

Abstract
In many experimental systems, proinflammatory stimuli exhibit proconvulsant properties. There are also accumulating data suggesting that inflammation may contribute to epileptogenesis in experimental models as well as in humans. Using two different models (Lithium-pilocarpine induced-status epilepticus (SE) and rapid kindling), we address this issue in the developing brain. Using P14 wistar rat pups, we showed that inflammation induced by LPS results, after SE, into a more severe disease in adulthood. The main histological feature was an active gliosis that was observed only when inflammation and SE was combined. The use of a kindling model at P14, a model where seizure progress without any neurodegeneration, permits to show that systemic inflammation is responsible of an enhancement of epileptogenesis. The role of inflammation should be further explored in immature brain to identify therapeutic targets that may be relevant to clinical practice where the association of inflammation and epileptic events is common.
Keywords: Developing brain, Epileptogenesis, Inflammation, Kindling, LPS, Status epilepticus
Introduction
Febrile seizures (FS) are seizures triggered by fever that occur in 3–5% of children between the age of 6 months and 5 years (Berg and Shinnar, 1996). In humans, retrospective analyses have considered FS, in particular prolonged FS, as a risk factor for the development of temporal lobe epilepsy (TLE) (Cendes et al., 1993;French et al., 1993). Currently, it is not proven that FS is responsible of epileptogenesis leading to TLE. FS can be a symptom of other factors that lead to the epileptogenic process. This question is particularly complex, because prospective studies in children with FS have not yet demonstrated the development of TLE.
Many experimental models of FS have used hyperthermia. However, very different physiologic mechanisms underlie fever and hyperthermia (Berg, 1993). A treatment with bacterial endotoxin lipopolysaccharide (LPS) permits to induce inflammatory mechanisms as with fever. In many experimental systems, proinflammatory stimuli exhibit proconvulsant properties (Vezzani and Granata, 2005). Recently, the involvement of interleukin-1 (IL-1) pathway in epileptogenesis has been suggested (Ravizza et al., 2008). The role of inflammation as a causative factor in human epileptogenesis has also been explored. An association of prolonged FS and TLE with hippocampal sclerosis with a polymorphism in the promoter of IL-1β gene (IL-1β-511T) was reported (Kanemoto et al., 2000;Kanemoto et al., 2003). These data have been challenged by other studies (Buono et al., 2001;Heils et al., 2000). However, a recent meta-analysis suggest the link between IL-1β gene polymorphism and TLE (Kauffman et al., 2008).
In order to mimic human disease condition, it seems important to understand how inflammation may affect epileptogenesis in immature brain. However, few data are available. Using the lithium-pilocarpine model of SE at P9 and P21, it has been shown that age-dependent brain inflammation induced by SE and vascular changes were associated with epileptogenesis, suggesting that these phenomena are implicated in the mechanisms underlying the occurrence of spontaneous seizures (Marcon et al., 2009). When LPS is given prior to systemic kainate injection or prior to short hyperthermic seizure, it results in a long term modification of brain excitability (Auvin et al., 2009;Heida et al., 2005). However, recent data suggest that inflammation in developing brain causes by itself a long lasting increase in seizure susceptibility (Galic et al., 2008).
Here, we studied the role of LPS injection on the epileptogenesis in the developing brain. For the first time, we have used two models responsible of epileptogenesis in immature brain: the lithium-pilocarpine model and a rapid kindling model. We used lithium-pilocarpine in P14 rats with or without LPS injections. Then we studied the rats 3 months after the initial SE to examine both epileptogenesis and long term histological changes. Moreover, we used a rapid kindling model to study epileptogenesis in immature brain without any acute neuronal injury. It is the first study in immature brain exploring the effect of induced-inflammation in models of epileptogenesis.
Materials and Methods
1/Lithium-Pilocarpine model
Animals, injection of inflammatory factor, and induction of seizures
Wistar male rat pups (Charles River Laboratories, Wilmington, MA, U.S.A.) were housed in standard laboratory conditions with controlled temperature/humidity, a 12-h light/dark cycle, and free access to food and water. Studies were approved by the Animal Research Committee at the University of California, Los Angeles.
At P13, animals were injected subcutaneously with 3 mEq/kg lithium chloride (Sigma, St. Louis, MO, U.S.A.). After 14–18 h, rats received i.p. injections of either LPS (50μg/kg, E. coli serotype 055:B5;Sigma), or vehicle (LiPC group). SE was induced 2 h later by s.c. injection of pilocarpine (PC, Sigma) in a dose of 60 mg/kg at P14. Only rats that demonstrated behavioral manifestations of seizures progressing to forelimbs clonus during at least 1h were used for further studies.
In order to investigate whether sustained inflammation affected the long term consequences of SE, a separate group of 2-week-old animals were given 50 μg/kg of LPS (Sigma) or vehicle 2 h preceding the injection of PC (60 mg/kg), followed by repeated injections of 50 μg/kg of LPS (3LPS+LiPC) or vehicle 24 and 48 h after the first LPS treatment (LPS+LiPC). A control group of three animals received lithium chloride followed by LPS and atropine, and finally LPS 24 and 48 hours after atropine. The table 1 represents the four studied groups.
Table 1.
Design of study: studied groups in the lithium-pilocarpine models
| 2 hours before pilocarpine s.c. | Pilocarpine | 24 hours after SE | 48 hours after SE | |
|---|---|---|---|---|
| Control (n=3) | 50μg/kg of LPS | 0 | 50μg/kg of LPS | 50μg/kg of LPS |
| LiPC (n=11) | Vehicle | 60mg/kg | vehicle | vehicle |
| LPS+LiPC (n=8) | 50μg/kg of LPS | 60mg/kg | vehicle | vehicle |
| 3LPS+LiPC (n=8) | 50μg/kg of LPS | 60mg/kg | 50μg/kg of LPS | 50μg/kg of LPS |
We use an i.p. injection of LPS two hours prior the procedure as we did in our previous studies (Auvin et al., 2007;Auvin et al., 2009). We originally based this choice on experiments that demonstrate that the higher proconvulsant effect of LPS using PTZ model was 2 hours and 8 hours after the LPS injection (Sayyah et al., 2003). Moreover, Heida et al. have used a LPS injection 2.5h prior a subconvulsant dose of kainate (Heida et al., 2004). A recent study has also reported that the levels of cytokines in brain after i.p. injection of LPS reached the highest level after 1h–2h (Kwon et al., 2010). The repetitive injection of LPS after lithium-pilocarpine SE (3LPS+LiPC) was design in order to evaluate the effect of sustained inflammation. We already reported a higher level of cell injury 72h after SE at P14 when LPS was given repetitively (Auvin et al., 2007).
EEG recordings (Figure 1)
Figure 1.

A: Table with the number of epileptic rats and the various type of seizures (Stage 1–2, Stage 3–4, reflex seizure) that were observed in the studied groups. B: example of a stage 1 seizure in a rat from the LiPC group. C: Example of a stage 4 reflex seizure in a rat from the 3LPS-LiPC group.
Three months after the initial SE, all animals were recorded 24hours a day for a 6-day period. Following isoflurane anesthesia administration, the two leads of the EEG transmitter were operatively implanted on the dura through two drilled bilateral burr holes in the skull on each side of the cranium, (AP: 4.5 mm; ML: 4 or - 4 mm; V: 0 mm to the bregma). After surgery, the rats were transferred to the cage that was placed on top of the telemetry receiving platform (Data Sciences International, Arden Hills, MN, USA) and continuous EEG monitoring was performed after a 24 hours period of recovery. We recorded each rat 24 hours a day during 6 consecutive days (i.e. 144 hours of continuous recording). Stellate software (Montreal, Quebec, Canada) was used to capture the data that were then manually reviewed offline for epileptiform events. Video recording was combined with the EEG for each rat for duration of 24 hours minimum. Analysis was carried out based on amplitude, frequency and time with EEG evidence of seizures.
Histology
All rats were euthanized with pentobarbital (100 mg/kg, i.p.) and underwent transcardiac perfusion-fixation with saline followed by 4% paraformaldehyde 24 h after the end of the video-EEG recording (n=11 LiPC group, n=8 LPS+LiPC, n=8 3LPS+LiPC and n=3 controls). Brains were removed, dehydrated, embedded in paraffin, cut at 8-μmthick coronal sections, and stained with either hematoxylin & eosin (H&E, Sigma)
To address the concern of acute neuronal injury, Fluoro- Jade B (F-JB, Histochem Inc., Jefferson, AR, U.S.A.) were used. Sections for Fluorojade-B were deparaffinized, rehydrated, incubated with KMnO4 followed by 0.001% Fluorojade-B. Injured (green fluorescent) cells were examined bilaterally in the CA1, CA3, hilus and dentate gyrus in three adjacent sections in the hippocampus approximating -3.6mm posterior to bregma. Cell counting using H&E staining was also performed in the same areas of the hippocampus (Paxinos and Watson, 1982). Quantification of cell density was performed with a 1-cm2 10 × 10-box microscopic grid. The grid of counting was placed on a well-defined area of the cerebral structure of interest, and counting was carried out with a microscopic enlargement of 200- or 400-fold defined for each single cerebral structure. Cell counts were performed twice on each side of three adjacent sections for each region by a single observer unaware of the animal’s treatment (SA). The number of cells obtained in the 12 counted fields in each cerebral structure was averaged. Neurons touching the inferior and right edge of the grid were not counted. Counts involved only neurons with cell bodies larger than 10 μm. Smaller cells considered as glial cells were not counted.
Immunohistochemistery
GFAP immunostaining
The slides were deparaffined and rehydrated. The sections were then preincubated in 2% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) containing 0.3% Triton X-100 for 30 min and incubated with polyclonal anti-GFAP from rabbit diluted 1:200 in 2% BSA in PBS–Triton X-100 for 48 h at 4 °C. After washing several times, tissue sections were incubated in a rabbit PAP-conjugated anti-rabbit IgG diluted 1:50 in PBS at room temperature for 2 h. The immunohistochemical reaction was revealed by incubating the sections in a histochemical medium that contained 0.06% 3,3-diaminobenzidine dissolved in PBS for 10 min and then, in the same solution containing 1 μM of 3% H2O2 per milliliter of DAB medium for approximately 10 min. Three sections (six hippocampi) from each rat (sections every 80 μm) were used to perform optical density analysis.
Values of background staining were subtracted from the immunoreactive intensities of CA1 area
NeuN-GFAP double immunostaining
The slides were deparaffined and rehydrated. The sections were then preincubated in 2% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) containing 0.3% Triton X-100 for 30 min and incubated with polyclonal anti-GFAP (Chemicon) from rabbit diluted 1:200 and anti-NeuN from mouse (Chemicon) diluted 1:100 in 2% BSA in PBS–Triton X-100 for 48 h at 4 °C. After washing several times, fluorescence immunohistochemistry was performed using Alexa Fluor tagged secondary antibodies Alexa 488 (green) and Alexa 568 (red) (Molecular probes, Eugene, OR, USA).
2/Rapid kindling model
Surgery
At P13, the animals were anesthetized with Isoflurane and stereotaxically implanted with a twisted bipolar stimulating electrode (Plastics1 Inc., Roanoke, VA, U.S.A.) in the left ventral hippocampus. The coordinates with respect to Bregma were 3.0-mm posterior, 3.9-mm left, 4.2-mm ventral. A tripolar recording electrode (Plastics1 Inc.) was wrapped around skull screws using the nasal bone as the ground.
Kindling procedure
The rapid kindling protocol (RKP) was adapted for immature animals (Michelson and Lothman, 1991). Twenty-four h after electrode implantation, the animals were connected to the DS8000 electrical stimulator via DSI100 stimulus isolators (World Precision Instruments, Sanasota, FL, U.S.A.) and to the MP100/EEG100B acquisition system (BIOPAC, Santa Barbara, CA, U.S.A.). EEG was acquired using AcqKnowledge 3.8 software (BIOPAC) along with simultaneous digital video. Both EEG and behavioral responses were analyzed off-line in a blinded fashion. At the beginning of the experiment, afterdischarge threshold (ADT) and afterdischarge duration (ADD) were detected by applying electrical stimuli—10-s train duration, 20 Hz, 1-ms pulse duration, square wave monophasic stimuli, starting at 0.2 mA, with 0.1-mA increments, delivered every 10 min.
The RKP started 5 minutes after the determination of the afterdischarge. Kindling consisted of 60 trains delivered every 5 min using the parameters described above using a current of 100 μA over the ADT. Behavioral seizures were scored: 1—Motor arrest and twitching vibrissae; 2—chewing, head bobbing; 3—forelimb clonus; 4—forelimb clonus and rearing; 5—rearing and falling (Mazarati et al., 2007;Mazarati et al., 2008). Kindling progression was analyzed by calculating the number of stage 4–5 seizures and the duration of electrographic correlates of stage 4–5 convulsions. In order to examine kindling retention, 24 hours after the end of the RKP, animals were reconnected to the stimulating recording system and afterdischarge properties were studied again. Animals were considered kindled if they showed statistically significant decrease of ADT, prolongation of ADD and responded with a behavioral seizure of any stage to the threshold stimulation (Mazarati et al., 2007;Mazarati et al., 2008;Mazarati et al., 2009).
Design of the RKP study
All the experiments were done at room temperature 22–24°C. The rat pups were randomly divided into 2 groups: (1) sham group receiving saline i.p. 2h prior the RKP (n=12); (2) LPS treated rats receiving 50μg/kg i.p. 2h prior the RKP (n=12).
We also evaluate the ADT and ADD in 2 additional groups: (1) sham group receiving saline i.p. 24h prior the AD evaluation (n=7); (2) LPS treated rats receiving 50μg/kg i.p. 24h prior the AD evaluation (n=7). This last experiment was done to evaluate the role of the timing in LPS injection on the AD. We did not perform the whole kindling procedure in these groups.
Histological study
After the end of the experiments, three animals in each group were anesthetized with pentobarbital (100 mg/kg), and underwent intracardiac perfusion with saline followed by paraformaldehyde. Brains were removed, dehydrated, embedded in paraffin, cut at 8-μm thick coronal sections, and stained with hematoxylin & eosin (H&E, Sigma), fluorojade-B and GFAP immunostaining as described above.
Statistical analysis
Data are expressed as the mean ± standard error of the mean (SEM). Kruskall–Wallis one-way analysis of variance (ANOVA) with post hoc Dunn’s test, or Mann–Whitney rank sum test were performed. Categorical variables were analyzed by using either the χ2 test or the Fisher’s exact test (GraphPad Prism5 Software Inc, San Diego, CA, U.S.A.). p ≤ 0.05 was considered significant.
For the study part using the lithium-pilocarpine model of SE, three types of comparison were done. The first analysis compared the three studied groups. A second analysis was done in order to analyze the effect of LPS. We compared all LPS-treated animals (LPS+LiPC and 3LPS+LiPC) that had SE to those that experienced SE without any LPS injection (LiPC). The third analysis was done in order to identify if the findings could be related to the fact that animals have become epileptic. In this third analysis, we compared epileptic and non-epileptic animals irrespectively to their initial groups.
Results
In the lithium-pilocarpine model, LPS treated animals that had SE exhibit more severe epilepsy at adulthood (Figure 1)
Using a 6-day period EEG telemetry, we found that 3/11 rats of the LiPC group became epileptic while 5/8 and 4/8 became epileptic in LPS+LiPC (p=0.14 vs LiPC) and 3LPS+LiPC groups (p=0.3 vs LiPC), respectively (Figure 1). We observed a trend to observed more severe seizure in both LPS+LiPC and 3LPS+LiPC compare to LiPC (0/11 in LiPC vs 3/8 in LPS+LiPC; p=0.06 and 0/11 in LiPC vs 3/8 in 3LPS+LiPC; p=0.06). More severe seizures (stage 3–4) were only observed in groups that received LPS (LPS+LiPC and 3LPS+LiPC groups) (p=0.05) (Figure 1C). The mean number of seizure per animal per day was similar among the groups (9.6 Sz/day/animal in the LiPC group; 6.2 Sz/day/animal in the LPS+LiPC group and 4.7 Sz/day/animal in the 3LPS+LiPC group). None of the 3 controls exhibited either seizure or epileptiform activity during the recording.
Three months after the lithium-pilocarpine-SE, the number of pyramidal cell in CA-1 was comparable between the groups while we found active gliosis in LPS-treated that had experienced SE
We did not observe any difference in pyramidal cell counts in CA1 among the groups (Figure 2; upper left corner). There was also no difference in cell counts when the animals were compared according to the fact they were LPS-treated (LPS+LiPC (n=8) and 3LPS+LiPC (n=8) groups) or they had SE without LPS administration (LiPC (n=11)) (Figure 2; lower left corner). There was a trend of cell loss in CA-1 when epileptic animals were compared to non epileptic animals (Figure 2; upper right corner). This data suggest that cell loss may be linked to the epileptic state of the animals.
Figure 2.
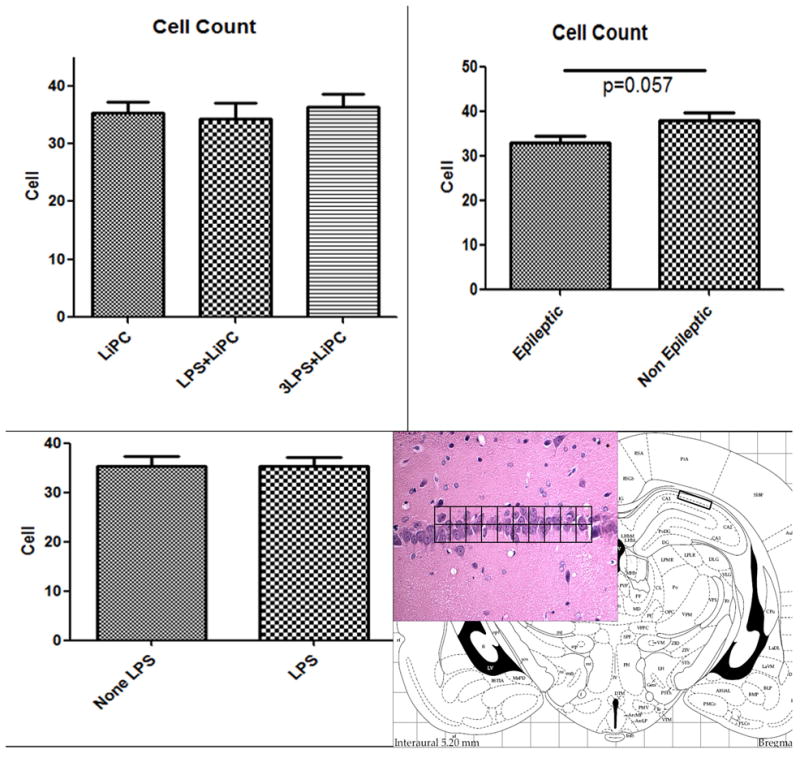
Histograms of the number (mean ± SEM) of pyramidal cell in CA-1 counted using a grid. A: pyramidal cell counts in CA-1 in the three studied groups. B: pyramidal cell counts in CA-1 comparing epileptic animals versus non-epileptic animals. C: pyramidal cell counts in CA-1 comparing LPS-treated versus non-LPS-treated animals. D: Hematoxylin & Eosin stained brain section taken at the level of the hippocampus representing the areas where the cell counts were performed.
All animals had a mild gliosis in CA-1 but we observed a very intense reactive gliosis in CA1 in all LPS-treated animals that experienced SE (Figure 3). None of the controls nor animals from LiPC group had such active gliosis (Figure 3A–B). These glial cells appeared to be more hypertrophied with thickened processes in the LPS-treated that experienced SE compared to both controls and rats from the LiPC group (Figure 3C–D). Using measurement of GFAP staining optical densities in CA-1, we found a mean density of 8.7±1.2 in the LiPC group, of 16.6±3 in the LPS+LiPC group and of 14.2±1.9 in the 3LPS+LiPC group. We found a significant difference between LiPC vs LPS+LiPC (p=0.03) and LiPC vs 3LPS+LiPC (p=0.05) while there was no difference between LPS-treated group (LPS+LiPC vs 3LPS+LiPC)
Figure 3.
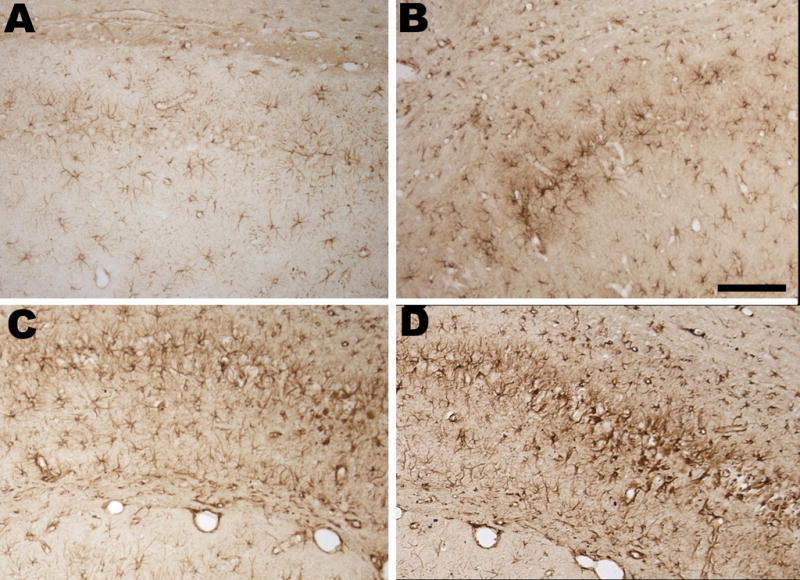
GFAP-Immunostaining in the hippocampus. A: CA-1 of a rat from control group. B: CA-1 of a rat from LiPC group at higher magnification showing a moderate gliosis. C: CA-1 of a rat from LPS+LiPC group at higher magnification showing strong reactive gliosis. D: CA-1 of a rat from 3LPS+LiPC group at higher magnification showing strong reactive gliosis. Note in both C and D panel the aspect of the glial cells that are hypertrophic with thick processes.
Three months after the initial SE, we did not observe any fluorojade-B positive cell in LiPC group while we observed fluorojade-B positive cells in CA-1 in 2 rats from LPS+LiPC group and in 3 rats from 3LPS+LiPC group. 4 of 9 LPS-treated rats that developed spontaneous seizures have fluorojade-B positive cells in CA-1 (Figure 4D) while none of the animals that had SE showed such findings. We did not observe any fluorojade-B positive cell in other area. Looking at the slides from the same brains with H&E staining, we observed gliosis reaction within CA-1 (Figure 4A). We also observed small acidophilic cells that were surrounded by glial reaction (Figure 4A). The double staining (NeuN-GFAP) showed that active gliosis surrounded small neuronal cells in CA-1 (Figure 4B–C).
Figure 4.
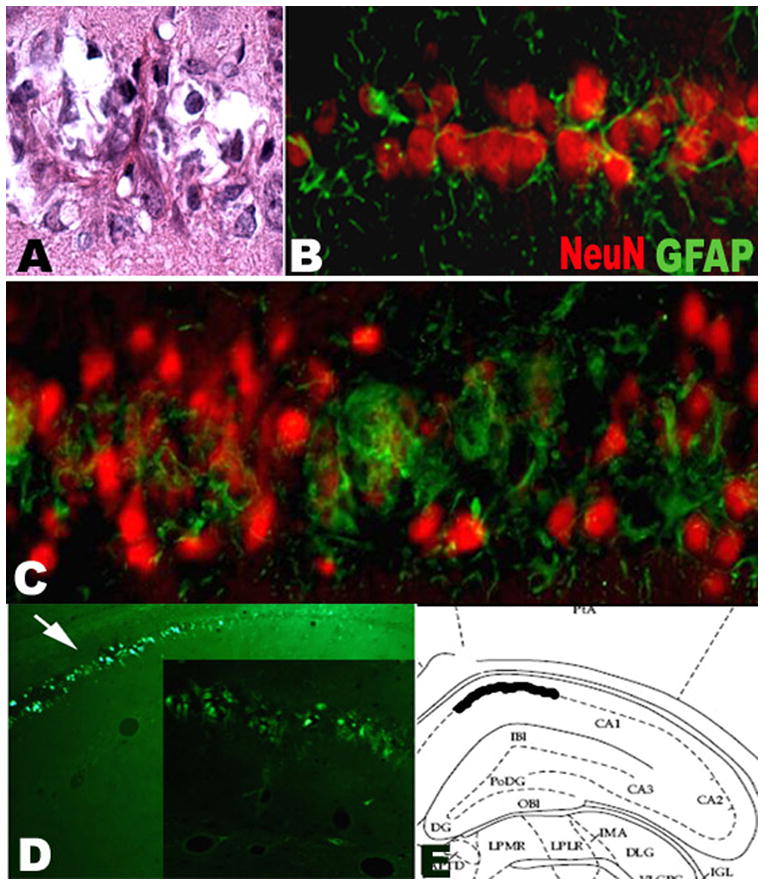
A: H&E showing damage of CA-1 in a rat from LPS+LiPC group. A small cell with acidophilic cytoplasm is surrounded by a fibrillar structure. B: Double immunostaining NeuN-GFAP in CA-1 area from LiPC group showing a mild gliosis within CA-1; C: Double immunostaining NeuN-GFAP in CA-1 with strong gliosis in a rat from LPS+LiPC. The gliosis surounds the neurons. Note the decrease of the size of the NeuN staining in the area of gliosis. D: Fluorojade-B staining in CA-1 area that was observed only in LPS treated animals that exhibit spontaneous recurrent seizures. E: Representation in a stereotaxic atlas of the location of the positive Fluorojade-B staining.
LPS enhances epileptogenesis in kindling model at P14 (Table 2)
Table 2.
Results of the kindling protocol in the two studied groups
| SSI + kindling (n=12) | LPS + kindling (n=12) | |
|---|---|---|
| Initial ADT, mA | 1.6±0.15 | 1.3±0.16 |
| Initial ADD, s | 26.3±2.4 | 33.3±2.8 |
| Number of stimulation to the first St. 4 seizure | 25.5±1.3 | 10.7±0.6 * |
| Number of stage 4 seizure | 17.2±1.0 | 36.2±0.7 * |
| Mean duration of the seizure EEG, s | 59.6±3.6 | 140.8±6.2 * |
| ADT 24 hrs after kindling, mA | 0.9±0.06 | 0.6±0.06 * |
| ADD 24 hrs after kindling, s | 51.8±1.7 | 162.5±7.3 * |
| Score of seizure induced by the retest | 2.7±0.2 | 3.7±0.2* |
Using a kindling model at P14, we found that the systemic injections of LPS 2h prior the kindling procedure have no effect on both baseline afterdischarge threshold (ADT) and afterdischarge duration (ADD). During the kindling process, the rat pups that received LPS experienced a higher number of stage 4 seizure (10.7±0.6 in LPS+RKP (n=12) vs 25.5±1.3 in RKP (n=12); p<0.05) and a longer mean duration of the seizure as well (140.8sec±6.2 in LPS+RKP vs 59.6sec±3.6 in RKP; p<0.05). The enhancement of epileptogenesis induced by LPS was sustained at the time of the retest. The ADT was lower (0.6mA±0.06 in LPS+RKP vs 0.9mA±0.06;p<0.05) while the ADD (162.5sec±7.3 vs 51.8sec±1.7;p<0.05) was increased 24 hours in the LPS+RKP group compared to the RKP group. In addition, the retest induced stronger seizure with a score at 3.7±0.2 in LPS+RKP and at 2.7±0.2 in RKP. No cell injury was observed in both groups.
When LPS was given 24 hours prior the AD evaluation (n=7 in each group), we did not observe any difference between the groups in both ADT (2mA±0.4 in saline 24 hours prior AD evaluation (n=7) vs 1.4mA±0.2 in LPS 24h prior AD evaluation (n=7); p=0.4) and ADD (83.1 ±10.9 saline vs 68.6 ±7.5 LPS 24 h prior; p=0.17).
We did not observe any cell injury (H&E or Fluorojade-B) or any significant gliosis in the different groups using the RKP.
Discussion
We showed that inflammation enhances the severity of the disease after epileptogenesis following lithium-pilocarpine induced-SE in immature rat brain. We also observed a strong reactive gliosis three months after the initial SE in LPS-treated animals. Our previous data showed that injection of LPS at 22–24 RT did not result in the increase of body temperature or increase of the duration of SE (Auvin et al., 2007); therefore the observed changes cannot be attributed to hyperthermia or to a longer duration of seizure. We also study, for the first time, the effect of induced-inflammation using a rapid kindling model in immature brain. By using this model that does not induce acute cell injury, we observed an enhancement of epileptogenesis by a systemic injection of LPS without any modification of baseline excitability.
It is now well established that consequences of SE in experimental models vary according to the maturity of the brain and to the different models (Haas et al., 2001;Sankar et al., 1998). In the lithium-pilocarpine model, acute cell injury pattern is different depending when SE is induced. Damage to the CA1 neurons was maximal in the 2- and 3-week-old pups and decreased as a function of age (Sankar et al., 1998). We recently showed that LPS exacerbated hippocampal injury in both P7 and P14 rat pups (Auvin et al., 2007). Regarding the long term consequences, few animals that underwent SE at 2 weeks of age developed spontaneous seizures later in life while most of the animals that underwent SE at 3 or 4 weeks of age exhibit spontaneous seizures (Dubé et al., 2001;Priel et al., 1996;Sankar et al., 1998). Here, we showed that the number of epileptic animals increases in LPS-treated animals suggesting that inflammation concomitant to SE enhance epileptogenesis. However, the number of epileptic animal did not reach the significant. We found a significant difference only for Stage 3–4 seizure suggesting a more severe disease in the combined LPS-treated groups.
The role of inflammation in epileptogenesis was first suggested by the fact that inflammatory mediators were upregulated in both astrocytes and neurons after SE until the onset of spontaneous seizures (Ravizza et al., 2008). These studies had particularly highlighted the potential role of IL-1 system (Ravizza et al., 2005;Ravizza et al., 2007). It has been also suggested that IL-1β is involved in epileptogenesis using a selective inhibition of Interleukin Converting Enzyme (ICE) cleaving the biologically active form of IL-1β (Ravizza et al., 2008). In humans, a polymorphism in the promoter region of the IL-1β gene is linked to temporal lobe epilepsy (TLE) (Kanemoto et al., 2000;Kanemoto et al., 2003;Kauffman et al., 2008).
In immature brain, inflammation and microvasculature changes were observed after lithium-pilocarpine induced-SE in P21 but not in P9 rats. These changes were seen only in animals showing spontaneous seizures in adulthood. This study has suggested that inflammation becomes self-sustained and chronic only in a fraction of animals (Marcon et al., 2009). In the developing brain, it has been shown that neuroinflammation by itself in early development or a combination of LPS injection and a model of seizure causes a long-lasting increase in seizure susceptibility (Auvin et al., 2009;Galic et al., 2008;Heida et al., 2005). Here, we showed that combination of neuroinflammation and lithium-pilocarpine induced SE had aggravated epileptogenesis by a modification of the severity of the disease. In the lithium-pilocarpine model, our findings may be compared to other studies that combined factors to produce a ‘double-hit injury’ (Koh et al., 2004;Scantlebury et al., 2005;Schmid et al., 1999;Setkowicz et al., 2006). The double hit hypothesis suggested that a combination of two factors is needed in immature brain to be responsible of a significant epileptogenesis. The association of hypoxia, freeze-induced cortical microgyric lesion, cortical mechanical injury or repetitive neonatal seizure with a latter prolonged seizure is responsible of the increase of SE-induced consequences. Here, the induced-neuroinflammation associated with lithium-pilocarpine induced-SE at P14 may be considered as a double injury model.
The increase of the number of animal with epilepsy and the increase of the severity in epilepsy in our study may be related to the increase of SE-induced cell injury by inflammation after lithium-pilocarpine at P14 (Auvin et al., 2007). Using a perinatal Hypoxia-Ischemia model to induce post-stroke epilepsy, it has been strongly suggested that a significant injury is required to induce epileptogenesis with spontaneous recurrent seizures (Kadam et al., 2010). Looking at the cell count in CA-1 in adulthood, we did not observe any difference among the groups. However, a trend was observed in the comparison of epileptic versus non-epileptic animals. This discrepancy between our results and our previous study may be due to the methods of cell count. We can not also exclude that the process of cell injury was still ongoing because we observed fluorojade-B positive cells.
The development of glial activation after experimental SE is also well known. It has been shown in several models of epilepsy after SE (Aronica et al., 2000;Immonen et al., 2008). An active astrogliosis has been also described in other models of epileptogenesis without cell degeneration such as kindling procedure (Khurgel et al., 1995). After SE onset, hippocampal glia activation, cytokine expression, and neuronal damage seem to be an age-dependent phenomena (Marcon et al., 2009). In the hippocampus, neuronal injury occurs only when cytokines are induced in glia, and cytokine synthesis precedes the appearance of degenerating neurons. It has also been shown, in immature brain, that neuroinflammation itself may result in glial activation in adulthood (Galic et al., 2008). The combination of LPS and lithium-pilocarpine induced-SE may explain the observed strong active gliosis (LPS-SE and 3LPS-SE groups). A mild gliosis was observed in LPS and in SE group. Undoubtedly, the most conspicuous property of astrocytes is their morphological transformation in response to virtually any type of neural insult. This aspect of astrocytic function has been studied extensively, but its functional significance is vague in most types of CNS perturbations. Studies of tissue from patients with various forms of epilepsy and the experimental work on animals reveal that an apparent gliosis is almost always present in brain regions which exhibit epileptiforrn activity. Astrogliosis is a common feature observed in patients with mesial temporal lobe epilepsy (Eid et al., 2008). The involvement of glial cells in seizure occurrence is now well established (Angulo et al., 2004;Tian et al., 2005). Several studies showed the implication of the number and distribution of astrocytes to enhance seizure susceptibility in a number of seizure models (Oberheim et al., 2008;Somera-Molina et al., 2007). An increasing number of studies have shown that astrocytes can be significant sources of extracellular glutamate (Eid et al., 2008). Astrocytes can also synthesize and release proinflammatory cytokines like microglia (Dong and Benveniste, 2001;Kipp et al., 2008;Vezzani et al., 2008;Wetherington et al., 2008).
Three months after the initial SE, we observed fluorojade-B positive cells in CA-1. This was observed only in 4/9 epileptic animals form the LPS-treated and SE groups (LPS+LiPC and 3LPS+LiPC). Several studies have shown that neuronal cell deaths are induced by the initial SE but not by the repeated spontaneous seizure that occurred after the epileptogenesis phase (Du et al., 1995;Nevander et al., 1985;Schwob et al., 1980). However, it has been described that few FJ positive stained cells may be observed 6 weeks after SE in CA1, in CA-3 and in the piriform cortex. This was not observed in rats 3 months after they had SE (Gorter et al., 2003). The majority of cells that die as a consequence of the initial SE, degenerate in the course of the first weeks after SE (Covolan and Mello, 2000;Gorter et al., 2003;Ingvar et al., 1988;Poirier et al., 2000). In our study, it seems that the reactive gliosis in CA-1 surround neuron and may be responsible of cell suffering or cell injury. We are currently unable to describe the evolution of such cells. Since we observed such cell only in rat that exhibit strong reactive gliosis, we should consider that the amplification of glutamate toxicity by glia might be involved (Eid et al., 2008).
Using a kindling model in P14 rats, we were able to study the effect of inflammation on epileptogenesis in the developing brain in a model where seizure progression occurs in the absence of neurodegeneration. We found that systemic injection of LPS enhances epileptogenesis. The systemic injection of LPS did not result in any significant modification of the baseline excitability when LPS is given 2hours or 24 hours prior the AD evaluation. We can then exclude a facilitation of the epiletogenesis by a prior hippocampal hyperexcitability. During the kindling process, the acquisition of the kindling was easier. These effects were still observed on the afterdischarge retest. We conclude that systemic inflammation by LPS injection is responsible of increased epileptogenesis in immature brain. As previously described in this model, we did not observe any acute cell injury (Mazarati et al., 2007;Mazarati et al., 2008;Mazarati et al., 2009). The absence of significant gliosis is probably related to the short duration of this procedure. we cannot exclude the involvement of glial cells only on the absence of histological change.
In conclusion, neuroinflammation induced by systemic injection of LPS in immature brain is responsible of worsen of the consequences of the lithium-pilocarpine model. Regarding the recent data on neuroinflammation in immature brain, we conclude that inflammation combine with SE should be considered as a double hit injury model. The use of a kindling model a P14 permits to show that systemic inflammation is responsible of an enhancement of epileptogenesis. The role of inflammation should be further explored in immature brain to identify therapeutic targets that may be relevant to clinical practice where the association of inflammation and epileptic events is common.
Acknowledgments
This work was supported by NS059505, MH079933 and the Epilepsy Foundation of America
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, Gorter JA. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci. 2000;12:2333–2344. doi: 10.1046/j.1460-9568.2000.00131.x. [DOI] [PubMed] [Google Scholar]
- Auvin S, Porta N, Nehlig A, Lecointe C, Vallee L, Bordet R. Inflammation in rat pups subjected to short hyperthermic seizures enhances brain long-term excitability. Epilepsy Res. 2009;86:124–130. doi: 10.1016/j.eplepsyres.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Auvin S, Shin D, Mazarati A, Nakagawa J, Miyamoto J, Sankar R. Inflammation exacerbates seizure-induced injury in the immature brain. Epilepsia. 2007;48(Suppl 5):27–34. doi: 10.1111/j.1528-1167.2007.01286.x. [DOI] [PubMed] [Google Scholar]
- Berg AT. Are febrile seizures provoked by a rapid rise in temperature? Am J Dis Child. 1993;147:1101–1103. doi: 10.1001/archpedi.1993.02160340087020. [DOI] [PubMed] [Google Scholar]
- Berg AT, Shinnar S. Unprovoked seizures in children with febrile seizures: short-term outcome. Neurology. 1996;47:562–568. doi: 10.1212/wnl.47.2.562. [DOI] [PubMed] [Google Scholar]
- Buono RJ, Ferraro TN, O’Connor MJ, Sperling MR, Ryan SG, Scattergood T, Mulholland N, Gilmore J, Lohoff FW, Berrettini WH. Lack of association between an interleukin 1 beta (IL-1beta) gene variation and refractory temporal lobe epilepsy. Epilepsia. 2001;42:782–784. doi: 10.1046/j.1528-1157.2001.42900.x. [DOI] [PubMed] [Google Scholar]
- Cendes F, Andermann F, Gloor P, Lopes-Cendes I, Andermann E, Melanson D, Jones-Gotman M, Robitaille Y, Evans A, Peters T. Atrophy of mesial structures in patients with temporal lobe epilepsy: cause or consequence of repeated seizures? Ann Neurol. 1993;34:795–801. doi: 10.1002/ana.410340607. [DOI] [PubMed] [Google Scholar]
- Covolan L, Mello LE. Temporal profile of neuronal injury following pilocarpine or kainic acid-induced status epilepticus. Epilepsy Res. 2000;39:133–152. doi: 10.1016/s0920-1211(99)00119-9. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Du F, Eid T, Lothman EW, Kohler C, Schwarcz R. Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. J Neurosci. 1995;15:6301–6313. doi: 10.1523/JNEUROSCI.15-10-06301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubé C, da Silva Fernandes MJ, Nehlig A. Age-dependent consequences of seizures and the development of temporal lobe epilepsy in the rat. Dev Neurosci. 2001;23:219–223. doi: 10.1159/000046147. [DOI] [PubMed] [Google Scholar]
- Eid T, Williamson A, Lee TS, Petroff OA, de Lanerolle NC. Glutamate and astrocytes--key players in human mesial temporal lobe epilepsy? Epilepsia. 2008;49(Suppl 2):42–52. doi: 10.1111/j.1528-1167.2008.01492.x. [DOI] [PubMed] [Google Scholar]
- French JA, Williamson PD, Thadani VM, Darcey TM, Mattson RH, Spencer SS, Spencer DD. Characteristics of medial temporal lobe epilepsy: I. Results of history and physical examination. Ann Neurol. 1993;34:774–780. doi: 10.1002/ana.410340604. [DOI] [PubMed] [Google Scholar]
- Galic MA, Riazi K, Heida JG, Mouihate A, Fournier NM, Spencer SJ, Kalynchuk LE, Teskey GC, Pittman QJ. Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci. 2008;28:6904–6913. doi: 10.1523/JNEUROSCI.1901-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter JA, Goncalves Pereira PM, van Vliet EA, Aronica E, Lopes da Silva FH, Lucassen PJ. Neuronal cell death in a rat model for mesial temporal lobe epilepsy is induced by the initial status epilepticus and not by later repeated spontaneous seizures. Epilepsia. 2003;44:647–658. doi: 10.1046/j.1528-1157.2003.53902.x. [DOI] [PubMed] [Google Scholar]
- Haas KZ, Sperber EF, Opanashuk LA, Stanton PK, Moshe SL. Resistance of immature hippocampus to morphologic and physiologic alterations following status epilepticus or kindling. Hippocampus. 2001;11:615–625. doi: 10.1002/hipo.1076. [DOI] [PubMed] [Google Scholar]
- Heida JG, Boisse L, Pittman QJ. Lipopolysaccharide-induced febrile convulsions in the rat: short-term sequelae. Epilepsia. 2004;45:1317–1329. doi: 10.1111/j.0013-9580.2004.13704.x. [DOI] [PubMed] [Google Scholar]
- Heida JG, Teskey GC, Pittman QJ. Febrile convulsions induced by the combination of lipopolysaccharide and low-dose kainic acid enhance seizure susceptibility, not epileptogenesis, in rats. Epilepsia. 2005;46:1898–1905. doi: 10.1111/j.1528-1167.2005.00286.x. [DOI] [PubMed] [Google Scholar]
- Heils A, Haug K, Kunz WS, Fernandez G, Horvath S, Rebstock J, Propping P, Elger CE. Interleukin-1beta gene polymorphism and susceptibility to temporal lobe epilepsy with hippocampal sclerosis. Ann Neurol. 2000;48:948–950. [PubMed] [Google Scholar]
- Immonen RJ, Kharatishvili I, Sierra A, Einula C, Pitkanen A, Grohn OH. Manganese enhanced MRI detects mossy fiber sprouting rather than neurodegeneration, gliosis or seizure-activity in the epileptic rat hippocampus. Neuroimage. 2008;40:1718–1730. doi: 10.1016/j.neuroimage.2008.01.042. [DOI] [PubMed] [Google Scholar]
- Ingvar M, Morgan PF, Auer RN. The nature and timing of excitotoxic neuronal necrosis in the cerebral cortex, hippocampus and thalamus due to flurothyl-induced status epilepticus. Acta Neuropathol. 1988;75:362–369. doi: 10.1007/BF00687789. [DOI] [PubMed] [Google Scholar]
- Kadam SD, White AM, Staley KJ, Dudek FE. Continuous electroencephalographic monitoring with radio-telemetry in a rat model of perinatal hypoxia-ischemia reveals progressive post-stroke epilepsy. J Neurosci. 2010;30:404–415. doi: 10.1523/JNEUROSCI.4093-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemoto K, Kawasaki J, Miyamoto T, Obayashi H, Nishimura M. Interleukin (IL)1beta, IL-1alpha, and IL-1 receptor antagonist gene polymorphisms in patients with temporal lobe epilepsy. Ann Neurol. 2000;47:571–574. [PubMed] [Google Scholar]
- Kanemoto K, Kawasaki J, Yuasa S, Kumaki T, Tomohiro O, Kaji R, Nishimura M. Increased frequency of interleukin-1beta-511T allele in patients with temporal lobe epilepsy, hippocampal sclerosis, and prolonged febrile convulsion. Epilepsia. 2003;44:796–799. doi: 10.1046/j.1528-1157.2003.43302.x. [DOI] [PubMed] [Google Scholar]
- Kauffman MA, Moron DG, Consalvo D, Bello R, Kochen S. Association study between interleukin 1 beta gene and epileptic disorders: a HuGe review and meta-analysis. Genet Med. 2008;10:83–88. doi: 10.1097/GIM.0b013e318161317c. [DOI] [PubMed] [Google Scholar]
- Khurgel M, Switzer RC, III, Teskey GC, Spiller AE, Racine RJ, Ivy GO. Activation of astrocytes during epileptogenesis in the absence of neuronal degeneration. Neurobiol Dis. 1995;2:23–35. doi: 10.1006/nbdi.1995.0003. [DOI] [PubMed] [Google Scholar]
- Kipp M, Norkute A, Johann S, Lorenz L, Braun A, Hieble A, Gingele S, Pott F, Richter J, Beyer C. Brain-region-specific astroglial responses in vitro after LPS exposure. J Mol Neurosci. 2008;35:235–243. doi: 10.1007/s12031-008-9057-7. [DOI] [PubMed] [Google Scholar]
- Koh S, Tibayan FD, Simpson JN, Jensen FE. NBQX or topiramate treatment after perinatal hypoxia-induced seizures prevents later increases in seizure-induced neuronal injury. Epilepsia. 2004;45:569–575. doi: 10.1111/j.0013-9580.2004.69103.x. [DOI] [PubMed] [Google Scholar]
- Kwon MS, Seo YJ, Choi SM, Won MH, Lee JK, Park SH, Jung JS, Sim YB, Suh HW. The time-dependent effect of lipopolysaccharide on kainic acid-induced neuronal death in hippocampal CA3 region: possible involvement of cytokines via glucocorticoid. Neuroscience. 2010;165:1333–1344. doi: 10.1016/j.neuroscience.2009.11.060. [DOI] [PubMed] [Google Scholar]
- Marcon J, Gagliardi B, Balosso S, Maroso M, Noe F, Morin M, Lerner-Natoli M, Vezzani A, Ravizza T. Age-dependent vascular changes induced by status epilepticus in rat forebrain: implications for epileptogenesis. Neurobiol Dis. 2009;34:121–132. doi: 10.1016/j.nbd.2008.12.018. [DOI] [PubMed] [Google Scholar]
- Mazarati A, Shin D, Auvin S, Sankar R. Age-dependent Effects of Topiramate on the Acquisition and the Retention of Rapid Kindling. Epilepsia. 2007;48:765–773. doi: 10.1111/j.1528-1167.2007.00987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarati A, Shin D, Sankar R. Bumetanide inhibits rapid kindling in neonatal rats. Epilepsia. 2009;50:2117–2122. doi: 10.1111/j.1528-1167.2009.02048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarati A, Wu J, Shin D, Kwon YS, Sankar R. Antiepileptogenic and antiictogenic effects of retigabine under conditions of rapid kindling: an ontogenic study. Epilepsia. 2008;49:1777–1786. doi: 10.1111/j.1528-1167.2008.01674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson HB, Lothman EW. An ontogenetic study of kindling using rapidly recurring hippocampal seizures. Brain Res Dev Brain Res. 1991;61:79–85. doi: 10.1016/0165-3806(91)90116-z. [DOI] [PubMed] [Google Scholar]
- Nevander G, Ingvar M, Auer R, Siesjo BK. Status epilepticus in well-oxygenated rats causes neuronal necrosis. Ann Neurol. 1985;18:281–290. doi: 10.1002/ana.410180303. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Tian GF, Han X, Peng W, Takano T, Ransom B, Nedergaard M. Loss of astrocytic domain organization in the epileptic brain. J Neurosci. 2008;28:3264–3276. doi: 10.1523/JNEUROSCI.4980-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; Sydney: 1982. [DOI] [PubMed] [Google Scholar]
- Poirier JL, Capek R, De KY. Differential progression of Dark Neuron and Fluoro-Jade labelling in the rat hippocampus following pilocarpine-induced status epilepticus. Neuroscience. 2000;97:59–68. doi: 10.1016/s0306-4522(00)00026-9. [DOI] [PubMed] [Google Scholar]
- Priel MR, dos Santos NF, Cavalheiro EA. Developmental aspects of the pilocarpine model of epilepsy. Epilepsy Res. 1996;26:115–121. doi: 10.1016/s0920-1211(96)00047-2. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2007 doi: 10.1016/j.nbd.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Noe F, Zardoni D, Vaghi V, Sifringer M, Vezzani A. Interleukin Converting Enzyme inhibition impairs kindling epileptogenesis in rats by blocking astrocytic IL-1beta production. Neurobiol Dis. 2008;31:327–333. doi: 10.1016/j.nbd.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Rizzi M, Perego C, Richichi C, Veliskova J, Moshe SL, De Simoni MG, Vezzani A. Inflammatory response and glia activation in developing rat hippocampus after status epilepticus. Epilepsia. 2005;46(Suppl 5):113–117. doi: 10.1111/j.1528-1167.2005.01006.x. [DOI] [PubMed] [Google Scholar]
- Sankar R, Shin DH, Liu H, Mazarati A, Pereira d V, Wasterlain CG. Patterns of status epilepticus-induced neuronal injury during development and long-term consequences. J Neurosci. 1998;18:8382–8393. doi: 10.1523/JNEUROSCI.18-20-08382.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyah M, Javad-Pour M, Ghazi-Khansari M. The bacterial endotoxin lipopolysaccharide enhances seizure susceptibility in mice: involvement of proinflammatory factors: nitric oxide and prostaglandins. Neuroscience. 2003;122:1073–1080. doi: 10.1016/j.neuroscience.2003.08.043. [DOI] [PubMed] [Google Scholar]
- Scantlebury MH, Gibbs SA, Foadjo B, Lema P, Psarropoulou C, Carmant L. Febrile seizures in the predisposed brain: a new model of temporal lobe epilepsy. Ann Neurol. 2005;58:41–49. doi: 10.1002/ana.20512. [DOI] [PubMed] [Google Scholar]
- Schmid R, Tandon P, Stafstrom CE, Holmes GL. Effects of neonatal seizures on subsequent seizure-induced brain injury. Neurology. 1999;53:1754–1761. doi: 10.1212/wnl.53.8.1754. [DOI] [PubMed] [Google Scholar]
- Schwob JE, Fuller T, Price JL, Olney JW. Widespread patterns of neuronal damage following systemic or intracerebral injections of kainic acid: a histological study. Neuroscience. 1980;5:991–1014. doi: 10.1016/0306-4522(80)90181-5. [DOI] [PubMed] [Google Scholar]
- Setkowicz Z, Nowak B, Janeczko K. Neocortical injuries at different developmental stages determine different susceptibility to seizures induced in adulthood. Epilepsy Res. 2006;68:255–263. doi: 10.1016/j.eplepsyres.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Somera-Molina KC, Robin B, Somera CA, Anderson C, Stine C, Koh S, Behanna HA, Van Eldik LJ, Watterson DM, Wainwright MS. Glial activation links early-life seizures and long-term neurologic dysfunction: evidence using a small molecule inhibitor of proinflammatory cytokine upregulation. Epilepsia. 2007;48:1785–1800. doi: 10.1111/j.1528-1167.2007.01135.x. [DOI] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Ravizza T, Balosso S, Aronica E. Glia as a source of cytokines: implications for neuronal excitability and survival. Epilepsia. 2008;49(Suppl 2):24–32. doi: 10.1111/j.1528-1167.2008.01490.x. [DOI] [PubMed] [Google Scholar]
- Wetherington J, Serrano G, Dingledine R. Astrocytes in the epileptic brain. Neuron. 2008;58:168–178. doi: 10.1016/j.neuron.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]