

1. Introduction
Corneal transparency maintenance depends on continuous renewal of its outermost epithelial layer. This process replaces the uppermost terminally differentiated layers that are being sloughed off into the tears. Their replacement assures preservation of epithelial integrity and its smooth optical properties. In addition, epithelial renewal preserves other needed functions for visual clarity that include: 1) tight junction barrier intactness, which protects the cornea from becoming translucent due to tissue swelling caused by exposure to environmental stresses such as pathogens and anisosmotic challenges (Lu et al., 2001); 2) innate immune responsiveness, which detects the presence of pathogens and provides signals that activate the corneal defense system (Zhang et al., 2005); 3) aldehyde dehydrogenase expression, which protects this layer against UV- and 4-hydroxynonenal-induced cellular damage (Pappa et al., 2005).
As epithelial turnover is modulated by a host of cytokines, extensive effort is devoted to identifying cognate receptor-linked cell signaling drug targets for hastening this process subsequent to corneal injury (Reinach and Pokorny, 2008). These studies employ injury-induced in vitro epithelial and in vivo wound healing models to determine which receptor-linked cell signaling pathways mediate control of cell proliferation, migration, differentiation, inflammation and apoptosis. In addition to improving our understanding of corneal biology, these studies have identified potential novel drug targets to lessen corneal scarring and inflammation that can persist subsequent to wound closure. These complications may be severe enough to prevent restoration of corneal transparency and visual function (Saika et al., 2007). In human corneal epithelial cells (HCEC), epidermal growth factor receptors (EGFRs) contribute to mediating corneal epithelial renewal. EGFR activation by EGF results in stimulation of cell proliferation and migration through activation of: a) the three member pathways of the mitogen-activated protein kinase (MAPK) signaling cassette (i.e., ERK, p38 and JNK); b) the phosphoinositide 3-kinase (PI3-K)/Akt/GSK-3 pathway; c) adenylate cyclase and phospholipase C (PLC)-induced Ca2+ signaling as well as phospholipase D (PLD)-mediated phosphatidic acid formation (Islam and Akhtar, 2000; Kang et al., 2000; Kang et al., 2001; Mazie et al., 2006; Wang et al., 2006; Wang et al., 2009; Yang et al., 2005; Yang et al., 2001; Yin and Yu, 2009; Zhang and Akhtar, 1998). EGFR activation also can occur through transactivation by other receptors and mediators (Block and Klarlund, 2008; Lyu et al., 2006; Spix et al., 2007; Xu et al., 2006; Xu et al., 2007). In this process, agonists other than EGF activate their cognate receptors, which leads to matrix metalloproteinase activation and scission of EGF from membrane bound heparin. Therefore, the EGFR-linked cell signaling pathways serve as a conduit for eliciting tissue responses to a number of different mediators besides EGF.
Members of the transient receptor potential (TRP) protein superfamily are polymodal in that they are activated by numerous different stimuli. In the corneal epithelium, some members of the vanilloid (V) TRP subfamily were identified. In HCEC, there is functional expression of TRPV1, 3 and 4 (Pan et al., 2008; Yamada et al., 2010; Zhang et al., 2007). TRPV1 is a nonselective ion channel which is activated by injury-induced endogenous mediators such as endocannabinoids, endovanilloids, declines in pH, elevated temperature and hypertonicity as well as capsaicin, which is present in red pepper extracts. Capsaicin (CAP) is a selective TRPV1 agonist and in HCEC induces increases in the release of proinflammatory cytokine mediators, such as interleukin (IL)-6 and the chemoattractant, IL-8. MAPK activation is a contributor to their increases (Zhang et al., 2007). These rises induced by CAP have physiological relevance since TRPV1 activation by injury in a mouse corneal wound healing model contributes to the development of severe inflammation that persists subsequent to wound closure. Evidence of its role stems from our finding that in homozygous TRPV1(−/−) knockout mice the wound healing response to injury is more favorable. This is apparent since inflammation and scarring are less severe at the time of wound closure (Okada et al., 2008). Even though EGFR-linked pathways are activated by CAP, it is not known if EGFR transactivation contributes to the development of inflammation and scarring.
The cannabinoid receptor subtype 1 (CB1) modulates, through the GTP binding protein (Gi), a number of important physiological processes in different tissues including neurotransmitter release, pain and analgesia, energy homeostasis regulation, and control of immune cell function (Graham et al., 2009; Howlett, 2005; Kress and Kuner, 2009; Pertwee, 2006; Stephens, 2009). CB1 activation by cannabinoids has immunosuppressive effects, which have beneficial effects in the treatment of autoimmune disorders. These results suggest that the cannabinoid system has various roles in disease pathologies and provides potential therapeutic targets. A functional role for CB1 in the human corneal epithelium has not yet been described even though CB1 expression was detected in the corneas of isolated human eyes (Straiker et al., 1999). In some other tissues, TRPV1 and CB1 are coexpressed and functionally interact with one another. Such is the case in the colonic epithelium, in neuronal enriched mesencephalic cultures, primary sensory neurons and myometrial smooth muscle cells (Brighton et al., 2009; Kim et al., 2008; Mahmud et al., 2009; Sibaev et al., 2006). The coexpression of TRPV1 and CB1 in the corneal epithelium prompted us to probe for a functional interaction between them in HCEC.
We show in HCEC that there is a functional interaction between TRPV1 and CB1. Together they mediate increases in cell proliferation and migration through EGFR transactivation and MAPK/Akt-linked signaling. On the other hand, other EGFR independent TRPV1-linked pathway(s) contribute to mediating TRPV1 stimulation of IL-6 and IL-8 release. In contrast, CB1 activation counters TRPV1-induced increases in IL-8. It is conceivable in a clinical setting that drugs targeted to activate CB1 receptors may be effective in reducing TRPV1-induced inflammation caused by corneal injury.
2. Methods
2.1. Materials
The following chemicals were purchased from Sigma-Aldrich (St. Louis, MO): WIN55,212-2 (WIN), anandamide (AEA), capsazepine (CPZ), CAP, AM251, AG1478, GM6001, CRM197, EGF, bovine insulin, gentamicin and TrypLE™ Express Stable Trypsin-Like Enzyme. Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium fetal bovine serum (FBS) and fura-2 acetoxymethyl ester were purchased from Invitrogen (Carlsbad, CA). Anti-CB1, anti-EGFR, phospho-EGFR, anti-Erk1/2, phospho-Erk1/2, phospho-Akt, phospho-GSK, phospho-p38; goat anti-mouse IgG-HRP, goat anti-rabbit IgG-HRP antibody, anti (H196) actin, anti-Erk1/2, anti-p38, and β-actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-EGFR neutralizing clone, LA1, was purchased from Millipore Corporate (Billerica, MA).
2.2. Cell culture
SV40-adenovirus immortalized HCEC were obtained as a generous gift from Dr. Kaoru Araki-Sasaki. The cells were cultured at 37°C in an incubator with 5% CO2 and 95% ambient air in DMEM/F12 medium, supplemented with 6% FBS, 5 ng/ml EGF and 5 μg/ml insulin. Cell cycle arrest was achieved by culturing cells in serum-free and EGF-free DMEM/F12 medium for 24 h before experimentation.
2.3. Single cell fluorescence imaging
Cells grown on 40-mm circular coverslips (Bioptechs Inc, Butler, PA) were loaded with 2 μM fura-2 AM at room temperature for 30 min and then washed with NaCl Ringer’s solution containing (in mM): NaCl (141), KCl (4.2), CaCl2 (0.8), KH2PO4 (2), MgCl2 (1), glucose (5.5), and HEPES (10) with osmolarity 300 mOsm and pH 7.4. Cells were continuously superfused at 34°C in a Focht Closed System 2 (FCS2) perfusion chamber with temperature control (Bioptechs Inc, Butler, PA) and placed on the stage of an inverted microscope (Nikon Diaphot 200). Cells were then alternately illuminated at 340 and 380 nm, and emission was monitored every 5 sec at 510 nm using a Roper Scientific CCD camera. Each field of interest contained 15~20 cells. Changes in intracellular Ca2+ levels, [Ca2+]i, were analyzed using Ratio Tool software (Isee Imaging, Durham, NC). The n values provided indicate the number of experiments per data point.
2.4. Western blot analysis
HCEC were gently washed twice in cold phosphate-buffered saline (PBS) and harvested in 0.5 ml cell lysis buffer. Cell lysates were centrifuged and supernatants were collected for measuring proteins with a bichinchoninic acid assay (BCA) protein assay kit (Pierce Biotechnology, Chicago, IL). Twenty to 50 μg of denatured protein was electrophoresed on 10% polyacrylamide sodium dodecylsulphate (SDS) minigels and polyvinylidene difluoride (PVDF) membranes were blocked with nonfat dry milk. The blots were exposed to the appropriate primary antibody overnight at 4°C and then exposed to an appropriate secondary antibody (e.g., anti-rabbit, anti-goat or anti-mouse) HRP labeled IgG for 1 h at room temperature. The immunoreactive bands were detected with an Amersham ECL Plus kit and band density was quantified using SigmaScan Pro 5.0 software. The monoclonal anti β-actin antibody tested for protein loading equivalence.
2.5. Immunocytochemistry
CB1 immunocytochemical localization was determined as described (Yang et al., 2005). Briefly, cells were seeded onto a Lab-Tek chamber slide system (Nunc, Naperville, IL) and after reaching confluence, they were washed twice with HEPES-buffered Ringer’s solution, fixed on ice for 30 min in 4% paraformaldehyde, washed three times with HEPES Ringer’s solution, and then rendered permeable using 0.1% Triton solution. After blotting with 10% normal goat serum, cells were exposed to anti-CB1 antibody (sc-20754, 1:100; Santa Cruz Biotechnology) plus 1% bovine serum albumin (BSA) overnight at 4°C. After three washes with HEPES Ringer’s solution, cells were incubated with goat anti-rabbit IgG TR (sc-3842, 1:800; Santa Cruz Biotechnology) for 30 min and 1 μM SYTO16 green fluorescent nucleic acid stain (Invitrogen) for 5 min at room temperature. Fluorescence was visualized using a Nikon fluorescence microscope with a 60x oil objective lens. Images were processed using Adobe® Photoshop 5.5 software (Adobe Systems, Inc., San Diego, CA).
2.6. Scratch wound assay
Cells were grown to confluence in 35-mm culture plate wells. They were then washed twice with PBS and placed in the appropriate serum-free medium. A small wound spanning the diameter of the culture was made with a sterile cell scraper and was marked. The monolayer was washed twice with basic medium to remove suspended cells and re-fed with medium in the presence or absence of EGF (10 ng/ml) immediately after wounding. Hydroxyurea (2.5 mM) was also added to the medium to reduce proliferation during the experiment. This inhibitor reduced cell proliferation by about 95% with minimal effects on cell viability (data not shown). Time-dependent wound closure was recorded for 24 h after wound creation. Images were obtained using a Roper Scientific CCD camera attached to Nikon Diaphot inverted-stage microscope (Nikon Inc., Morton Grove, IL). The remaining denuded area of each field was measured using SigmaScan Pro 5 software. Statistical analyses were performed using unpaired Student’s t-test. A P value less than 0.05 was assumed to be significant. Data are shown as mean ± SEM.
2.7. Cell proliferation
[3H] thymidine incorporation was performed as described (Wang et al., 2009). Following 20 h of serum starvation in medium supplemented with 0.5% BSA, the cells were incubated at 37°C for 1 h with 1μCi/ml [3H] thymidine (3.3 to 4.8 TBq/mmol). They were then washed twice with cold PBS, three times with ice-cold 5% trichloroacetic acid (TCA) and twice with cold 90% alcohol. Cell lysis was obtained with 0.2 N NaOH/2% SDS. The radioactivity was monitored in a Tri-Carb 2900TR Liquid Scintillation Analyzer (Perkin-Elmer, Boston, MA) and the data were normalized to cellular protein content determined with a BCA Protein Assay Kit.
2.8. Enzyme-linked immunosorbent assay (ELISA)
IL-6 and IL-8 levels in the supernatants were measured according to the manufacturer’s instructions using ELISA kits (QuantiGlo Human IL-8 or IL-6 Chemiluminescent Immunoassay; R&D Systems, Minneapolis, MN). The cells were washed with basic medium and then exposed to CPZ, or AM251, for 30 min before exposing them for 24 h to either CAP or EGF. Supernatants were harvested on ice and centrifuged at 2,000 rpm for 5 min at 4°C to remove cell debris. The supernatants were stored at −80°C until analysis. Their amounts in the culture medium were normalized to the total amount of cellular protein lysed with 2% SDS and 0.2 N NaOH and measured using a Micro BCA protein assay (Pierce, Rockford, IL). Results are expressed as mean picograms of IL-6 or IL-8 per mg cell protein ± SEM (n=3).
3. Results
3.1. CB1 expression
To ascertain whether there is CB1 expression in HCEC, immunocytochemistry and Western blot analysis were performed. CB1 expression is evident in Fig. 1A based on bright diffuse staining around the cell periphery. Fig. 1B validates primary antibody selectivity since following its preadsorption to an epitope blocking peptide there is no staining. Western blot analysis was performed on a subcellular fraction to validate CB1 protein expression. Fig. 1C shows a band with an apparent molecular weight of 63 kDa, which closely corresponds to the reported apparent molecular weight of CB1 in rat, monkey and human bladder (Gratzke et al., 2009).
Fig. 1.

CB1 protein expression in HCEC. (A) Following permeabilization with Triton X-100, CB1 expression was detected using a primary antibody (i.e., anti-rabbit polyclonal IgG) followed by incubation with a conjugated secondary antibody (i.e., goat anti-rabbit IgG-TR). Calibration bar is 50 μm. (B) Selectivity of the primary antibody was documented through its omission. (C) Western blot shows CB1 protein expression with an apparent molecular mass of 63 kDa.
3.2. CB1 functional expression
To probe for CB1 functional expression, we determined if WIN, a CB1 agonist, induced a rise in [Ca2+]i. Fig. 2A indicates that 10 μM WIN caused a transient rise in [Ca2+]i after about 10 min that was approximately 4-fold above the control. This transient then slightly declined and stabilized after another 10 min at a level that was nearly 3-fold above the baseline level. On the other hand, preincubation with 10 μM AM251, a CB1 antagonist, blocked the subsequent rise in [Ca2+]i by 93%. In a Ca2+-free NaCl Ringers supplemented with 2 mM EGTA, the responses were essentially the same as those in the Ca2+-containing counterpart (data not shown). Figure 2B shows that AEA (10 μM), an endogenous cannabinoid analogue which is a mixed CB1 and TRPV1 agonist, elicited after 10 min a 2-fold [Ca2+]i transient, which then gradually fell after another 10 min to reach a level that remained about 65% above the control level. A component of this rise is attributable to TRPV1 activation since preincubation with a TRPV1 antagonist, 10 μM CPZ suppressed the transient by about 65%.
Fig. 2.

CB1 functional expression in HCEC. (A) HCEC were loaded with fura2-AM (2μM). WIN55, 212-2 (WIN, 10 μM) induced [Ca2+]i transient whereas preincubation with AM251 (10 μM) blocked CB1 activation by 95%. (B) Anandamide (AEA), induced a [Ca2+]i transient. Following exposure to 10 μM capsazepine (CPZ) this response was suppressed. The data represents the means ± SEM (n=3, P < 0.05).
3.3. CB1 and TRPV1 stimulation mediate EGFR transactivation
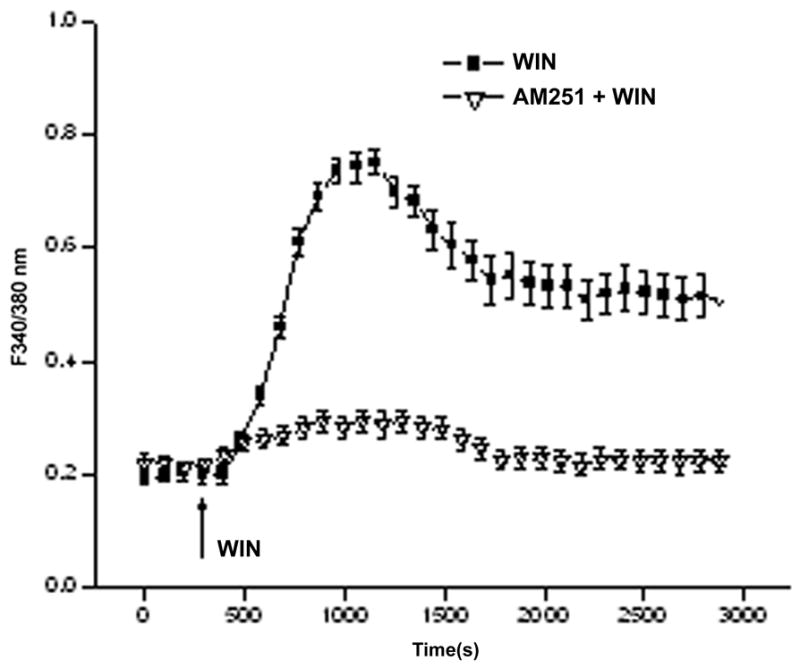
EGFR transactivation by ligands activating growth factor receptors and GTP binding coupled receptors induces a host of different responses that are dependent on the duration and magnitude of activation of EGFR linked signaling pathways. EGFR transactivation by CB1 activation was evaluated by determining if preincubation with the specific EGFR inhibitor, AG1478 (5 μM) suppressed 10 μM WIN-induced rises in [Ca2+]i (Levitzki and Gazit, 1995; Osherov and Levitzki, 1994). The results shown in Fig. 3A indicate that WIN induced a transient rise in the F340/F380 nm ratio after 6 min that reached more than 3-fold above the baseline followed by stabilization at a level that remained about 2.5-fold above its baseline level prior to WIN stimulation. On the other hand, in the continued presence of AG1478, WIN only initially increased the F340/F380 nm ratio to a level that was about 25% above the baseline. Subsequently, this ratio decreased to a level that was about 10% above the control level. This diminution suggests a large portion of CB1-induced increases in plasma membrane Ca2+ influx is attributable to EGFR transactivation by CB1. Transactivation of EGFR by direct TRPV1 stimulation was similarly assessed. Fig. 3B shows that 10 μM CAP increased the F340/F380 nm ratio maximally by about 70% whereas preincubation with AG1478 suppressed this rise by 42%. These partial declines induced during AG1478 exposure suggest that a component of the overall increase in Ca2+ influx elicited by WIN or CAP are accounted for EGFR transactivation by CB1 and TRPV1 agonists.
Fig. 3.

CB1 and TRPV1 mediate EGFR transactivation. (A) WIN55, 212-2 (WIN; 10 μM) induced [Ca2+]i transients in the presence and absence of AG1478 (5 μM). (B) Similarly, capsaicin (CAP; 10 μM) induced [Ca2+]i transients in the presence and absence of AG1478 (5 μM). The data represents the means ± SEM (n=3, P < 0.05).
3.4. CB1 and TRPV1 induce EGFR phosphorylation
Western blot analysis of changes in the phosphorylation status of EGFR was used to assess for its transactivation by CB1 and TRPV1. This was done by determining if WIN and CAP induced phosphorylation of EGFR at Tyr1078. Fig. 4A indicates that exposure for 5 min to either 10 μM WIN or 10 μM CAP increased EGFR phosphorylation status by 3-fold whereas EGF (10 ng/ml) caused it to rise 5-fold. The effect of the CB1 agonist was fully suppressed by preincubating cells with 10 μM AM251. Similarly, during continuous exposure to 10 μM CPZ the CAP-induced rises in EGFR phosphorylation status declined by about 90%. Pre-exposure to 5 μM AG1478 also completely blocked increases in the EGFR phosphorylation status induced by either 10 ng/ml EGF, 10 μM CAP or 10 μM WIN. These results provide further substantiation that either CB1 or TRPV1 activation induces EGFR transactivation.
Fig. 4.

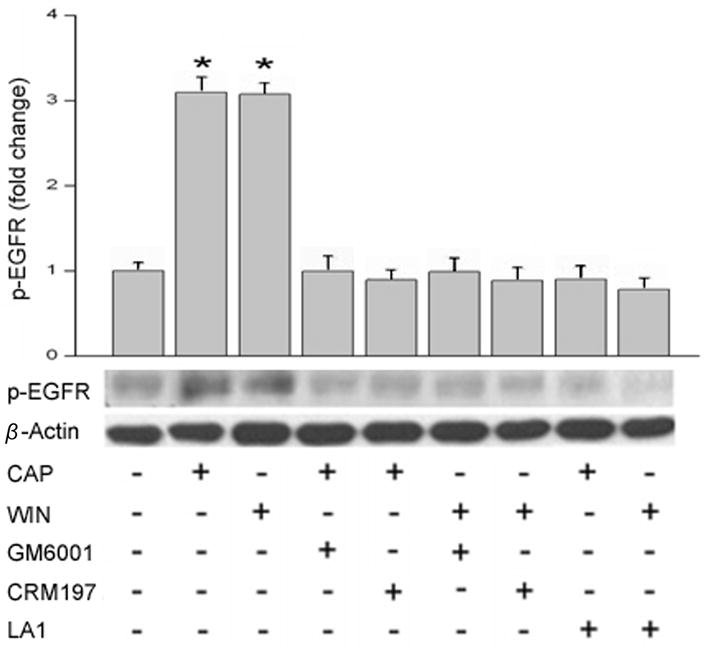
CB1 and TRPV1 induce EGFR phosphorylation. (A) Growth factor starved cells were preincubated 30 min with capsazepine (CPZ; 10 μM), AM251 (10 μM) or AG1478 (5 μM) prior to being exposed to either EGF (10 ng/ml), WIN55, 212-2 (WIN; 10 μM) or capsaicin (CAP; 10 μM). Western Blot was used to evaluate EGFR phosphorylation status. (B) HCEC were exposed to WIN55, 212-2 (WIN; 10 μM) or capsaicin (CAP; 10 μM) with or without GM6001 (50 μM) or CRM197 (10 μg/ml) preincubation for 30 min. Anti-EGFR, neutralizing clone LA1 (10 μg/ml) preincubation occurred for 2 h. Cells were lysed at the end of stimulation and equal amounts of cell lysate were blotted by an antibody against phosphorylated EGFR. The data represents the means ± SEM (n=3, *P < 0.05).
To determine whether CAP and/or WIN induces EGFR transactivation through HB-EGF shedding, cells were pretreated 30 min with either GM6001 (50 μM), a matrix metalloproteinase (MMP) inhibitor, CRM197 (10 μg/mL), a heparin binding EGF-like growth factor inhibitor, or for 2 h with a EGFR ligand-binding domain antibody LA1 (10 μg/ml) (Block et al., 2010). Figure 4B shows that GM6001 and CRM197 eliminated CAP and WIN stimulated EGFR phosphorylation. Furthermore, the anti-EGFR neutralizing clone LA1 abolished EGFR phosphorylation (n=3).
3.5. CB1 and TRPV1 induce EGFR-linked signaling activation
As CB1 and TRPV1 stimulation induced EGFR transactivation, we determined if this response induced phosphorylation of some of the EGFR-linked signaling pathways. Fig. 5A, shows that EGFR stimulation with either 10 ng/ml EGF, 10 μM CAP or 10 μM WIN at 10 min increased the phosphorylation status of Akt, p38 and Erk1/2 with similar magnitudes. This time point was chosen since we previously found that these signaling mediators were maximally stimulated at 10 min (Wang et al., 2009). In all cases, these increases were diminished if the cells were instead exposed at the same time to either AG1478, CPZ or AM251. The patterns of the time-dependent changes in the phosphorylation status of Erk1/2, p38 and JNK1/2 during exposure to either EGF or CAP shown in Figs. 5B and C are essentially the same. On the other hand, EGFR is not the sole route for their activation since pre-exposure to AG1478 only partially attenuated the CAP-induced increases in MAPK phosphorylation.
Fig. 5.

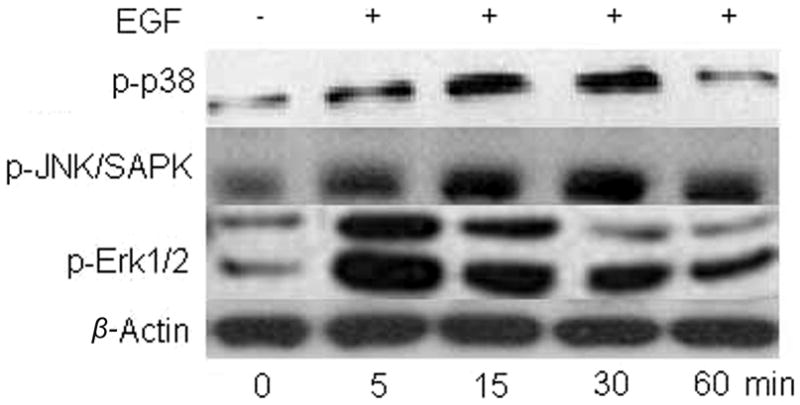

CB1 and TRPV1 induce EGFR-linked signaling activation. (A) Changes in Akt, p38 and Erk1/2 phosphorylation status induced by either EGF, WIN55, 212-2 (WIN; 10 μM) or capsaicin (CAP; 10 μM). HCEC were incubated with either 5 μM AG1478, 10 μM AM251 or 10 μM capsazepine (CPZ) for 30 min; then exposed to 10 ng/ml EGF, 10 μM CAP or 10 μM WIN. In some cases, the cells were exposed to EGF, CAP or WIN alone. Following the incubation, the cells were exposed to either an anti p-Akt, p-p38 or p-Erk1/2 antibody and phosphorylation status was detected based on Western blot analysis. (B) Time-dependent changes in the phosphorylation status of p-p38, p-JNK/SAPK and p-Erk1/2 induced by exposure to 10 ng/ml EGF. (C) Time-dependent changes in the phosphorylation status of p-p38, p-JNK/SAPK and p-Erk1/2 induced by exposure to 10 μM CAP. Equal loading of proteins in each lane was always confirmed by reprobing the same blot with an anti β-actin antibody. The data represent the means ± SEM (n=3, P < 0.05).
3.6. CB1 and TRPV1 stimulate mitogenesis through EGFR transactivation
Since selective CB1 and TRPV1 activation induced EGFR-linked pathway stimulation, we assessed if such changes are linked to increases in cell proliferation. Fig. 6 compares the respective effects of selective stimulation of either CB1 or TRPV1 with 10 μM WIN or 10 μM CAP on [3H] thymidine incorporation with that of EGF (10 ng/ml). Following 24 h serum starvation and exposure to each of these agonists for another 20 h, both EGF and CAP increased proliferation by nearly 2-fold. The rise induced by WIN was about 1.4-fold. The selectivity of these agonist-induced responses was documented by showing that preincubation for 30 min to either 5 μM AG1478, 10 μM AM251 or 10 μM CPZ fully suppressed each of their effects on proliferation. Therefore, CB1 and TRPV1 activation mediates increases in cell proliferation through EGFR transactivation.
Fig. 6.
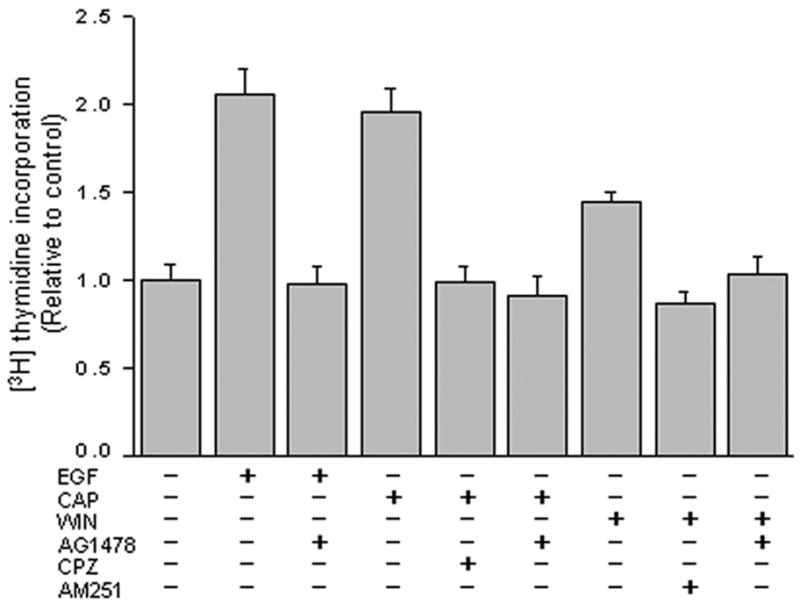
CB1 and TRPV1 stimulate mitogenesis through EGFR transactivation. HCEC were pretreated for 30 min with either 5 μM AG1478, 10 μM capsazepine (CPZ), or 10 μM AM251. Under some conditions, cells were then exposed for an additional 20 h to either 10 ng/ml EGF 10 μM capsaicin (CAP) or WIN55, 212-2 (WIN; 10 μM). Cells were then incubated for 1 h with 1 μCi/ml [3H] thymidine. Protein content was determined with a BCA protein assay kit. The data represent the means ± SEM (n=3, P < 0.05).
3.7. CB1 and TRPV1 stimulate cell migration through EGFR transactivation
The scratch wound assay determined if CB1 and TRPV1 activation stimulates cell migration through EGFR transactivation. The inset to Fig. 7 (upper panel) shows micrographs of the extent of closure at 6 and 24 h obtained under control conditions compared to those with either 10 ng/ml EGF, 1 μM CAP or 1 μM WIN. The extent of wound closure is compared to the control during exposure for 24 h to either EGF, CAP or WIN. Lower panel shows CAP and WIN stimulated wound closure by 1.65 and 1.52-fold, respectively, relative to the untreated control. These increases are comparable to that of EGF, which increased this response by 2-fold. These responses are dependent on EGFR transactivation since 5 μM AG1478 fully blocked them. The selectivity of AM251 and CPZ are indicated by their lack of inhibition of the respective increases in migration induced by CAP and WIN. On the other hand, the somewhat smaller increases in migration induced by WIN and CAP may be due to cytotoxicity based on some rounding up of cell morphology after 24 h exposure to these two agonists. A recent report shows a similar result in which the mixed CB1 and TRPV1 agonist, 1 μM AEA, reduced smooth muscle cell viability and inhibited cell migration (Brighton et al., 2009).
Fig. 7.
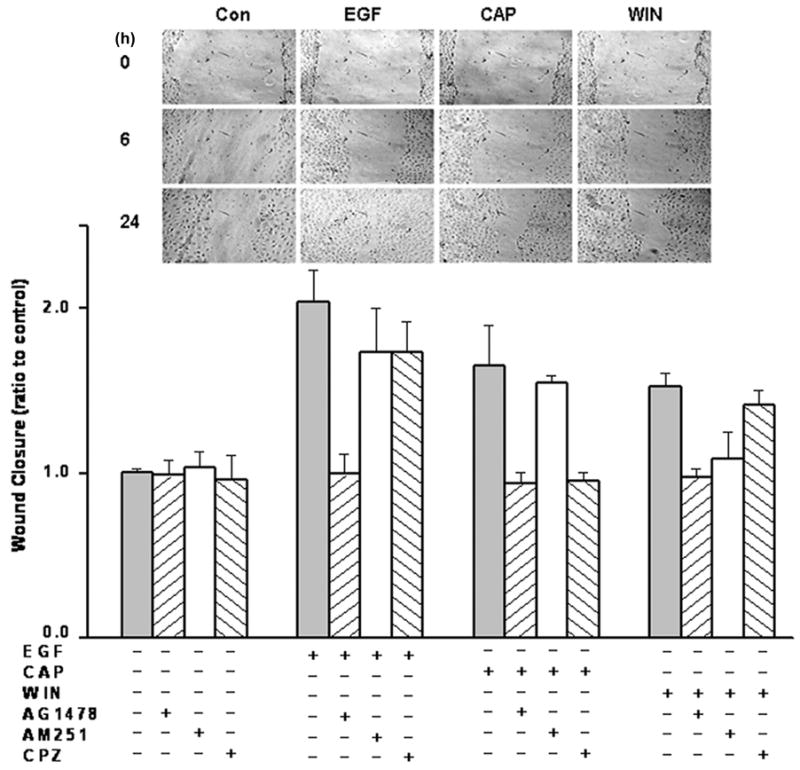
CB1 and TRPV1 stimulate cell migration through EGFR transactivation. HCEC confluent monolayers were preincubated with either AM251 (2 μM), capsazepine (CPZ; 1 μM) or AG1478 (5 μM) for 30 min and scratches were created across the culture diameter. The same conditioned medium was then supplemented with either 10 ng/ml EGF, 1 μM capsaicin (CAP) or 1 μM WIN55, 212-2 (WIN) for up to another 24 h. Inset shows representative micrographs of wound closure extent at the indicated times for untreated cells or in the presence of either EGF, CAP or WIN. The data represent the means ± SEM (n=3, P < 0.05).
3.8. Independent effects of TRPV1 and EGFR activation on IL-6 and IL-8 release
We have preliminary evidence that TRPV1 activation is an important player in mediating inflammation to injury in vivo. In a mouse alkali burn cornea wound healing model, loss of TRPV1 gene function markedly improved corneal wound healing outcome due to more complete restoration of corneal transparency (Okada et al., 2008). This difference between the wound healing outcomes in wildtype and TRPV1 knockout mice prompted us to compare the effects of selective TRPV1 and EGFR activation on IL-6 and IL-8 release. This was done to determine if there is any disconnect between the effects of EGF and CAP on this response even though both EGF and CAP stimulate the JNK pathway, which in many other tissues is linked to inducing increases in the release of these proinflammatory cytokines and pain (Ji et al., 2009). The ELISA results shown in Fig. 8 indicate that exposure to 10 μM CAP for 24 h induced about 3 and 6-fold increases in IL-6 and IL-8 levels whereas 10 ng/ml EGF did not stimulate their release. The selectivity of CAP to induce these increases through TRPV1 activation is indicated by the full blockage by 10 μM CPZ of these effects. The dependence was determined of increases in IL-6 and IL-8 release by CAP-induced EGFR transactivation. Pretreatment with AG1478 (1 μM) partially blocked CAP induced rises in IL-6 and IL-8 secretion by 21% in both cases. This partial decline suggests that EGFR linked signaling does not fully account for all of the pathways mediating CAP-induced increases in IL-6 and IL-8 release.
Fig. 8.

Independent effects of TRPV1 and EGFR activation on IL-6 and IL-8 release. ELISA was performed after 24 h to evaluate increases in IL-6 and IL-8 release into DMEM/F12 in the presence or absence of either EGF (10 ng/ml) or CAP (10 μM). Values were normalized to the respective control. In some cases, cells were first exposed to 10 μM capsazepine (CPZ) or 1 μM AG1478 for 30 min before supplementation with capsaicin (CAP; 10 μM) or EGF (10 ng/ml). Cytokine levels were measured in the supernatants using ELISA kits according to the manufacturer’s instructions. The data represent the means ± SEM (n=3, *P < 0.001).
3.9. CB1 activation dampens TRPV1 induced increases in IL-8 release
In vivo, costimulation by AEA of CB1 and TRPV1 in inflammatory conditions elicited decreases in the electrical activity stemming from TRPV1-induced nociceptive activity and subsequent pain-related behavior (Mahmud et al., 2009). We probed with ELISA for an interaction between CB1 and TRPV1 by determining if CB1 activation altered CAP-induced increases in IL-8 release. Fig. 9 shows that without modulating CB1 activity CAP by itself induced after 24 h about a 3-fold increase in IL-8 release. On the other hand, 10 μM AEA fully suppressed the CAP-induced increase in IL-8 release. Interestingly, preincubating with a selective CB1 antagonist AM251 (5 μM), instead enhanced the 10 μM CAP induced rise. IL-8 release rose about 8-fold above the control level. Therefore, blocking the CB1 component of the overall response to AEA with AM251 enhanced the CAP-induced increase in IL-8 release. This result suggests that CB1 activation counters the increases in IL-8 release induced by CAP.
Fig. 9.
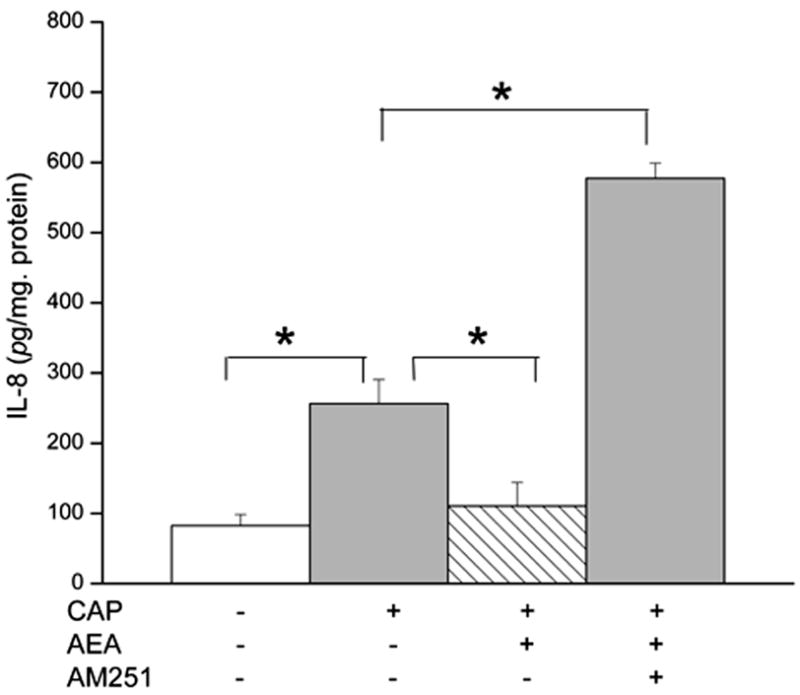
CB1 activation eliminates TRPV1 induced increases in IL-8 release. HCEC were pretreated with Anandamide (AEA; 10 μM) and/or AM251 (5 μM) for 1 h and then incubated with capsaicin (CAP; 10 μM). At the end of the incubation period, the levels of IL-8 were determined by ELISA. The data represent the means ± SEM (n=3, *P < 0.01).
4. Discussion
Our results show that TRPV1 activation elicited increases in HCEC proliferation, migration, IL-6 and IL-8 release through EGFR transactivation. However, the predominant contributor to eliciting rises in IL-6 and IL-8 levels is attributable to activation of another EGFR-independent pathway. TRPV1 control of the first two responses is dependent on EGFR-mediated MAPK as well as PI3-K/Akt signaling. CB1 receptor activation also induced increases in cell proliferation and migration through the same process as that induced by TRPV1. However, CB1 activation instead suppressed the increase in IL-8 release induced through TRPV1 activation by CAP. Suppression by CB1 activation of a TRPV1-mediated response was also described in myometrial smooth muscle cells, rat primary sensory neurons and in mesencephalic dopamine neurons (Brighton et al., 2009; Kim et al., 2008; Mahmud et al., 2009). This study broadens our understanding of the roles played by TRPV1 and CB1 in eliciting through EGFR transactivation and linked signaling pathways responses underlying injury-induced corneal epithelial wound healing.
CB1 and TRPV1 receptors elicit control of cell proliferation through three MAPK parallel signaling pathways as well as the Akt system linked to EGFR. Such control is indicated by the finding that either CB1 or TRPV1 activation induced changes in the duration and magnitude of increases in the phosphorylation status of the signaling components that were similar to those induced by EGF. Furthermore, the magnitudes of the resulting increases in cell proliferation and migration were also similar to one another. TRPV1 and CB1 receptors may also be linked to other cell signaling pathways than those linked to EGFR. This is possible because TRPV1 and CB1 receptor activation modulated IL-8 release whereas EGF had no effect on this response. Another indication that EGFR-linked signaling makes only a small contribution to mediating TRPV1 control of IL-6 and IL-8 release is that AG1478 only slightly decreased CAP-induced such rises. Recently, we obtained preliminary data suggesting that a component of this alternate pathway may be transforming growth factor-beta activated kinase 1 (TAK1), which is linked to TNF-α receptor control of cell survival in corneal epithelial cells (Wang et al., 2005). Such a pathway is referred to as a non-canonical TAK1 dependent MAPK independent pathway and is inhibited by a selective inhibitor, 5Z-7-oxozeaenol (Ninomiya-Tsuji et al., 2003). We found that 5Z-7-oxozeaenol suppressed CAP-induced increases in IL-6 and IL-8 release (data not shown).
CB1 receptors are seven transmembrane-domain neuronal receptors coupled to pertussis toxin (PTX)-sensitive Gi/o proteins. CB1 activation can inhibit adenylate cyclase and/or activate as well as inhibit ion channels. They are activated by endogenously activated lipidic compounds such as N-arachidonoylethanolamine (AEA) and 2-arachidonoylglycerol (2-AG). In addition, CB1 receptor coupling to PLC-dependent Ca2+ mobilization from intracellular stores has been reported in different cell types (De Petrocellis et al., 2007; Fimiani et al., 1999; McIntosh et al., 2007; Netzeband et al., 1999). The WIN-induced [Ca2+]i transients shown in Fig. 2A indicate that CB1 activation by this selective CB1 agonist induced mobilization from intracellular Ca2+ stores as these responses were essentially the same irrespective of the presence or absence of Ca2+ in the bathing solution. The specificity of this response was validated by showing that preincubation with AM251 nearly fully attenuated this response. Fig. 2B provides further indication of CB1 coupling to TRPV1-induced Ca2+ signaling. The mixed CB1 and TRPV1 agonist, AEA, also induced a rise in [Ca2+]i levels that was attenuated by about 65% during exposure to CPZ. Therefore, CB1 and TRPV1 induced increases in Ca2+ occur independently of one another since inhibition of CB1 activation does not fully suppress TRPV1 activation. As CB1 activation suppressed TRPV1-induced increases in IL-8, selective CB1 activation in a clinical setting may provide a novel drug strategy to reduce TRPV1-induced proinflammatory cytokine release.
EGFR transactivation in HCEC has been described in response to exposure to lysophosphatidic acid (LPA), purinergic receptor activation and exposure to either ATP or insulin (Block and Klarlund, 2008; Lyu et al., 2006; Spix et al., 2007; Xu et al., 2006; Xu et al., 2007). Fig. 3A and B are supportive of the notion that the Ca2+ transients induced by either CB1 or TRPV1 stimulation are in part accounted for by EGFR transactivation. This is evident since AG1478 suppressed these maximal rises by about 90% and 35%, respectively. As the inhibitions were only partial, some of the Ca2+ rise induced by CB1 activation may not be dependent on EGFR transactivation. Nevertheless, EGFR transactivation by exposure to either WIN or CAP is evident based on the increases in EGFR phosphorylation shown in Fig. 4A. The selectivity of the CB1 and TRPV1 agonists is indicated by the finding that either AM251 or CPZ suppressed EGFR phosphorylation to a level similar to that obtained with AG1478. EGFR transactivation by CB1 and TRPV1 agonists occurs through the activation of the same matrix metalloproteinases that elicit this response in the corneal epithelium during exposure to either lysophosphatidic acid, pseudomonas aeruginosa or following its wounding (Xu et al., 2004; Xu et al., 2007; Zhang et al., 2004). This correspondence is shown by the fact that their inhibition with either GM6001 or CRM197 blocked EGFR phosphorylation (c.f. Figure 4B). Similarly, functional blockage of EGFR activation with LA1 eliminated WIN and CAP-induced EGFR phosphorylation.
EGFR activation induces control of linked responses through sequential transient changes in the MAPK phosphorylation status. Such regulation is modulated through changes in the duration and magnitude of these phosphorylation events. The level of phosphorylation increases and their duration are under the control of dual specific protein phosphatases (DUSPs) (Patterson et al., 2009). DUSP1 is broad spectrum in that it elicits a negative feedback on Erk1/2, p38 and JNK1/2 MAPK phosphorylation. We previously showed that phosphorylation of DUSP1 in HCEC alters the balance between EGF-induced increases in proliferation and migration (Wang et al., 2009). Specifically, prolongation of Erk1/2 phosphorylation resulting from declines in DUSP1 (or MKP-1) levels resulting from GSK-3 inhibition reduced the mitogenic response to EGF whereas cell migration was enhanced. In the current study, we found that even though CB1 and TRPV1-induce EGFR transactivation followed by Erk1/2, p38 and JNK pathway signaling (Fig. 5A and C), only TRPV1 and CB1 activation modulate proinflammatory IL-6 and IL-8 release. Fig. 8 shows that CPZ blocked CAP-induced increases in IL-6 and IL-8 whereas EGF had no effect on the control. Our results indicating an interaction between CB1 and TRPV1 shown in Figure 9 are consistent with another study in which toll-like receptor 4 (TLR4) activation is controlled by CB1. In this report, the lipopolysaccharide-induced increase in IL-8 was enhanced 2-fold through blockage of CB1 activation by AM251. Such an augmentation reveals an interaction between CB1 and TLR4 in regulating hyperinflammatory reactions by periodontal tissues (Nakajima et al., 2006). It remains to be determined if CB1 blunts through changes in GSK-3 activation CAP-induced increases in IL-8. Such changes could modulate MAPK or TAK1 signaling control of IL-8 release.
In summary, CB1 and TRPV1 activation induces in HCEC through EGFR transactivation increases in proliferation and migration. However, only TRPV1 activation induced increases IL-6 and IL-8 release, which are blunted through CB1 activation. These differences between the effects of CB1, TRPV1 and EGFR activation on IL-6 and IL-8 release suggest that the EGFR-linked pathway does not solely mediate CB1 and TRPV1 control of this response.
Acknowledgments
The authors thank Dr. Jin Zhao support for immunocytochemistry work. This work was supported in part by grants from National Institutes of Health (EY04795) and Department of Defense (W81XWH-09-2-0162).
Footnotes
Disclosure: Yang, Hua, none; Wang, Zheng, none; Capó-Aponte, José E., none; Zhang, Fan, none; Pan, Zan, none; and Reinach, Peter S., none
Disclaimer:
The views, opinions and/or findings contained in this report are those of the author(s) and should not be construed as an official Department of the Army position, policy or decision, unless so designated by other official documentation. Citation of trade names in this report does not constitute an official Department of the Army endorsement or approval of the use of such commercial items.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Block ER, Klarlund JK. Wounding sheets of epithelial cells activates the epidermal growth factor receptor through distinct short- and long-range mechanisms. Mol Biol Cell. 2008;19:4909–17. doi: 10.1091/mbc.E08-01-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ER, Tolino MA, Klarlund JK. Pyk2 activation triggers epidermal growth factor receptor signaling and cell motility after wounding sheets of epithelial cells. J Biol Chem. 2010;285:13372–9. doi: 10.1074/jbc.M109.083089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brighton PJ, McDonald J, Taylor AH, Challiss RA, Lambert DG, Konje JC, Willets JM. Characterization of anandamide-stimulated cannabinoid receptor signaling in human ULTR myometrial smooth muscle cells. Mol Endocrinol. 2009;23:1415–27. doi: 10.1210/me.2009-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Marini P, Matias I, Moriello AS, Starowicz K, Cristino L, Nigam S, Di Marzo V. Mechanisms for the coupling of cannabinoid receptors to intracellular calcium mobilization in rat insulinoma beta-cells. Exp Cell Res. 2007;313:2993–3004. doi: 10.1016/j.yexcr.2007.05.012. [DOI] [PubMed] [Google Scholar]
- Fimiani C, Mattocks D, Cavani F, Salzet M, Deutsch DG, Pryor S, Bilfinger TV, Stefano GB. Morphine and anandamide stimulate intracellular calcium transients in human arterial endothelial cells: coupling to nitric oxide release. Cell Signal. 1999;11:189–93. doi: 10.1016/s0898-6568(98)00060-6. [DOI] [PubMed] [Google Scholar]
- Graham ES, Ashton JC, Glass M. Cannabinoid receptors: a brief history and “what’s hot”. Front Biosci. 2009;14:944–57. doi: 10.2741/3288. [DOI] [PubMed] [Google Scholar]
- Gratzke C, Streng T, Park A, Christ G, Stief CG, Hedlund P, Andersson KE. Distribution and function of cannabinoid receptors 1 and 2 in the rat, monkey and human bladder. J Urol. 2009;181:1939–48. doi: 10.1016/j.juro.2008.11.079. [DOI] [PubMed] [Google Scholar]
- Howlett AC. Cannabinoid receptor signaling. Handb Exp Pharmacol. 2005:53–79. doi: 10.1007/3-540-26573-2_2. [DOI] [PubMed] [Google Scholar]
- Islam M, Akhtar RA. Epidermal growth factor stimulates phospholipase cgamma1 in cultured rabbit corneal epithelial cells. Exp Eye Res. 2000;70:261–9. doi: 10.1006/exer.1999.0783. [DOI] [PubMed] [Google Scholar]
- Ji RR, Gereau RWt, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–48. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SS, Li T, Xu D, Reinach PS, Lu L. Inhibitory effect of PGE2 on EGF-induced MAP kinase activity and rabbit corneal epithelial proliferation. Invest Ophthalmol Vis Sci. 2000;41:2164–9. [PubMed] [Google Scholar]
- Kang SS, Wang L, Kao WW, Reinach PS, Lu L. Control of SV-40 transformed RCE cell proliferation by growth-factor-induced cell cycle progression. Curr Eye Res. 2001;23:397–405. doi: 10.1076/ceyr.23.6.397.6965. [DOI] [PubMed] [Google Scholar]
- Kim SR, Bok E, Chung YC, Chung ES, Jin BK. Interactions between CB(1) receptors and TRPV1 channels mediated by 12-HPETE are cytotoxic to mesencephalic dopaminergic neurons. Br J Pharmacol. 2008;155:253–64. doi: 10.1038/bjp.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress M, Kuner R. Mode of action of cannabinoids on nociceptive nerve endings. Exp Brain Res. 2009;196:79–88. doi: 10.1007/s00221-009-1762-0. [DOI] [PubMed] [Google Scholar]
- Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–8. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- Lu L, Reinach PS, Kao WW. Corneal epithelial wound healing. Exp Biol Med (Maywood) 2001;226:653–64. doi: 10.1177/153537020222600711. [DOI] [PubMed] [Google Scholar]
- Lyu J, Lee KS, Joo CK. Transactivation of EGFR mediates insulin-stimulated ERK1/2 activation and enhanced cell migration in human corneal epithelial cells. Mol Vis. 2006;12:1403–10. [PubMed] [Google Scholar]
- Mahmud A, Santha P, Paule CC, Nagy I. Cannabinoid 1 receptor activation inhibits transient receptor potential vanilloid type 1 receptor-mediated cationic influx into rat cultured primary sensory neurons. Neuroscience. 2009;162:1202–11. doi: 10.1016/j.neuroscience.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Mazie AR, Spix JK, Block ER, Achebe HB, Klarlund JK. Epithelial cell motility is triggered by activation of the EGF receptor through phosphatidic acid signaling. J Cell Sci. 2006;119:1645–54. doi: 10.1242/jcs.02858. [DOI] [PubMed] [Google Scholar]
- McIntosh BT, Hudson B, Yegorova S, Jollimore CA, Kelly ME. Agonist-dependent cannabinoid receptor signalling in human trabecular meshwork cells. Br J Pharmacol. 2007;152:1111–20. doi: 10.1038/sj.bjp.0707495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Furuichi Y, Biswas KK, Hashiguchi T, Kawahara K, Yamaji K, Uchimura T, Izumi Y, Maruyama I. Endocannabinoid, anandamide in gingival tissue regulates the periodontal inflammation through NF-kappaB pathway inhibition. FEBS Lett. 2006;580:613–9. doi: 10.1016/j.febslet.2005.12.079. [DOI] [PubMed] [Google Scholar]
- Netzeband JG, Conroy SM, Parsons KL, Gruol DL. Cannabinoids enhance NMDA-elicited Ca2+ signals in cerebellar granule neurons in culture. J Neurosci. 1999;19:8765–77. doi: 10.1523/JNEUROSCI.19-20-08765.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M, Mihara M, Tsuchiya M, Matsumoto K. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem. 2003;278:18485–90. doi: 10.1074/jbc.M207453200. [DOI] [PubMed] [Google Scholar]
- Okada Y, Saika S, Shirai K, Miyajima M, Kao WW, Reinach P. Reduced Inflammation and Scarring During. Corneal Wound Healing in TRPV1 Knockout Mice. Invest Ophthalmol Vis Sci. 2008:49. [Google Scholar]
- Osherov N, Levitzki A. Epidermal-growth-factor-dependent activation of the src-family kinases. Eur J Biochem. 1994;225:1047–53. doi: 10.1111/j.1432-1033.1994.1047b.x. [DOI] [PubMed] [Google Scholar]
- Pan Z, Yang H, Mergler S, Liu H, Tachado SD, Zhang F, Kao WW, Koziel H, Pleyer U, Reinach PS. Dependence of regulatory volume decrease on transient receptor potential vanilloid 4 (TRPV4) expression in human corneal epithelial cells. Cell Calcium. 2008;44:374–85. doi: 10.1016/j.ceca.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappa A, Brown D, Koutalos Y, DeGregori J, White C, Vasiliou V. Human aldehyde dehydrogenase 3A1 inhibits proliferation and promotes survival of human corneal epithelial cells. J Biol Chem. 2005;280:27998–8006. doi: 10.1074/jbc.M503698200. [DOI] [PubMed] [Google Scholar]
- Patterson KI, Brummer T, O’Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 2009;418:475–89. doi: 10.1042/bj20082234. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. The pharmacology of cannabinoid receptors and their ligands: an overview. Int J Obes (Lond) 2006;30(Suppl 1):S13–8. doi: 10.1038/sj.ijo.0803272. [DOI] [PubMed] [Google Scholar]
- Reinach PS, Pokorny KS. The corneal epithelium: clinical relevance of cytokine-mediated responses to maintenance of corneal health. Arq Bras Oftalmol. 2008;71:80–6. doi: 10.1590/s0004-27492008000700016. [DOI] [PubMed] [Google Scholar]
- Saika S, Yamanaka O, Okada Y, Miyamoto T, Kitano A, Flanders KC, Ohnishi Y, Nakajima Y, Kao WW, Ikeda K. Effect of overexpression of PPARgamma on the healing process of corneal alkali burn in mice. Am J Physiol Cell Physiol. 2007;293:C75–86. doi: 10.1152/ajpcell.00332.2006. [DOI] [PubMed] [Google Scholar]
- Sibaev A, Massa F, Yuce B, Marsicano G, Lehr HA, Lutz B, Goke B, Allescher HD, Storr M. CB1 and TRPV1 receptors mediate protective effects on colonic electrophysiological properties in mice. J Mol Med. 2006;84:513–20. doi: 10.1007/s00109-006-0040-x. [DOI] [PubMed] [Google Scholar]
- Spix JK, Chay EY, Block ER, Klarlund JK. Hepatocyte growth factor induces epithelial cell motility through transactivation of the epidermal growth factor receptor. Exp Cell Res. 2007;313:3319–25. doi: 10.1016/j.yexcr.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens GJ. G-protein-coupled-receptor-mediated presynaptic inhibition in the cerebellum. Trends Pharmacol Sci. 2009;30:421–30. doi: 10.1016/j.tips.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Straiker AJ, Maguire G, Mackie K, Lindsey J. Localization of cannabinoid CB1 receptors in the human anterior eye and retina. Invest Ophthalmol Vis Sci. 1999;40:2442–8. [PubMed] [Google Scholar]
- Wang L, Reinach P, Lu L. TNF-alpha promotes cell survival through stimulation of K+ channel and NFkappaB activity in corneal epithelial cells. Exp Cell Res. 2005;311:39–48. doi: 10.1016/j.yexcr.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yang H, Tachado SD, Capo-Aponte JE, Bildin VN, Koziel H, Reinach PS. Phosphatase-mediated crosstalk control of ERK and p38 MAPK signaling in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006;47:5267–75. doi: 10.1167/iovs.06-0642. [DOI] [PubMed] [Google Scholar]
- Wang Z, Yang H, Zhang F, Pan Z, Capo-Aponte J, Reinach PS. Dependence of EGF-induced increases in corneal epithelial proliferation and migration on GSK-3 inactivation. Invest Ophthalmol Vis Sci. 2009;50:4828–35. doi: 10.1167/iovs.08-2983. [DOI] [PubMed] [Google Scholar]
- Xu KP, Ding Y, Ling J, Dong Z, Yu FS. Wound-induced HB-EGF ectodomain shedding and EGFR activation in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2004;45:813–20. doi: 10.1167/iovs.03-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu KP, Yin J, Yu FS. SRC-family tyrosine kinases in wound- and ligand-induced epidermal growth factor receptor activation in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006;47:2832–9. doi: 10.1167/iovs.05-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu KP, Yin J, Yu FS. Lysophosphatidic acid promoting corneal epithelial wound healing by transactivation of epidermal growth factor receptor. Invest Ophthalmol Vis Sci. 2007;48:636–43. doi: 10.1167/iovs.06-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Ueda T, Ugawa S, Ishida Y, Imayasu M, Koyama S, Shimada S. Functional expression of transient receptor potential vanilloid 3 (TRPV3) in corneal epithelial cells: involvement in thermosensation and wound healing. Exp Eye Res. 2010;90:121–9. doi: 10.1016/j.exer.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Yang H, Mergler S, Sun X, Wang Z, Lu L, Bonanno JA, Pleyer U, Reinach PS. TRPC4 knockdown suppresses epidermal growth factor-induced store-operated channel activation and growth in human corneal epithelial cells. J Biol Chem. 2005;280:32230–7. doi: 10.1074/jbc.M504553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang Z, Miyamoto Y, Reinach PS. Cell signaling pathways mediating epidermal growth factor stimulation of Na:K:2Cl cotransport activity in rabbit corneal epithelial cells. J Membr Biol. 2001;183:93–101. doi: 10.1007/s00232-001-0057-6. [DOI] [PubMed] [Google Scholar]
- Yin J, Yu FS. ERK1/2 mediate wounding- and G-protein-coupled receptor ligands-induced EGFR activation via regulating ADAM17 and HB-EGF shedding. Invest Ophthalmol Vis Sci. 2009;50:132–9. doi: 10.1167/iovs.08-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Yang H, Wang Z, Mergler S, Liu H, Kawakita T, Tachado SD, Pan Z, Capo-Aponte JE, Pleyer U, Koziel H, Kao WW, Reinach PS. Transient receptor potential vanilloid 1 activation induces inflammatory cytokine release in corneal epithelium through MAPK signaling. J Cell Physiol. 2007;213:730–9. doi: 10.1002/jcp.21141. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li H, Wang J, Dong Z, Mian S, Yu FS. Role of EGFR transactivation in preventing apoptosis in Pseudomonas aeruginosa-infected human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2004;45:2569–76. doi: 10.1167/iovs.03-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wu XY, Yu FS. Inflammatory responses of corneal epithelial cells to Pseudomonas aeruginosa infection. Curr Eye Res. 2005;30:527–34. doi: 10.1080/02713680590968150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Akhtar RA. Epidermal growth factor stimulates phospholipase D independent of phospholipase C, protein kinase C or phosphatidylinositol-3 kinase activation in immortalized rabbit corneal epithelial cells. Curr Eye Res. 1998;17:294–300. doi: 10.1076/ceyr.17.3.294.5223. [DOI] [PubMed] [Google Scholar]