, Poussin MA, Christadoss P. Mapping myasthenia gravis-associated T cell epitopes on human acetylcholine receptors in HLA transgenic mice. J Clin Invest. 2002;109:1111–1120. doi: 10.1172/JCI14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.