

Abstract
The goal of this study was to characterize the bone phenotype and molecular alterations in Col3.6-HSD2 mice in which a 3.6-kb Col1a1 promoter fragment drives 11β-HSD2 expression broadly in the osteoblast lineage to reduce glucocorticoid signaling. Serum corticosterone was unchanged in transgenic females exluding a systemic effect of the transgene. Adult transgenic mice showed reduced vertebral trabecular bone volume and reduced femoral and tibial sub-periosteal and sub-endosteal areas as assessed by microCT. In adult female transgenic mice, histomorphometry showed that vertebral bone mass and trabecular number were reduced but that osteoblast and osteoclast numbers and the mineral apposition and bone formation rates were not changed, suggesting a possible developmental defect in the formation of trabeculae. In a small sample of male mice, osteoblast number and percent osteoid surface were increased but the mineral apposition bone formation rates were not changed, indicating subtle sex-specific phenotypic differences in Col3.6-HSD2 bone. Serum from transgenic mice had decreased levels of the C-terminal telopeptide of α1(I) collagen but increased levels of osteocalcin. Transgenic calvarial osteoblast and bone marrow stromal cultures showed decreased alkaline phosphatase and mineral staining, reduced levels of Col1a1, bone sialoprotein and osteocalcin mRNA expression, and decreased cell growth and proliferation. Transgenic bone marrow cultures treated with RANKL and M-CSF showed greater osteoclast formation; however, osteoclast activity as assessed by resorption of a calcium phosphate substrate was decreased in transgenic cultures. Gene profiling of cultured calvarial osteoblasts enriched in the Col3.6-HSD2 transgene showed modest but significant changes in gene expression, particularly in cell cycle and integrin genes. In summary, Col3.6-HSD2 mice showed a low bone mass phenotype, with decreased ex vivo osteogenesis. These data further strengthen the concept that endogenous glucocorticoid signaling is required for optimal bone mass acquisition and highlight the complexities of glucocorticoid signaling in bone cell lineages.
Keywords: Glucocorticoids, 11-beta-hydroxysteroid dehydrogenase type 2, transgenic mice, osteoblasts, osteoclasts
Introduction
Bone remodeling, the coupled process of osteoclastic resorption and osteoblastic formation, maintains bone mass and integrity during adulthood. Glucocorticoids exhibit complex regulatory effects on bone remodeling. Glucocorticoid excess leads to substantial bone loss, due to decreased osteoblast proliferation, increased osteoblast apoptosis and heightened osteoclast resorption [1, 2]. A variety of in vitro models, however, show that glucocorticoids are osteogenic since they promote osteoblast differentiation and mineralized nodule formation [3, 4]. This paradox has yet to be resolved, although recent studies suggest that endogenous glucocorticoids are pro-osteogenic [5, 6].
Glucocorticoids signal via the classical steroid-hormone receptor pathway. Glucocorticoids bind to receptors (GR) in the cytoplasm to form a complex, which translocates into the nucleus to regulate downstream targets, either by binding to glucocorticoid response elements (GRE) [7, 8], or by protein-protein interaction [9, 10]. Regulation of glucocorticoid signaling occurs prior to receptor binding by the 11β-hydroxysteroid dehydrogenase (11β-HSD) type 1 (11β-HSD1) and type 2 (11β-HSD2) enzymes. 11β-HSD1 is a bidirectional enzyme, but in the presence of sufficient NADP+ cofactor, it functions primarily as a reductase, activating cortisone to cortisol. By contrast, 11β-HSD2 is a unidirectional dehydrogenase that metabolizes cortisol to cortisone [11].
To determine the functions of endogenous glucocorticoids in bone, we previously developed Col2.3-HSD2 transgenic mice, in which a 2.3-kb Col1a1 promoter fragment drives expression of 11β-HSD2 in mature osteoblasts. Col2.3-HSD2 mice showed reduced vertebral trabecular and femoral cortical bone content due in part to impairment of osteoblast differentiation, suggesting that endogenous glucocorticoid signaling in mature osteoblasts is required for cortical bone acquisition and full differentiation [5, 6].
In the present study, we characterized the bone phenotype of Col3.6-HSD2 mice in which a 3.6-kb Col1a1 promoter fragment drives gene expression broadly in the osteoblast lineage. Similar to Col2.3-HSD2 mice, Col3.6-HSD2 mice showed a low bone mass phenotype and impaired ex vivo osteoblast proliferation and differentiation. In addition, Col3.6-HSD2 bone marrow cultures showed enhanced ex vivo osteoclast formation but interestingly, the osteoclasts were less active. To gain further insight into the molecular events associated with the disrupted glucocorticoid signaling, Col3.6-HSD2 mice were bred with pOBCol3.6-driven green fluorescent protein (pOBCol3.6-GFP) transgenic mice. Calvarial osteoblast cultures were subjected to fluorescent activated cell sorting (FACS) based on GFP. Gene profiling of sorted cells by microarray showed that transgenic cells had alterations in cell cycle and integrin pathway genes. Taken together, these data demonstrate that endogenous glucocorticoid signaling is required for optimal bone mass acquisition by affecting both the osteoblast and osteoclast lineages.
Materials and Methods
Animals
A rat 11β-HSD2 cDNA was cloned downstream of a 3.6-kb fragment of the rat Col1a1 promoter and upstream of the bovine GH polyadenylation sequence to produce the construct Col3.6-HSD2 [12]. Transgenic founder mice founders were developed in the CD-1 outbred background standard using pronuclear injection and maintained as outbred lines. From multiple microinjections, only one founder was identified indicating that high levels of transgene expression might have been lethal. The founder was were bred to wild-type mice to establish Col3.6-HSD2 line 211. For all of the experiments described in this study, hemizygous males were bred with wild type CD-1 females to produce litters containing hemizygous transgenic mice and wild type littermates. For microarray studies, hemizygous Col3.6-HSD2 mice were bred with homozygous pOBCol3.6-driven green fluorescent protein (pOBCol3.6-GFP) transgenic mice [13, 14]. All progeny were hemizygous for the pOBCol3.6-GFP transgene and either wild type or hemizygous for the Col3.6-HSD2 transgene. All animal care protocols were reviewed and approved by the University of Connecticut Health Center Animal Care Committee.
Bone density and morphometry
Mice were sacrificed at 7 weeks of age. The bone mineral content (BMC), bone mineral density (BMD) and percent body fat were measured by dual energy X-ray absorptiometry (DEXA) using a PIXImus densitometer (GE-Lunar, Madison, WI). Morphometry of calvaria and the trabecular and cortical compartments of femurs, tibiae, and third lumbar vertebrae (L3) were quantified at 12 μm resolution (578,704 isometric voxels/mm3) using cone beam X-ray computed microtomography (μCT40, Scanco Medical AG, Brüttisellen, Switzerland) as described previously [15]. In addition, 3-day-old skulls were imaged separately at 16 μm resolution (244,141 isometric voxels/mm3). All images were acquired at 55 kV, 145 μA, 1000 projections/revolution, and 300 ms integration time. Segmentation of bone from background and soft tissue employed a three-dimensional Gaussian filter, applying density thresholds of 120, 470, 620, and 710 mg/cm3 for newborn skulls, trabecular bone, calvaria, and cortical bone, respectively.
Trabecular morphometry measurements included the bone volume fraction (BV/TV%), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular spacing (Tb.Sp). Cortical morphometry was quantified through a 600-μm long span extending distally from the diaphyseal mid-point between proximal and distal growth plates. Cortical morphometry measurements included average cortical thickness, sub-periosteal cross-sectional area, sub-endosteal (marrow) area and cortical bone area. Femur length was measured from the distal end of the condyles to the proximal aspect of the femoral head. Calvaria morphometry was measured bilaterally within the central third (~2 mm) of each parietal bone between the sagittal and squamous sutures. Calvaria width was measured and averaged at three locations (center and ends) of the central region.
Histomorphometry
Static and dynamic histomorphometry were performed on wild type and Col3.6-HSD2 mice as previously described with a few modifications [16]. Mice were weighed and injected intraperitoneally (IP) with 0.1 ml of calcein in 2% NaHCO3 to give a final dose of 20 mg/kg body weight. Five days later, mice were injected IP with 0.1 ml of demeclocycline in 2% NaHCO3 to give a final dose of 20 mg/kg body weight. Mice were euthanized 2 days later. Bones were dissected, fixed in 4% paraformaldehyde and embedded in methyl methacrylate. Five μm thick sections were stained with 0.1% toluidine blue or von Kossa reagent. Static and dynamic histomorphometric measurements of bone formation and resorption were made with an OsteoMeasure morphometry system (Osteometrics, Atlanta, GA). Dynamic measurements were made on unstained sections under UV light using a triple diamidino-2-phenylindole, fluorescein isothiocyanate and Texas red long-pass filter. Histomorphometric measurements were expressed using terminology and units suggested by the Histomorphometry Nomenclature Committee of the American Society for Bone and Mineral Research [17].
Alizarin red/alcian blue staining
Alizarin red/alcian blue staining was performed by a method similar to one previously described [18]. Briefly, the skin and organs were removed from 1- and 7-day-old mice, which were stored in 100% ethanol for 7 days, followed by acetone for 2 days. The samples were then incubated with fresh stain for 3 days at 37°C. The staining solution was composed of 1 volume of 0.3% alcian blue (Sigma, St. Louis, MO) in 70% ethanol, 1 volume of 0.1% alizarin red-S (Sigma, St. Louis, MO) in 95% ethanol, 1 volume of acetic acid and 17 volumes of 70% ethanol. The samples were then washed in distilled water, placed in 1% KOH in 20% glycerol until cleared and stored in 100% glycerol. For alizarin red staining of bone marrow stromal cultures, cells were rinsed with PBS and fixed with 70% ethanol then stained with a 40 mM alizarin red solution at room temperature for 10 min. The plates were rinsed with water to remove unbound alizarin red, and 10% w/v cetylpyridinium chloride was added to extract alizarin red at room temperature for 30 min. The alizarin red content of the cultures was determined measuring absorbance at 562 nm on a multiplate reader, using an alizarin red standard curve prepared in the same solution. The absorbance values were converted to calcium content using the following formula: calcium (ng/ml) = 0.685 × (OD562) + 0.0503.
Primary calvarial osteoblast cultures
Hemicalvaria from 6- to 8-day-old neonatal mice were dissected and serially digested with collagenase P (Roche Diagnostics, Indianapolis, IN) to obtain calvarial osteoblasts as described previously [6]. Calvarial cells were plated in 35 mm wells at a density of approximately 15,000 cells/cm2 in DMEM containing 10% heat inactivated fetal calf serum (HI-FCS; Hyclone, Logan, UT) and antibiotics. On days 1 and 4, the medium was changed. On day 7, cells were switched to differentiation medium (αMEM containing 10% HI-FCS, 4 mM β-glycerol phosphate, and 50 μg/ml ascorbic acid). Cells were refreshed with differentiation medium every other day for the duration of the experiment.
Bone marrow stromal cultures
Bone marrow was flushed from femurs and tibias of 6- to 8-week-old mice and cell cultures were established as previously described [6]. Briefly, cells were plated in 6-well dishes at 3.2 × 106 cells/well and cultured in αMEM supplemented with 10% HI-FCS for up to 28 days. On day 3 of culture, half of the medium was changed. Starting on day 7, the medium was supplemented with 50 μg/ml ascorbic acid and 8 mM β-glycerol phosphate. Cells were given fresh supplemented medium every other day until the end of the culture.
Alkaline phosphatase, von Kossa and crystal violet staining, and GFP imaging
Alkaline phosphatase, von Kossa and crystal violet staining were performed sequentially as previously described [6]. Alkaline phosphatase staining was performed using a kit (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s instructions. Alkaline phosphatase-stained plates were then stained with von Kossa reagent using a 5% AgNO3 solution. Following von Kossa staining, plates were rinsed with PBS and stained with 0.05% crystal violet (Sigma-Aldrich, St. Louis, MO). All plates were imaged using a flatbed scanner.
Alkaline phosphatase activity assay
Cells were scraped from the culture plates with 1 mM MgCl2/0.2% NP-40 and incubated at 37°C for 15 min. Cells were centrifuged at 4°C for 10 min. Ten μl of the resulting supernatant was added in duplicate to individual wells of a 96-well plate and incubated with an assay mixture of alkaline buffer solution and stock substrate solution (Sigma-Aldrich, St. Louis, MO). Absorbance at 410 nm was measured every 5 min for 30 min. Alkaline phosphatase activity was calculated according to the standard curve created with known concentrations of alkaline phosphatase. The protein content in each sample was determined using bicinchoninic acid [19]. Alkaline phosphatase activity was expressed as nM/min/mg protein.
Bromodeoxyuridine (BrdU) incorporation
Cell proliferation was assessed using a BrdU Cell Proliferation Assay kit (Oncogene Research Products, San Diego, CA). Primary calvarial cells were plated at 10,000/well in 96-well dishes in DMEM supplemented with 10% HI-FBS and antibiotics. Cells were allowed to attach overnight, labeled with BrdU (1:2000) at 37°C for 6 h and fixed with fixative/denaturing solution at room temperature for 1 h. After washing with PBS, cells were incubated with anti-BrdU antibody solution at room temperature for 1 h. Cells were washed and incubated with 100 μl goat anti-mouse IgG-horseradish peroxidase (HRP) conjugate for 30 min at room temperature. After washing with deionized water, 100 μl of a substrate solution were added to each well and plates were incubated in the dark at room temperature for 15 min for color development. A stop solution was added and absorbance was measured in a plate reader at 450 and 540 nm.
Osteoclast formation in bone marrow cultures and tartrate resistant acid phosphatase (TRAP) staining
Femurs and tibiae from 7-week-old mice were dissected from surrounding tissues. The epiphyses were removed and marrow was flushed with serum-free α-MEM. Cells were plated at 80,000/well in 96-well plates and cultured in αMEM containing 10% HI-FBS, 1% antibiotics, and M-CSF and RANKL, each at 30 ng/ml. The medium was changed on day 3. Osteoclast formation was measured daily on days 3 to 5 as the number of TRAP positive cells containing 3 or more nuclei as previously described [20]. To assess the osteoclast resorption in vitro, bone marrow cells were plated at 1.5 × 105/well with M-CSF and RANKL (30 ng/ml each), on BD BioCoat Osteologic MultiTest slides (BD Biosciences, Bedford, MA). Resorbed areas were analyzed on day 10 as previously described [21].
Serum markers
Blood sample were obtained from 7-week old females by thoracic puncture following the euthanasia. After centrifugation, serum was collected and assayed for corticosterone levels with an enzyme-linked immunosorbent assay (ELISA) kit (Assay design, Ann Arbor, Michigan). Serum osteocalcin was measured by an ELISA assay with a mouse osteocalcin EIA kit (Biomedical Technologies Inc, Stoughton, MA). Serum levels of CTX, a degradation product released during bone resorption [22], were measured by an ELISA using a RatLaps kit (Nordic Bioscience Diagnostics, Fountain Hill, AZ).
Cell sorting and microarray analysis
Hemizygous Col3.6-HSD2 mice were bred with homozygous pOBCol3.6-GFP mice as described above to yield progeny that were all hemizygous for GFP but either wild type (WT) or hemizygous transgenic (TG) for Col3.6-HSD2. Calvarial osteoblasts were prepared from neonates and cultured for 7 days as previously described [23]. Cultures were sorted based on the intensity of GFP fluorescence on a FACS Vantage Sorter (BD Biosciences, Franklin Lakes, NJ). RNA was immediately extracted from WT/GFP+ and TG/GFP+ sorted cells. Microarray was performed with the Mouse 6 Illumina chip and results analyzed using BeadStudio software. Genes with expression intensities >100 and diff scores of >7 or <−7 (calculated by Illumine Custom model) were considered significantly regulated. These genes were clustered according to the major pathways known to modulate bone mass using Pathway Studio software. The microarray analysis was performed using three independent biological samples for each genotype (WT/GFP+ and TG/GFP+).
RNA Extraction and real time RT-PCR
RNA was extracted from tissues using TRIzol reagent (Life Technologies, Grand Island, NY) as previously described [12]. Reverse transcription was carried out with 3 μg RNA using Superscript™ II reverse transcriptase reagent (Invitrogen, Carlsbad, CA). For real time PCR, cDNA product was diluted in 600 μl H2O and 5 μl was used with a Bio-Rad MyiQ thermocycler and a SYBR Green detection system. Amplification reactions were performed in 11 μl containing 0.5 μM primers and 5 μl SYBR-green supermix. Primer sequences are shown in Supplementary Table S1.
Statistics
Data points for each experimental group were expressed as the mean ± standard deviation (SD). Data was analyzed with t-tests or two-way ANOVA followed by Bonferroni post-tests as indicated in the text and individual figure legends with p<0.05 considered significant.
Results
At 7-weeks of age, wild type and Col3.6-HSD2 mice had comparable body weights (Fig 1A). However, Col3.6-HSD2 mice showed a reduction in the percentage of whole body fat (Fig 1B), a change that was not seen in Col2.3-HSD2 mice (data not shown). Wild type and transgenic females had comparable serum corticosterone levels (Fig 1C), indicating that the bone-directed transgene did not have a systemic effect. Transgenic mice showed reductions of whole body BMD (Fig 1D), and BMD of femurs, tibiae and vertebrae (Fig 1E–G). These data suggest that the Col3.6-HSD2 transgene led to a low bone mass phenotype.
Figure 1.
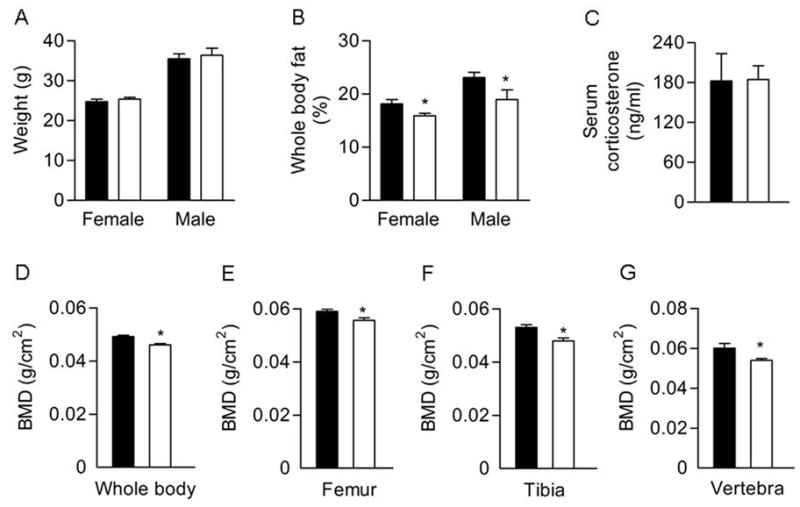
Phenotypic analysis of 7-week-old Col3.6-HSD2 (TG) and wild type (WT) littermates. A) Body weight. B) Percent body fat analyzed by DEXA. C) Serum corticosterone levels in female mice. D–G) BMD of whole body, femurs, tibiae and vertebrae in female mice assessed by DEXA. Each value is the mean ± SD for 5–10 mice. Black bars, WT; White bars, TG. *Different than WT, p<0.05.
Bone morphometry of 7-week-old female mice was analyzed by microCT. In vertebrae, the percent bone volume (BV/TV%) and trabecular number (Tb.N) were reduced and trabecular spacing (Tb.Sp) was increased. The trabecular bone of transgenic femurs and tibiae showed similar trends, but the changes were not significant (Table 1).
Table 1.
MicroCT trabecular bone parameters in 7-week-old female wild type (WT) and Col3.6-HSD2 transgenic (TG) mice.
| Vertebrae | Femur | Tibia | ||||
|---|---|---|---|---|---|---|
| WT | TG | WT | TG | WT | TG | |
| BV/TV% | 19.6 ± 6.0 | 15.7 ± 6.8* | 17.1 ± 4.3 | 14.0 ± 6.0 | 13.1 ± 5.6 | 12.1 ± 6.7 |
| Tb.N, μm | 4.96 ± 0.32 | 4.13 ± 0.47* | 5.33 ± 0.31 | 4.55 ± 0.73 | 5.56 ± 0.53 | 5.24 ± 0.70 |
| Tb.Sp, 1/mm | 203 ± 10 | 248 ± 29* | 188 ± 11 | 226 ± 45 | 177 ± 16 | 197 ± 34 |
Each value is the mean ± SD of 12–18 bones.
Different than WT, p<0.05.
Transgenic femurs and tibiae showed a slight but significant increase in length (Table 2). The cortical bone of transgenic femurs showed a reduction in sub-periosteal area and sub-endosteal area, together resulting in an increase cortical thickness but no change in cortical bone area. Transgenic tibiae showed a decrease in sub-periosteal area and sub-endosteal area, together resulting in a reduction in cortical bone area. Thus, the femurs and tibiae of transgenic mice were longer but slimmer at the mid-diaphysis with a reduced amount of tibial cortical bone.
Table 2.
MicroCT cortical bone parameters in 7-week-old female wild type (WT) and Col3.6-HSD2 transgenic (TG) mice.
| Femur | Tibia | |||
|---|---|---|---|---|
| WT | TG | WT | TG | |
| Bone length, mm | 15.02 ± 0.39 | 15.35 ± 0.28* | 17.68 ± 0.29 | 18.22 ± 0.27* |
| Sub-periosteal area, mm2 | 1.78 ± 0.15 | 1.59 ± 0.14* | 0.89 ± 0.09 | 0.74 ± 0.07* |
| Sub-endosteal area, mm2 | 0.94 ± 0.09 | 0.74 ± 0.08* | 0.28 ± 0.06 | 0.16 ± 0.03* |
| Cortical thickness, mm | 0.22 ± 0.002 | 0.24 ± 0.01* | 0.25 ± 0.01 | 0.27 ± 0.02 |
| Cortical bone area, mm2 | 0.81 ± 0.06 | 0.82 ± 0.08 | 0.59 ± 0.03 | 0.55 ± 0.04* |
Each value is the mean ± SD of 12–18 samples.
Different from WT, p<0.05.
Calvaria showed a similar low bone mass phenotype. Calvaria from 7-week-old male and female transgenic mice had significantly reduced cortical width and bone area. In transgenic females, the calvarial marrow spaces were increased, but this was unchanged in transgenic males (Table 3). MicroCT imaging of 3-day-old transgenic skulls revealed increased surface pitting and enlarged occipital fontanelles (Fig 2A). The staining of 1- and 7-day-old transgenic skulls with alizarin red/alcian blue confirmed this finding and in addition, showed an increased amount of cartilage (Fig 2B).
Table 3.
MicroCT calvarial morphometry in 7-week-old wild type (WT) and Col3.6-HSD2 transgenic (TG) mice.
| Females | Males | |||
|---|---|---|---|---|
| WT | TG | WT | TG | |
| Cortical width, mm | 0.137 ± 0.010 | 0.117 ± 0.002* | 0.153 ± 0.016 | 0.119 ± 0.015* |
| Bone area, mm2 | 0.251 ± 0.027 | 0.198 ± 0.008* | 0.245 ± 0.038 | 0.170 ± 0.033* |
| Marrow area, mm2 | 0.018 ± 0.007 | 0.036 ± 0.013* | 0.037 ± 0.011 | 0.024 ± 0.011 |
Each value is the mean ± SD of 6 samples.
Different from WT, p<0.05.
Figure 2.
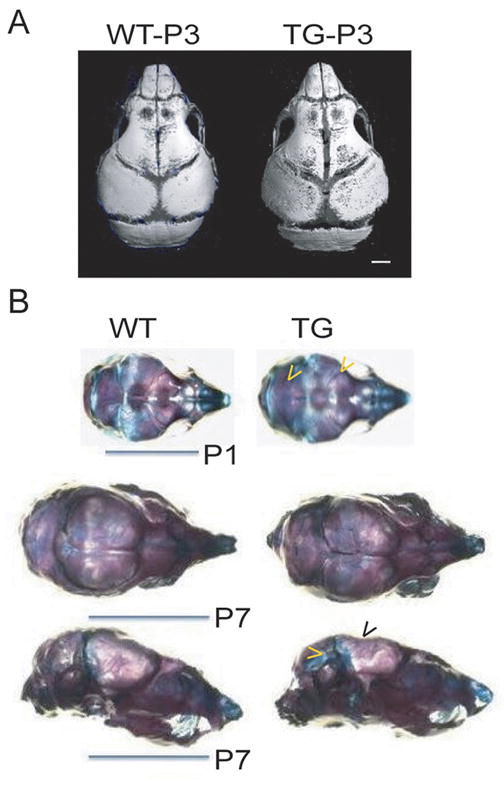
Cranial defects and increased cartilage formation in Col3.6-HSD2 (TG) compared to wild type (WT) littermates. A) MicroCT of 3-days-old (P3) transgenic skulls showed increased surface pitting and an enlarged occipital fontanelle. B) Alizarin red/alcian blue staining in 1- and 7-day-old mice (P1 and P7, respectively) showed thin calvaria (black arrowhead) and an increased amount of cartilage (yellow arrowheads) in transgenics.
Static and dynamic histomorphometry were performed to examine the cellular mechanisms responsible for the reduction in bone mass in transgenic mice (Table 4). Both female and male transgenic mice at 7 weeks of age showed a significant reduction in vertebral bone mass, a decrease in trabecular number and increase in trabecular spacing. These data concur with the microCT results for female transgenic mice (males were not examined by microCT). In female transgenic mice osteoblast and osteoclast number per bone perimeter (Nob/BPm and Noc/BPm, respectively), the mineral apposition rate (MAR) and the bone formation rate (BFR) were not changed. In the small sample of male mice analyzed in this study, osteoid surface (OS/BS%) was increased in transgenic mice, similar to previous findings in Col2.3-HSD2 mice [5]. In addition, male transgenic mice showed an increase in Nob/BPm but no change in MAR and BFR. These data indicate that there are subtle sex-specific phenotypic differences in Col3.6-HSD2 mice.
Table 4.
Histomorphometry of vertebrae from wild type (WT) and Col3.6-HSD2 (TG) 7-week-old female and male mice.
| Females | Males | |||
|---|---|---|---|---|
| WT | TG | WT | TG | |
| BV/TV, % | 14.50 ± 2.16 | 9.81 ± 2.60** | 18.32 ± 4.67 | 9.64 ± 1.21* |
| TbN, 1/mm | 4.83 ± 5.2 | 3.44 ± 0.64** | 5.68 ± 1.04 | 3.69 ± 0.61* |
| TbTh, μm | 29.99 ± 2.27 | 28.20 ± 2.51 | 32.03 ± 2.26 | 26.29 ± 1.38* |
| TbSp, μm | 179.24 ± 23.74 | 270.99 ± 58.08** | 147.79 ± 32.07 | 249.85 ± 41.34* |
| OS/BS, % | 5.40 ± 3.19 | 7.36 ± 3.88 | 1.05 ± 0.50 | 4.25 ± 0.61** |
| Nob/BPm, 1/mm | 37.10 ± 6.59 | 36.60 ± 4.72 | 21.98 ± 1.83 | 32.14 ± 3.65* |
| Noc/BPm, 1/mm | 3.38 ± 0.74 | 3.77 ± 0.79 | 3.05 ± 0.78 | 3.05 ± 0.17 |
| ES/BS, % | 16.05 ± 3.42 | 17.39 ± 4.76 | 14.60 ± 3.40 | 13.83 ± 1.48 |
| MAR, μm/day | 1.82 ± 0.17 | 1.91 ± 0.19 | 1.67 ± 0.08 | 1.77 ± 0.05 |
| BFR, μm3/μm2/day | 0.10 ± 0.02 | 0.09 ± 0.04 | 0.11 ± 0.03 | 0.07 ± 0.03 |
Values are the mean ± SD for 6 mice (females) and 3 mice (males), except for the MAR and BFR values for males (n=2). Significant difference compared to sex-matched WT group,
p<0.05;
p<0.01.
Osteoblast differentiation was assessed in primary neonatal calvarial cell cultures. Alkaline phosphatase staining (Fig 3A) and activity (Fig 3B) were reduced in transgenic cultures on days 7, 14 and 21. Real time RT-PCR showed that the mRNA levels of osteoblast marker genes α1(I) collagen (Col1a1), bone sialoprotein (BSP) and osteocalcin (OCN) were decreased by 2- to 3-fold in transgenic cultures (Fig 3C). These changes in gene expression were confirmed by Northern blot analysis (data not shown). In transgenic cultures, cell growth was reduced on days 3, 5 and 7 after plating (Fig 3D). Transgenic cultures showed a significant reduction in BrdU incorporation on days 3, 5 and 7 indicating that proliferation was reduced (Fig 3E). Surprisingly, serum levels of osteocalcin, a molecule secreted by mature osteoblasts, were increased in transgenic mice (Fig 3F). Ex vivo transgenic bone marrow stromal cell cultures also showed a significant reduction in alkaline phosphatase and von Kossa staining on days 14 and 21. Transgenic cultures showed reduced levels of crystal violet staining on days 7, 14, and 21, indicating reduced cellularity (Supplementary Fig S1 A). Transgenic cultures also showed a 70% and 80% reduction of alizarin red staining at days 14 and 21, respectively, indicating reduced calcium incorporation and mineralization (Supplementary Fig S1 B, C).
Figure 3.

Cell proliferation and differentiation were reduced in female Col3.6-HSD2 (TG) osteoblast cultures. A) Alkaline phosphatase (red) and von Kossa (black) staining of primary calvarial osteoblast cultures from 6- to 8-day old WT and TG mice. B) Alkaline phosphatase activity at days 7, 14 and 21 from WT and TG neonatal osteoblast cultures. C) Expression of mRNA of Col1a1, bone sialoprotein (BSP), and osteocalcin (OCN) in WT and TG primary neonatal osteoblast cultures at days 7, 14 and 21. D) Cell growth in osteoblast cultures from neonatal WT and TG mice. E) Incorporation of BrdU in primary osteoblast cultures from neonatal WT and TG mice. F) Serum osteocalcin (OCN) level in females measured by ELISA. Each value is the mean ± SD for 3 (A–C) or 6 (D–F) samples per group and each experiment has been performed at least twice with similar results. Black bars, WT; White bars, TG. *Different than WT, *p<0.05, **p<0.01.
To determine what molecular pathways were affected in transgenic osteoblasts, gene expression studies were performed on a cell population enriched in the Col3.6-HSD2 transgene. Heterozygous Col3.6-HSD2 mice were bred with homozygous pOBCol3.6-GFP mice (Fig 4A) yielding progeny that were all GFP positive and either wild type (WT) or transgenic (TG) for Col3.6-HSD2. Primary calvarial osteoblast cultures were established from these genotypes. GFP expression was lower in TG cultures compared to WT cultures on days 14 and 21 (Fig 4B) in accordance with the block in osteogenic differentiation seen previously. Next, GFP+ cells were isolated by FACS from 7-day WT and TG undifferentiated cultures (Fig 5A). The TG cultures contained a slightly lower percentage of GFP+ cells than the WT cultures, 36.9% versus 41.0%, respectively (Fig 4C). As expected, the 11β-HSD2 transgene was highly expressed in TG/GFP+ but not in WT/GFP+ sorted cells (Fig 4D). The transgene was also seen in TG/GFP- sorted cells (data not shown). This may be due to a slightly different spatial and/or temporal pattern of expression of the promoters used to drive the 11β-HSD2 transgene (Col3.6) and GFP (pOBCol3.6, which contains part of the Col1a1 first intron). Col1a1 expression was not statistically different in TG/GFP+ cells (although there was a trend toward increased expression, p<0.1), while BSP and OCN expression was significantly decreased compared to WT/GFP+ cells (Fig 4E, F, G).
Figure 4.
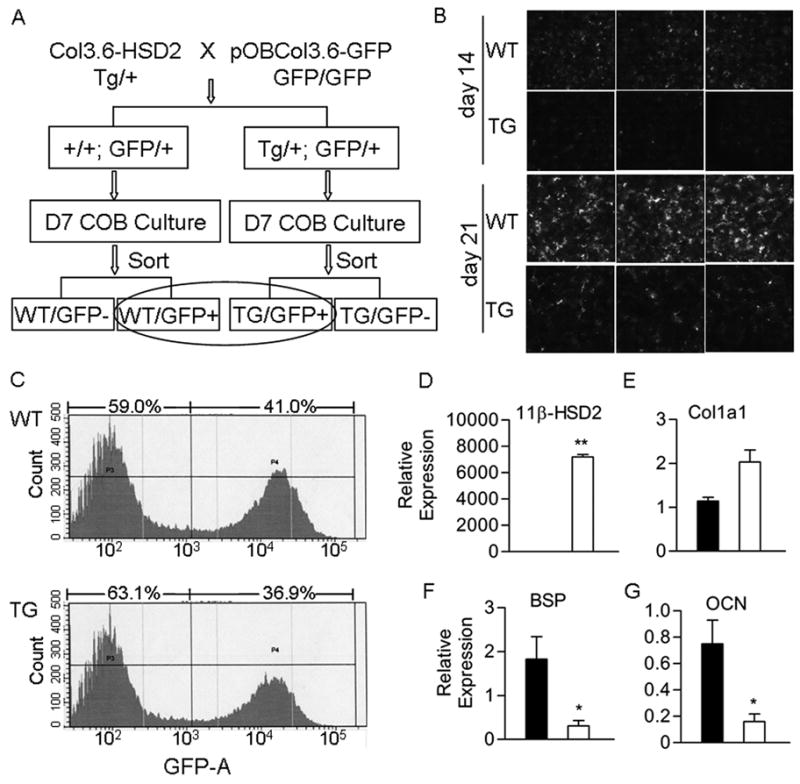
Gene expression in primary osteoblast cultures carrying a pOBCol3.6-GFP transgene. A) Heterozygous Col3.6-HSD2 mice were crossed with homozygous pOBCol3.6-GFP mice to yield mice that were all heterozygous for the pOBCol3.6-GFP transgene and either wild type (WT) or transgenic (TG) for the Col3.6-HSD2 transgene. Primary calvarial osteoblasts (COB) were prepared from the mice and cultured 7 days. Cells were sorted based on GFP and the WT/GFP+ and TG/GFP+ populations were used for gene expression studies. B) GFP fluorescence in day 14 and 21 cultures of unsorted COB cells prepared from WT and TG female mice. C) FACS analysis of GFP expression in sorted WT/GFP+ and TG/GFP+ day 7 cultures. D–G) Expression of the 11β-HSD2 transgene and osteoblast genes in sorted WT/GFP+ and TG/GFP+ cells. Black bars, WT/GFP+ cells; White bars, TG/GFP+ cells. Each value is the mean ± SD for 3 samples and each experiment has been performed at least twice with similar results. *Different than WT/GFP+, *p<0.05, **p<0.01.
Figure 5.

Osteoclast formation was increased but osteoclast activity reduced in Col3.6-HSD2 bone marrow cultures. A) Bone marrow cells were cultured with M-CSF and RANKL, each at 30 ng/ml, for 3–5 days. Osteoclast formation was measured as the number of TRAP-positive cells containing 3 or more nuclei. B) Bone marrow cells were cultured on BD Biocoat Osteologic discs with M-CSF and RANKL each at 30 ng/ml for 10 days. The resorbed area was quantified using Photoshop software after von Kossa staining. C) Serum levels of the C-terminal telopeptide of α1(I) collagen (CTX) in females measured by ELISA. D–G) the M-CSF, RANKL, OPG, and RANKL/OPG ratio in sorted WT/GFP+ and TG/GFP+ cells as measured by real time RT-PCR. For panels A–C, black bars, WT; White bars, TG. For panels D–G, black bars, WT/GFP+ sorted cells; White bars, TG/GFP+ sorted cells. Each value is the mean ± SD for at least 6 (A–C) or 3 (D–F) samples per group and each experiment has been performed at least twice with similar results. *Different than WT or WT/GFP+, *p<0.05, **p<0.01.
RNA from WT/GFP+ and TG/GFP+ cells was subjected to microarray analysis using the Illumina Mouse 6 platform. Significant changes were detected in 302 genes (Supplementary Table S2). Genes showing significant changes in expression included cyclin D1 (Ccnd1), cyclinD2 (Ccnd2), E2F transcription factor 1 (E2f1), histones H2 and H4, transforming growth factor beta 1 induced transcript 4 (Tgfb1i4), integrin beta 5 (Itgb5), sphingosine-1-phosphate phosphatase 1 (Sgpp1), secreted phosphoprotein 1 (Spp1), annexin A1 (Anxa1), caveolae protein (Cav1), matrix metalloproteinase 3 (MMP3) and cysteine and glycine-rich protein 1 (Crps1) (Supplementary Tables S3 and S4).
To assess the osteoclast potential of the marrow, transgenic bone marrow cells were cultured with RANKL and M-CSF for up to 5 days. Transgenic cultures showed a greater number of osteoclasts on days 3, 4 and 5 compared to wild type cultures (Fig 5A). Osteoclast activity was assessed by measuring the ability of bone marrow cultures induced with RANKL and M-CSF to resorb the surface of BD BioCoat Osteologic discs, which are coated with a calcium phosphate substrate. Transgenic bone marrow cells showed a small but significant reduction in their ability to resorb the substrate (Fig 5B). Thus, despite the increase in osteoclast number, osteoclast activity as assessed by this assay was decreased. Consistent with this finding, serum levels of CTX, a degradation product released during resorption, were decreased in transgenic mice, indicating reduced bone resorption in vivo (Fig 5C). Finally, the expression of factors associated with osteoclast formation in WT/GFP+ and TG/GFP+ osteoblast cells was measured by real-time RT-PCR. RANKL and M-CSF expression were increased in TG/GFP+ cells while OPG expression was not changed (Fig 5D–F); thus, the RANKL/OPG ratio was significantly increased in TG/GFP+ cells (Fig 5G).
Discussion
The goal of the present study was to characterize the bone phenotype of Col3.6-HSD2 mice and gain insight into the molecular and cellular alterations that might account for the observed changes in bone morphometry. In our previous studies, Col2.3-HSD2 mice, in which 11β-HSD2 is driven by the Col2.3 promoter in mature osteoblasts, showed decreased vertebral trabecular and femoral cortical bone mass [5, 6]. We had not previously characterized Col3.6-HSD2 mice, which show broader spatial and temporal expression within the osteoblast lineage by contrast to the more restricted expression of Col2.3-HSD2. Col3.6-HSD2 transgenic mice showed no change in serum corticosterone levels. This indicates that the activity of the hypothalamus-pituitary-adrenal gland (HPA) axis was unchanged or there was compensation by direct or indirect feedback from downstream targets. Nonetheless, this finding suggests that the bone phenotype was not secondary to an endocrine effect of glucocorticoids.
Col3.6-HSD2 mice showed a low bone mass phenotype, similar to Col2.3-HSD2 mice. The similarity in bone phenotypes of these models provides strong evidence that endogenous glucocorticoid signaling is required for optimal bone mass acquisition. Primary osteogenic cultures established from Col2.3-HSD2 and Col3.6-HSD2 mice both showed impaired differentiation. In bone marrow cultures established from both models, there was a significant reduction in overall cellularity and alkaline phosphatase staining. Despite these similarities, several differences exist between the bone phenotype of Col2.3-HSD2 and Col3.6-HSD2 mice. Femoral and tibial lengths were increased in Col3.6-HSD2 mice, but not in Col2.3-HSD2 mice. Because the Col3.6 promoter fragment drives gene expression in the early stages of the osteoblast lineage, it is possible that reduced glucocorticoid levels in the local bone microenvironment affected chondrocytes or their precursors by a cell non-autonomous mechanism. Since glucocorticoids enhance chondrocyte apoptosis [24, 25], it is possible that a reduction in local glucocorticoid levels in Col3.6-HSD2 mice might have reduced chondrocyte apoptosis resulting in longer bones. Another different between Col3.6-HSD2 and Col2.3-HSD2 mice is cortical bone morphometry. The sub-endosteal area of femurs and tibias was decreased in Col3.6-HSD2 but not in Col2.3-HSD2 femurs, while the sub-periosteal area was decreased in both Col3.6-HSD2 and Col2.3-HSD2 mice. As a result, cortical thickness was increased in Col3.6-HSD2 mice but decreased in Col2.3-HSD2 mice. The reduced sub-endosteal area in Col3.6-HSD2 mice suggests that endocortical resorption may have been decreased. This would be consistent with the ex vivo osteoclast culture and serum CTX data showing that Col3.6-HSD2 transgenic osteoclasts have reduced resorptive ability compared to wild type controls.
Neonatal transgenic skulls had reduced bone mass with thinner parietal bones and enlarged occipital fontanelles. In addition, there was an increase in the cartilage template in young mice. These findings are similar those published by Zhou and coworkers [26] using our Col2.3-HSD2 strain [5, 6]. These authors showed that Col2.3-HSD2 calvarial osteoblasts had reduced expression of Wnt 9a and 10b, which in turn led to reduced MMP14 expression by cranial chondrocytes and impaired cartilage removal. The microarray analysis of Col3.6-HSD2 calvarial osteoblasts, however, showed no change in the expression of β-catenin, Wnt 9a or Wnt 10b (Supplementary Table S4). The reason for these disparate results is not clear although it may be due to differences in the mouse strains used and/or methodological differences (for example, our study utilized sorted cells from early calvarial cultures).
Col3.6-HSD2 transgenic mice showed lower whole body fat than wild type controls. It has been reported that glucocorticoids stimulate fat differentiation [27, 28]. In Col3.6-HSD2 mice, there was no change in circulating corticosterone levels, excluding an endocrine role of glucocorticoids in the fat phenotype. Transgenic mice with adipose-targeted 11β-HSD2 are partially protected against an increase in body fat when fed a high fat diet [29]. Therefore, it is possible that the Col3.6-HSD2 transgene is expressed at a low level in adipocyte precursors, leading to a reduction in tissue levels of glucocorticoids and adipocyte differentiation.
To our surprise, Col3.6-HSD2 mice had increased serum levels of osteocalcin, which was unexpected given the low bone mass phenotype. Northern blot analysis of freshly excised transgenic calvaria also revealed a 2.6-fold increase in osteocalcin mRNA compared to wild type controls (data not shown). Since glucocorticoids negatively regulate osteocalcin transcription [30], increased osteocalcin expression in transgenic mice may have been due to the loss of the repressive action of glucocorticoids. It is intriguing to note that Lee et al [31] have reported that uncarboxylated osteocalcin increases pancreatic β-cell proliferation and insulin secretion and that mice receiving injections or infusions of under-carboxylated osteocalcin show dose-dependent decreases in fat pad mass [32]. Thus, we speculate that increased levels of circulating osteocalcin in Col3.6-HSD2 mice may have contributed to the reduction in whole body fat. It will be important to determine whether a component of the circulating osteocalcin in these mice is uncarboxylated.
The low bone mass phenotype of Col3.6-HSD2 mice was verified by both microCT and histomorphometry of vertebrae. However, the cellular mechanisms that resulted in this phenotype are complex and not yet clear. In female transgenic mice, neither osteoblast nor osteoclast number was affected, nor were there changes in MAR, BFR or eroded surface. One possible explanation is that Col3.6-HSD2 mice have a developmental defect in the formation of trabeculae, and once these have formed, trabecular bone mass is little unaffected. However, CTX was reduced suggesting that resorption is decreased in transgenic mice. As discussed, serum osteocalcin levels were increased in transgenic mice and we believe this reflects the glucocorticoid signaling status of the osteoblasts rather than bone formation per se. Taken together, these data suggest that Col3.6-HSD2 mice may have low bone turnover and the histomorphometry methodology may not be sufficiently precise to measure small changes in formation and resorption parameters, or more importantly, the changes may have been transient. Nevertheless, the expression of Col3.6-HSD2 broadly in the osteoblast lineage produced mice with a low bone mass phenotype, supporting a role for glucocorticoids in bone anabolism in vivo.
The ex vivo studies present a complex story about how osteoclastogenesis and osteoblastogenesis are affected in Col3.6-HSD2 mice. Transgenic bone marrow cell cultures treated with saturating concentrations of RANKL and M-CSF showed greater osteoclast formation than wild type cultures indicating that the transgenic marrow contains a greater number of osteoclast precursors or that osteoclast precursors have an enhanced ability to fuse and differentiate. Furthermore, transgenic osteoblasts showed increased expression of RANKL and M-CSF suggesting that there might be increased osteoclast formation in vivo. However, histomorphometric indices of bone resorption were not altered in transgenic mice. Moreover, transgenic osteoclast cultures showed less resorption in vitro and consistent with the lower serum CTX levels in vivo. Overall, these data suggest that the Col3.6-HSD2 mice may have an accumulation of osteoclast progenitors and that the transgene may regulate other factors that limit osteoclast formation and activity in vivo. The ex vivo osteogenic culture data suggest that transgenic mice have decreased osteoprogenitors and a defect in progenitor/precursor proliferation. We did not carry the osteogenic cultures longer than 21 days. Thus, it is unknown whether the transgenic cultures are just delayed in their differentiation capacity and catch up to wild type levels of mineralization and osteoblast marker gene expression after longer times in culture.
The Col3.6 promoter has been reported to direct very low level expression of a GFP transgene in the osteoclast lineage [33], although some of these cells may be bone lining cells [34]. Some studies have reported that glucocorticoids exert direct effects on osteoclasts. Kim et al have shown that glucocorticoids directly reduce the proliferation of osteoclast progenitors in bone marrow monocyte cultures, while they inhibit the spreading and resorptive capacity of mature osteoclasts [35]. In the present study, we found no evidence for transgene protein or mRNA expression in osteoclasts (data not shown) excluding a direct effect of the transgene on osteoclast formation or function. In the Col3.6-HSD2 model, it is possible that a reduction of glucocorticoid levels in the bone microenvironment indirectly led to an increase in osteoclast progenitor number as reflected by the ex vivo marrow cultures.
By contrast to Col3.6-HSD2 mice described in the present study, and Col2.3-HSD2 mice previously characterized by us [5, 6], mice with osteocalcin promoter-driven 11β-HSD2 (OG2-11β-HSD2) have normal bone development and turnover [36]. These differences are very likely due to multiple factors including the temporal and spatial pattern of transgene expression, the level of transgene expression and the background strain of the mice. Col3.6-driven transgenes are highly expressed during bone development in the osteoblast lineage [14, 37], while the OG2-driven transgene become activated in the perinatal period and is expressed specifically in osteoblasts and osteoclasts [36]. Thus, the earlier and broader activation of the Col3.6-HSD2 transgene would be more likely to result in altered bone development. In our model, it would be informative to assess the levels of corticosterone in isolated populations enriched in preosteoblasts, mature osteoblasts and osteocytes to determine the precise target cells of the Col3.6-HSD2 transgene. Our findings, however, are not contradictory with many previous studies showing that high levels of glucocorticoids have deleterious effects on bone mass acquisition, bone remodeling and bone quality [38, 39]. In many systems, glucocorticoids at basal levels have permissive effects that potentiate hormone action, while at highly elevated levels they have suppressive effects, and both effects are integrated over a wide range of glucocorticoid concentrations [40, 41].
Recently, it has been shown that aging C57BL/6 mice have greater adrenal production of glucocorticoids, as indicated by increased serum corticosterone levels, and higher bone expression of 11β-HSD1, which would increase the local production of active glucocorticoids [42]. These alterations were also associated with several adverse phenotypes including decreased bone vasculature, interstitial fluid and strength [42]. This study also showed that OG2-11β-HSD2 mice were protected from these age-related phenotypes while pharmacological doses of glucocorticoids recapitulated the adverse effects on bone. Although we have not performed similar studies in aging Col3.6-HSD2 mice, we predict that the Col3.6-HSD2 transgene, which is expressed broadly in the osteoblast lineage, would confer some degree of protection from the deleterious effect of aging on bone vascularization depending on the level of 11β-HSD2 enzymatic activity achieved in mature osteoblasts and osteocytes. An examination of bone vasculature in Col3.6-HSD2 mice would be an important and fruitful area for future investigation.
Gene profiling by microarray revealed a broad range of cellular pathways affected by overexpression of the Col3.6-HSD2 transgene in osteoblasts (see Supplementary Tables S2-S4). Most notably, genes involved in cell cycle were regulated in transgenic cultures. Specifically, the levels of cyclin D1, D2 and E2F1 were reduced. These genes encode the regulatory subunits of a holoenzyme that phosphorylates and inactivates the retinoblastoma protein, thereby promoting progression through the G1-S phase of the cell cycle [43, 44]. Therefore, this suggests that endogenous glucocorticoids are important for maintaining normal cell cycle progression of early osteoblast precursors. The levels of histone H2 and H4 were reduced in transgenic cultures. These histones together with histone H3 form the octameric nucleosome core of chromatin and are integral in the formation of higher order chromatin structures [45, 46]. Histone H4 is critical for maintaining genome integrity throughout the cell cycle [47] while histone H2 plays a critical role in DNA replication [48, 49]. It is possible that the inactivation of glucocorticoid signaling in transgenic osteoblasts may have led to reduced histone H2 and H4 expression and decreased cell cycle progression, resulting in the lower level of proliferation seen in transgenic osteoblast cultures. Integrin β5 and Spp1 (osteopontin) expression were dramatically reduced in transgenic cultures. Integrin αvβ3 and αvβ5 regulate the adhesion of osteoblasts to osteopontin, a process that is regulated by glucocorticoids [50]. Interestingly, since Spp1 is also an important regulator of bone resorption [51, 52], decreased Spp1 in transgenic mice may also contribute to the lower levels of bone resorption. Annexin A1 (lipocortin 1) is expressed in osteoblasts and in cartilage and in matrix vesicles isolated from chondrocytes [53–55]. The importance of annexin A1 for mouse craniofacial bone development was seen in newborn annexin A1 null mice, which have delayed ossification of the skull and incomplete fusion of sutures [56]. The reduction of annexin A1 expression seen in the present study is consistent with the cranial phenotype of transgenic mice. Finally, the expression of Tgfb1i4 (TSC-22d1), a leucine zipper/TSC-box transcription factor, was markedly reduced in transgenic cells. TSC-22 is induced by dexamethasone in MC3T3-E1 cells and human trabecular meshwork cells [57, 58]. Low intensity pulsed ultrasound treatment increased TSC-22 expression in bone marrow stromal cells suggesting a positive role for TSC-22 in bone formation [59]. These findings suggest that the reduction in TSC-22 expression in Col3.6-HSD2 mice may have contributed to the low bone mass phenotype.
Supplementary Material
Acknowledgments
This work accomplished in the study was made possible by the generous support of the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) (grant number AR048602) and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (grant number DK045227). LBT was supported by grant award T32DE007302 from the National Institute of Dental and Craniofacial Research (NIDCR). The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the NIDCR, NIAMS, NIDDK or the National Institutes of Health. We acknowledge the excellent technical assistance of the University of Connecticut Health Center Microcomputed Tomography Facility, the Translational Genomics Core, specifically Anupinder Kaur, for performing the microarray experiments, and Kristen Parker for performing the histomorphometric analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Canalis E. Mechanisms of glucocorticoid-induced osteoporosis. Curr Opin Rheumatol. 2003;15:454–7. doi: 10.1097/00002281-200307000-00013. [DOI] [PubMed] [Google Scholar]
- 2.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–82. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellows CG, Aubin JE, Heersche JN. Physiological concentrations of glucocorticoids stimulate formation of bone nodules from isolated rat calvaria cells in vitro. Endocrinology. 1987;121:1985–92. doi: 10.1210/endo-121-6-1985. [DOI] [PubMed] [Google Scholar]
- 4.Rickard DJ, Sullivan TA, Shenker BJ, Leboy PS, Kazhdan I. Induction of rapid osteoblast differentiation in rat bone marrow stromal cell cultures by dexamethasone and BMP-2. Dev Biol. 1994;161:218–28. doi: 10.1006/dbio.1994.1022. [DOI] [PubMed] [Google Scholar]
- 5.Sher LB, Woitge HW, Adams DJ, Gronowicz GA, Krozowski Z, Harrison JR, Kream BE. Transgenic expression of 11beta-hydroxysteroid dehydrogenase type 2 in osteoblasts reveals an anabolic role for endogenous glucocorticoids in bone. Endocrinology. 2004;145:922–9. doi: 10.1210/en.2003-0655. [DOI] [PubMed] [Google Scholar]
- 6.Sher LB, Harrison JR, Adams DJ, Kream BE. Impaired cortical bone acquisition and osteoblast differentiation in mice with osteoblast-targeted disruption of glucocorticoid signaling. Calcif Tissue Int. 2006;79:118–25. doi: 10.1007/s00223-005-0297-z. [DOI] [PubMed] [Google Scholar]
- 7.DeVack C, Lupp B, Nichols M, Kowenz-Leutz E, Schmid W, Schutz G. Characterization of the nuclear proteins binding the CACCC element of a glucocorticoid-responsive enhancer in the tyrosine aminotransferase gene. Eur J Biochem. 1993;211:459–65. doi: 10.1111/j.1432-1033.1993.tb17571.x. [DOI] [PubMed] [Google Scholar]
- 8.Drouin J, Sun YL, Chamberland M, Gauthier Y, De Lean A, Nemer M, Schmidt TJ. Novel glucocorticoid receptor complex with DNA element of the hormone-repressed POMC gene. Embo J. 1993;12:145–56. doi: 10.1002/j.1460-2075.1993.tb05640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stocklin E, Wissler M, Gouilleux F, Groner B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature. 1996;383:726–8. doi: 10.1038/383726a0. [DOI] [PubMed] [Google Scholar]
- 10.Grosset C, Jazwiec B, Taupin JL, Liu H, Richard S, Mahon FX, Reiffers J, Moreau JF, Ripoche J. In vitro biosynthesis of leukemia inhibitory factor/human interleukin for DA cells by human endothelial cells: differential regulation by interleukin-1 alpha and glucocorticoids. Blood. 1995;86:3763–70. [PubMed] [Google Scholar]
- 11.Stewart PM, Krozowski ZS. 11 beta-Hydroxysteroid dehydrogenase. Vitam Horm. 1999;57:249–324. [PubMed] [Google Scholar]
- 12.Woitge H, Harrison J, Ivkosic A, Krozowski Z, Kream B. Cloning and in vitro characterization of alpha 1(I)-collagen 11 beta-hydroxysteroid dehydrogenase type 2 transgenes as models for osteoblast-selective inactivation of natural glucocorticoids. Endocrinology. 2001;142:1341–8. doi: 10.1210/endo.142.3.8044. [DOI] [PubMed] [Google Scholar]
- 13.Dacic S, Kalajzic I, Visnjic D, Lichtler AC, Rowe DW. Col1a1-driven transgenic markers of osteoblast lineage progression. J Bone Miner Res. 2001;16:1228–36. doi: 10.1359/jbmr.2001.16.7.1228. [DOI] [PubMed] [Google Scholar]
- 14.Kalajzic I, Kalajzic Z, Kaliterna M, Gronowicz G, Clark SH, Lichtler AC, Rowe D. Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J Bone Miner Res. 2002;17:15–25. doi: 10.1359/jbmr.2002.17.1.15. [DOI] [PubMed] [Google Scholar]
- 15.Liu F, Lee SK, Adams DJ, Gronowicz GA, Kream BE. CREM deficiency in mice alters the response of bone to intermittent parathyroid hormone treatment. Bone. 2007;40:1135–43. doi: 10.1016/j.bone.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gazzerro E, Pereira RC, Jorgetti V, Olson S, Economides AN, Canalis E. Skeletal overexpression of gremlin impairs bone formation and causes osteopenia. Endocrinology. 2005;146:655–65. doi: 10.1210/en.2004-0766. [DOI] [PubMed] [Google Scholar]
- 17.Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 18.McLeod MJ. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology. 1980;22:299–301. doi: 10.1002/tera.1420220306. [DOI] [PubMed] [Google Scholar]
- 19.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 20.Lee SK, Lorenzo JA. Parathyroid hormone stimulates TRANCE and inhibits osteoprotegerin messenger ribonucleic acid expression in murine bone marrow cultures: correlation with osteoclast-like cell formation. Endocrinology. 1999;140:3552–61. doi: 10.1210/endo.140.8.6887. [DOI] [PubMed] [Google Scholar]
- 21.Yang M, Kream BE. Calcitonin induces expression of the inducible cAMP early repressor in osteoclasts. Endocrine. 2008 doi: 10.1007/s12020-008-9092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eyre DR. Bone biomarkers as tools in osteoporosis management. Spine. 1997;22:17S–24S. doi: 10.1097/00007632-199712151-00004. [DOI] [PubMed] [Google Scholar]
- 23.Kalajzic I, Staal A, Yang WP, Wu Y, Johnson SE, Feyen JH, Krueger W, Maye P, Yu F, Zhao Y, Kuo L, Gupta RR, Achenie LE, Wang HW, Shin DG, Rowe DW. Expression profile of osteoblast lineage at defined stages of differentiation. J Biol Chem. 2005;280:24618–26. doi: 10.1074/jbc.M413834200. [DOI] [PubMed] [Google Scholar]
- 24.Mocetti P, Silvestrini G, Ballanti P, Patacchioli FR, Di Grezia R, Angelucci L, Bonucci E. Bcl-2 and Bax expression in cartilage and bone cells after high-dose corticosterone treatment in rats. Tissue Cell. 2001;33:1–7. doi: 10.1054/tice.2000.0144. [DOI] [PubMed] [Google Scholar]
- 25.Silvestrini G, Ballanti P, Patacchioli FR, Mocetti P, Di Grezia R, Wedard BM, Angelucci L, Bonucci E. Evaluation of apoptosis and the glucocorticoid receptor in the cartilage growth plate and metaphyseal bone cells of rats after high-dose treatment with corticosterone. Bone. 2000;26:33–42. doi: 10.1016/s8756-3282(99)00245-8. [DOI] [PubMed] [Google Scholar]
- 26.Zhou H, Mak W, Kalak R, Street J, Fong-Yee C, Zheng Y, Dunstan CR, Seibel MJ. Glucocorticoid-dependent Wnt signaling by mature osteoblasts is a key regulator of cranial skeletal development in mice. Development. 2009;136:427–36. doi: 10.1242/dev.027706. [DOI] [PubMed] [Google Scholar]
- 27.Kim J, Temple KA, Jones SA, Meredith KN, Basko JL, Brady MJ. Differential modulation of 3T3-L1 adipogenesis mediated by 11beta-hydroxysteroid dehydrogenase-1 levels. J Biol Chem. 2007;282:11038–46. doi: 10.1074/jbc.M606197200. [DOI] [PubMed] [Google Scholar]
- 28.Balachandran A, Guan H, Sellan M, van Uum S, Yang K. Insulin and dexamethasone dynamically regulate adipocyte 11{beta}-hydroxysteroid dehydrogenase type 1. Endocrinology. 2008 doi: 10.1210/en.2008-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kershaw EE, Morton NM, Dhillon H, Ramage L, Seckl JR, Flier JS. Adipocyte-specific glucocorticoid inactivation protects against diet-induced obesity. Diabetes. 2005;54:1023–31. doi: 10.2337/diabetes.54.4.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stromstedt PE, Poellinger L, Gustafsson JA, Carlstedt-Duke J. The glucocorticoid receptor binds to a sequence overlapping the TATA box of the human osteocalcin promoter: a potential mechanism for negative regulation. Mol Cell Biol. 1991;11:3379–83. doi: 10.1128/mcb.11.6.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, Zhang Z, Kim JK, Mauvais-Jarvis F, Ducy P, Karsenty G. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–69. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferron M, Hinoi E, Karsenty G, Ducy P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci U S A. 2008;105:5266–70. doi: 10.1073/pnas.0711119105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boban I, Jacquin C, Prior K, Barisic-Dujmovic T, Maye P, Clark SH, Aguila HL. The 3.6 kb DNA fragment from the rat Col1a1 gene promoter drives the expression of genes in both osteoblast and osteoclast lineage cells. Bone. 2006;39:1302–12. doi: 10.1016/j.bone.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 34.Khosla S. Re: “The 3.6 kb DNA fragment from the rat Col1a1 gene promoter drives the expression of genes in both osteoblast and osteoclast lineage cells” by Boban et al. (Bone 39:1302–1312, 2006) Bone. 2007;40:1671–2. doi: 10.1016/j.bone.2006.06.025. author reply 1673–4. [DOI] [PubMed] [Google Scholar]
- 35.Kim HJ, Zhao H, Kitaura H, Bhattacharyya S, Brewer JA, Muglia LJ, Ross FP, Teitelbaum SL. Glucocorticoids suppress bone formation via the osteoclast. J Clin Invest. 2006;116:2152–60. doi: 10.1172/JCI28084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–41. doi: 10.1210/en.2003-0990. [DOI] [PubMed] [Google Scholar]
- 37.Liu F, Woitge HW, Braut A, Kronenberg MS, Lichtler AC, Mina M, Kream BE. Expression and activity of osteoblast-targeted Cre recombinase transgenes in murine skeletal tissues. Int J Dev Biol. 2004;48:645–53. doi: 10.1387/ijdb.041816fl. [DOI] [PubMed] [Google Scholar]
- 38.Mazziotti G, Angeli A, Bilezikian JP, Canalis E, Giustina A. Glucocorticoid-induced osteoporosis: an update. Trends Endocrinol Metab. 2006;17:144–9. doi: 10.1016/j.tem.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 39.Canalis E, Mazziotti G, Giustina A, Bilezikian JP. Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int. 2007;18:1319–28. doi: 10.1007/s00198-007-0394-0. [DOI] [PubMed] [Google Scholar]
- 40.Munck A. Glucocorticoid receptors and physiology: a personal history. Steroids. 2005;70:335–44. doi: 10.1016/j.steroids.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 42.Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O’Brien CA, Thostenson J, Roberson PK, Boskey AL, Clemens TL, Manolagas SC. Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in aged mice. Aging Cell. 2010;9:147–61. doi: 10.1111/j.1474-9726.2009.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology. 2004;145:5439–47. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 44.Xiong Y, Connolly T, Futcher B, Beach D. Human D-type cyclin. Cell. 1991;65:691–9. doi: 10.1016/0092-8674(91)90100-d. [DOI] [PubMed] [Google Scholar]
- 45.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 46.Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat Rev Mol Cell Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- 47.Turner BM. Histone H4, the cell cycle and a question of integrity. Bioessays. 1995;17:1013–5. doi: 10.1002/bies.950171204. [DOI] [PubMed] [Google Scholar]
- 48.Kimura H, Cook PR. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J Cell Biol. 2001;153:1341–53. doi: 10.1083/jcb.153.7.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jackson V, Chalkley R. A reevaluation of new histone deposition on replicating chromatin. J Biol Chem. 1981;256:5095–103. [PubMed] [Google Scholar]
- 50.Cheng SL, Lai CF, Fausto A, Chellaiah M, Feng X, McHugh KP, Teitelbaum SL, Civitelli R, Hruska KA, Ross FP, Avioli LV. Regulation of alphaVbeta3 and alphaVbeta5 integrins by dexamethasone in normal human osteoblastic cells. J Cell Biochem. 2000;77:265–76. doi: 10.1002/(sici)1097-4644(20000501)77:2<265::aid-jcb9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 51.Tani-Ishii N, Tsunoda A, Umemoto T. Osteopontin antisense deoxyoligonucleotides inhibit bone resorption by mouse osteoclasts in vitro. J Periodontal Res. 1997;32:480–6. doi: 10.1111/j.1600-0765.1997.tb00563.x. [DOI] [PubMed] [Google Scholar]
- 52.Reinholt FP, Hultenby K, Oldberg A, Heinegard D. Osteopontin--a possible anchor of osteoclasts to bone. Proc Natl Acad Sci U S A. 1990;87:4473–5. doi: 10.1073/pnas.87.12.4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mohiti J, Caswell AM, Walker JH. Calcium-induced relocation of annexins IV and V in the human osteosarcoma cell line MG-63. Mol Membr Biol. 1995;12:321–9. doi: 10.3109/09687689509072434. [DOI] [PubMed] [Google Scholar]
- 54.Giner RM, Mancini L, Kamal AM, Perretti M. Uneven modulation of the annexin 1 system in osteoblast-like cells by dexamethasone. Biochem Biophys Res Commun. 2007;354:414–9. doi: 10.1016/j.bbrc.2006.12.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kirsch T, Harrison G, Golub EE, Nah HD. The roles of annexins and types II and X collagen in matrix vesicle-mediated mineralization of growth plate cartilage. J Biol Chem. 2000;275:35577–83. doi: 10.1074/jbc.M005648200. [DOI] [PubMed] [Google Scholar]
- 56.Damazo AS, Moradi-Bidhendi N, Oliani SM, Flower RJ. Role of annexin 1 gene expression in mouse craniofacial bone development. Birth Defects Res A Clin Mol Teratol. 2007;79:524–32. doi: 10.1002/bdra.20368. [DOI] [PubMed] [Google Scholar]
- 57.Shibanuma M, Kuroki T, Nose K. Isolation of a gene encoding a putative leucine zipper structure that is induced by transforming growth factor beta 1 and other growth factors. J Biol Chem. 1992;267:10219–24. [PubMed] [Google Scholar]
- 58.Leung YF, Tam PO, Lee WS, Lam DS, Yam HF, Fan BJ, Tham CC, Chua JK, Pang CP. The dual role of dexamethasone on anti-inflammation and outflow resistance demonstrated in cultured human trabecular meshwork cells. Mol Vis. 2003;9:425–39. [PubMed] [Google Scholar]
- 59.Sena K, Leven RM, Mazhar K, Sumner DR, Virdi AS. Early gene response to low-intensity pulsed ultrasound in rat osteoblastic cells. Ultrasound Med Biol. 2005;31:703–8. doi: 10.1016/j.ultrasmedbio.2005.01.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.